

Abstract
Aldo-keto reductase family 1 member C3 has recently been regarded as a potential therapeutic target in castrate-resistant prostate cancer. Herein, we investigated whether berberine delayed the progression of castrate-resistant prostate cancer by reducing androgen synthesis through the inhibition of Aldo-keto reductase family 1 member C3. Cell viability and cellular testosterone content were measured in prostate cancer cells. Aldo-keto reductase family 1 member C3 mRNA and protein level were detected by RT-PCR and Western bolt analyses, respectively. Computer analysis with AutoDock Tools explored the molecular interaction of berberine with Aldo-keto reductase family 1 member C3. We found that berberine inhibited 22Rv1 cells proliferation and decreased cellular testosterone formation in a dose-dependent manner. Berberine inhibited Aldo-keto reductase family 1 member C3 enzyme activity, rather than influenced mRNA and protein expressions. Molecular docking study demonstrated that berberine could enter the active center of Aldo-keto reductase family 1 member C3 and form π-π interaction with the amino-acid residue Phe306 and Phe311. In conclusion, the structural interaction of berberine with Aldo-keto reductase family 1 member C3 is attributed to the suppression of Aldo-keto reductase family 1 member C3 enzyme activity and the inhibition of 22Rv1 prostate cancer cell growth by decreasing the intracellular androgen synthesis. Our result provides the experimental basis for the design, research, and development of AKR1C3 inhibitors using berberine as the lead compound.
Keywords: aldo-keto reductase family 1 member C3, androgen, berberine, castration-resistant prostate cancer
INTRODUCTION
Recently, more studies have focused on castration-resistant prostate cancer (CRPC) because of high mortality rates.1,2 One of the pivotal reasons for prostate cancer progression to CRPC is their acquired ability for intratumoral steroidogenesis from cholesterol or adrenal androgens.3,4,5 Abiraterone acetate was designed to inhibit steroidogenesis mediated by CYP17A1 (cytochrome P450 17A1), a key enzyme for intratumoral de novo steroid synthesis from cholesterol and production of progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens in the steroidogenic pathway.6,7 Clinical trials have shown that chemotherapy combined with abiraterone acetate treatments prolonged survival among patients with metastatic CRPC.8 However, CYP17A1 inhibitors have been associated with adrenocortical suppression and have led to an adrenocortical insufficiency because of the upstream blockage of steroidogenesis.9,10 Therefore, more effective prostate cancer therapy that avoids this adverse effect may be targeting downstream of the de novo steroid synthesis from cholesterol.
Aldo-keto Reductase Family 1 Member C3 (AKR1C3) is known to be a type 5 17-hydroxysteroid dehydrogenase (17β-HSD5) and is involved in the final two steps of steroid synthesis in human prostate cancer cells, which possesses reductase activity for the catalysis of low activity hormone precursors, androstenedione and androsterone, to highly active testosterone (T) and dihydrotestosterone (DHT), as Figure 1 depicted.11 Other works have also shown that AKR1C3 is overexpressed in localized, advanced or recurrent prostate cancers and the CRPC,12,13,14 and the expression levels of AKR1C3 were closely correlated with the Gleason grade of prostate cancer progression.15 The up-regulation of AKR1C3 was likely to be a survival adaptation to the T/DHT-deprived environment. Moreover, in vitro studies have shown that AKR1C3-overexpressed LNCaP prostate cancer cells (LNCaP-AKR1C3) were prone to generating significantly higher amounts of testosterone.16 Therefore, AKR1C3 is regarded to be a vital therapeutic target in the treatment of CRPC through suppressing intratumoral production of androgen.
Figure 1.
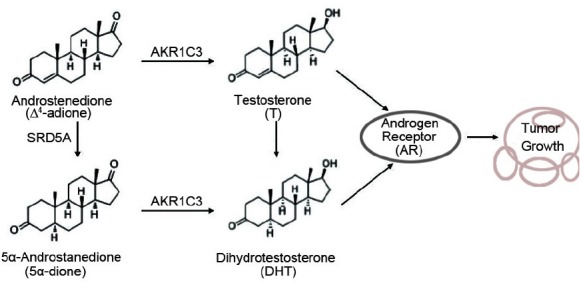
The synthesis of T/DHT under AKR1C3 catalysis. AKR1C3 catalyzes Δ4-adione to T and 5α-dione to DHT. SRD5A catalyzes Δ4-adione to 5α-dione and T to DHT. Then, T and DHT promote prostate cancer cell growth by activation of androgen receptor (AR).
In recent years, researchers have focused on the development of AKR1C3 inhibitors. A series of chemical compounds, such as medroxyprogesterone acetate (MPA), steroidal lactones, benzodiazepines, jasmonates, cinnamic acids, flavonoids, nonsteroidal anti-inflammatory drug (NSAIDs), and EM1404, were investigated for their efficacies in inhibiting the enzymatic activity of AKR1C3.17 However, more work needs to be done for these drugs in the clinical applications of CRPC. Therefore, the purpose of our study is to find out AKR1C3 inhibitor in “old,” nontoxic drugs. Berberine (2,3-methylenedioxy-9,10-dimenthoxyproto-berberine chloride; BBR), an isoquinoline alkaloid (Figure 2), was screened from a traditional Chinese medicine (TCM) monomer library and had been used as an anti-diarrheal agent for hundreds of years in China; Recently, it has been demonstrated possession of high antitumor activities against prostate cancer.18,19,20 Our prior study found that BBR could delay the latent periods to the progression of CRPC in castrated nu/nu mice bearing a subcutaneous LNCaP xenograft.19 However, the blocking mechanism by which BBR prevents AKR1C3-mediated intratumoral steroidogenesis in the inhibition of the development of CRPC has yet to be elucidated.
Figure 2.
Berberine (BBR) chemical structure.
Herein, we first investigated the inhibitory capacity of BBR on a human recombinant AKR1C3 enzyme in vitro and the effect of BBR on the cell proliferation. In addition, we studied androgen synthesis in the AKR1C3-overexpressing cell line 22Rv1. Finally, we used AutoDock Tools to elucidate the molecular interactions between BBR and AKR1C3. This work provides new insights on strategies for the prevention and treatment of CRPC.
MATERIALS AND METHODS
Materials
Androstendione, NADPH, and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA). Recombinant human AKR1C3 enzymes were obtained from (PROSPEC: Cat#enz-406, Ness-Ziona, Israel). The primary anti-AKR1C3 (NP6.G6.A6, mouse monoclonal antibody, 1:150) was purchased from Abcam Inc., (Cambridge, MA, USA) and anti-β-actin (mouse polyclonal antibody, 1:1000) was purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX, USA). The secondary antibody was HRP-conjugated anti-mouse IgG (1:10 000, Gaithersburg, MD, USA). Human LNCaP, PC3, PC3M cells and 22Rv1 were obtained from the American Type Culture Collection (Manassas, VA, USA). Tris–HCl gel, polyvinylidene difluoride (PVDF) membranes, and nonfat dry milk were purchased from Bio-Rad Laboratories, Inc., CA, USA.
Methods
Screening for AKR1C3 expression human prostate cancer cell
Cellular proteins were prepared from LNCaP cells, PC3 cells, PC3M cells and 22Rv1 cells in a modified RIPA buffer (50 mmol l−1 Tris–HCl, pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 mmol l−1 NaCl, 1 mmol l−1 EDTA and 2 mmol l−1 phenylmethylsulfonyl fluoride). Total soluble proteins (50 μg) were electrophoresed on a 12% Tris–HCl gel. Proteins were transferred onto PVDF membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 1% Tween 20. Antigens were detected by incubating with mouse anti-human AKR1C3 mAb (1:500) or anti-β-actin mAb (1:5000) at room temperature for 2 h followed by incubation with HRP-conjugated anti-mouse IgG (1:10 000, Gaithersburg, MD, USA) secondary antibody at room temperature for another 1 h. Immunoreactive protein was then detected using enhanced chemiluminescence reagent (Pierce, Rockford, IL, USA) according to the manufacturer's protocol.
Cell proliferation assay by MTT
The cells were maintained in RPMI 1640 medium supplemented with 10% FBS, 2 mmol l−1 glutamine, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin at 37°C in a humidified incubator containing 5% CO2. MTT assay was performed in 96-well plates in quintuple. Cells were seeded at a density of 1.5 × 104 cells/well overnight, and the medium was changed to RPMI 1640 with 10% charcoal-dextran stripped serum (CSS) and BBR (12.5 μmol l−1, 25 μmol l−1 and 50 μmol l−1) for 48 h with or without 0.1 μmol l−1 androstendione; 30 μmol l−1 indomethacin was used as positive control. Data are expressed as the means ± s.d. of at least three independent experiments.
Determination of testosterone formation
22Rv1 cells were seeded into 6-well plates in duplicate at a density of 2.5 × 105 cells/well overnight, and the cells were divided into three groups, including the negative control group (solvent only), the BBR group (25 μmol l−1) and the INN group (30 μmol l−1). In the cellular testosterone formation experiment, finasteride, an inhibitor of 5a-reductase (SRD5A1), was used to blocked metabolism of 0.1 μmol l−1 androstendione to 5a-andostane-3,17-dione and block T to the formation of DHT. Simply, cells were treated with chemicals for 24 h, and then androstendione was added to the medium with a final concentration of 0.1 μmol l−1 in each group. Then, the medium of the culture was separated, and the metabolism of androstendione was analyzed by determining the concentration of testosterone in the medium using chemiluminescence. Data are expressed as the means ± s.d. of at least three independent experiments.
AKR1C3 mRNA expression analysis using reverse transcription PCR (RT-PCR)
22Rv1 cells were divided into solvent control (DMSO) and BBR (25 μmol l−1). Total RNA was extracted from the cells using the Trizol reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions. First-strand cDNA was generated by reverse transcription of 5 μg RNA samples using a one-step gDNA removal and cDNA synthesis supermix (Transgene biotech, Beijing, China). One-tenth of the reverse-transcribed RNA was used in the PCR reaction. Absolute gene transcription was normalized to β-actin. The primer sequences were as follows: β-actin sense: 5’-dATCTGGCACCACACCTTCTACAATGAGCTGCG-3’ and antisense: 5’-dCGTCATACTCCTGCTTGCTGATCCACATCTGC-3’; AKR1C3 sense: 5’-dGTAAAGCTTTGGAGGTCAC-3’ and antisense: 5’-dCACCCATCGTTTGTCTCGT-3’. The PCR products were separated using electrophoresis on 1% agarose gels containing ethidium bromide. The PCR products were visualized using a Tanon-1600 figure gel image processing system and analyzed with a GIS 1D gel image system software (Tanon, Shanghai, China). For Western blot analysis, cellular proteins were prepared and performed as described in Screening for AKR1C3 expression human prostate cancer cell for 22Rv1 cells.
AKR1C3 protein expression by Western blot analysis
22Rv1 cells were divided into solvent control (DMSO) and BBR (25 μmol l−1). The protein was prepared from cells treated with BBR for 48 h. Protein extraction, quantification and Western blot steps were conducted the same as the screening for AKR1C3 expression human prostate cancer cells.
Enzyme activity assays
Enzyme activity was monitored in 200 μl volumes containing 6.25–100 μmol l−1 androstendione in 4% (v/v) DMSO, 0.2 mmol l−1 NADPH, 100 mmol l−1 potassium phosphate buffer (pH 7.0) and human recombinant AKR1C3 enzyme. The IC50 values for AKR1C3 inhibitors were carried out in a range of eight inhibitor concentrations (0.39–50 μmol l−1) with 50 μmol l−1 androstendione in the same reaction mixture mentioned above. All reactions were initiated by the addition of enzyme. The activities of the enzymes were determined at 37°C by measuring the rate of change in NADPH fluorescence (at 455 nm with an excitation wavelength of 340 nm). A standard curve was constructed by monitoring fluorescence changes with incremental additions of NADPH. Indomethacin was used as a positive control in the inhibition studies.
Molecular modeling
To estimate the potential interaction and the conformation of the protein-ligand complex, inhibitors were docked into the AKR1C3 protein using the AutoDock 4.2 program based on Lamarkian Genetic Algorithm (Scripps Research Institute, La Jolla, CA, USA). The amino acid sequence of AKR1C3 was collected from the UniProtKB (P42330), in which 323 amino acid residues were involved. The atomic coordinates for the AKR1C3-NADP+ -E1404 acid complex (PDB code 1ZQ5) were obtained from the RCSB Protein Data Bank. The 3D structure of inhibitors was acquired from the Internet (http://zinc.docking.org/). The predicted complexes were optimized and ranked according to the empirical scoring function, ScreenScore, which estimated the binding free energy of the ligand-receptor complex. Each docking was performed twice, and each operation screened 250 conformations for the protein-ligand complex that were advantageous for docking; each docking had 500 preferred conformations. The most stable conformation had the minimal binding energy as shown by Discovery Studio (DS) 3.5 Visualizer (Accelrys, Inc).
Statistical analysis
Statistical evaluation of the data was performed using independent Student's t-test and ANOVA followed by Fisher's test. A P value < 0.05 was considered statistically significant. IC50 was calculated by probit analysis using SPSS (SPSS Inc., Chicago, IL, USA).
RESULTS
Characterization of AKR1C3 protein expression in prostate cancer cell lines
To find a model to mimic the CRPC state, we screened for a prostate cancer cell line with high AKR1C3 expression. The characterization of AKR1C3 protein expression was conducted for a panel of prostate cancer cell lines, including LNCaP cells, PC3 cells, PC3M cells and 22Rv1 cells. Western blot analyses confirmed that the expression of AKR1C3 was relatively strong in PC3M cells and 22Rv1 cells (Figure 3). Because the 22Rv1 cell line was derived from the androgen-dependent 22Rv1 cells and has castration-resistant prostate cancer characteristics, the following experiments were carried out with 22Rv1 cells.
Figure 3.
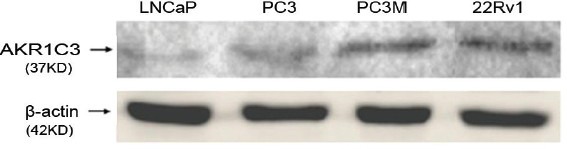
Protein expression of AKR1C3 in human prostate cancer cell lines.
Cell proliferation assay in 22Rv1 cells
To investigate the dose-effect relationship of BBR on 22Rv1 cell growth, an MTT assay was carried out using a 48 h treatment with three concentrations of BBR (12.5 μmol l−1, 25 μmol l−1 and 50 μmol l−1). The results showed that BBR exhibited a dose-dependent inhibition on the proliferation of 22Rv1 cells at 48 h. When 0.1 μmol l−1 androstenedione was added to the culture medium, the inhibitive effect of BBR on cell proliferation was partially counteracted, indicating that the additional androstenedione increased the synthesis of androgen and indirectly reversed the BBR effect (Figure 4).
Figure 4.
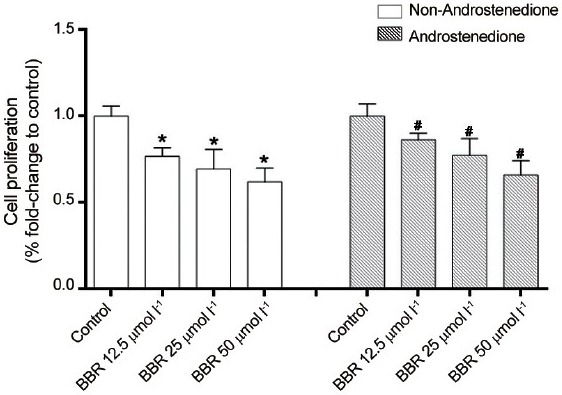
Effect of berberine (BBR) on 22Rv1 cell proliferation by MTT assay in conditions with or without androstenedione (4-adione) addition. *and #P < 0.05, compared with their control groups.
BBR on testosterone formation in 22Rv1 cells
To explore whether BBR influenced androgen synthesis, cellular T production was measured after the treatment with 25 μmol l−1 BBR for 48 h in 22Rv1 cells. As shown in Figure 1, T and DHT are the primary androgen species stimulating prostate cell growth, which are converted from Δ4 -adione and 5a-dione by AKR1C3 catalysis, respectively. Herein, we used a potent 5a-reductase inhibitor, finasteride, to block conversion of Δ4 -adione to 5a-dione and also block T to the formation of DHT, respectively. The aim is to confirm the efficacy of BBR on the inhibition of AKR1C3 activity. The results showed that after blockage of 5a-reductase, finasteride with the final concentration of 2 μmol l−1 increased T production approximately 2.19 folds to Control group. As shown in Figure 5, BBR and INN significantly reduced the formation of total testosterone, even slightly lower to the finasteride (P < 0.05).
Figure 5.
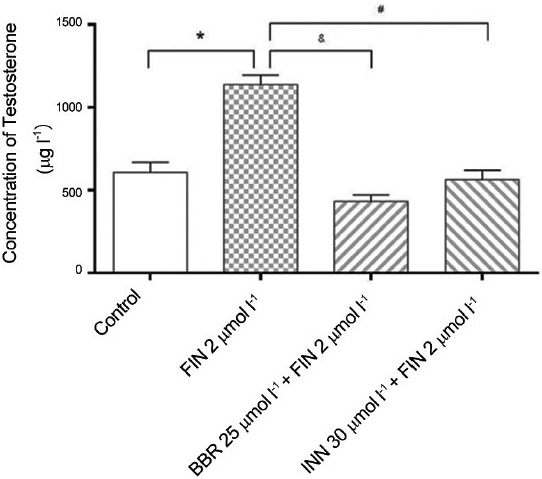
The influence of berberine on the formation of testosterone. *P < 0.05, compared with the control group. #and &P < 0.05, compared with the finasteride group.
Effects of BBR on AKR1C3 gene expression
To investigate whether BBR influences gene expression, RT-PCR and Western blot analysis were carried out. The results showed that BBR did not change the mRNA level or the protein level after the treatment of 22Rv1 cells with 25 μmol l−1 BBR for 48 h (Figure 6). This indicates that BBR did not influence the transcription or expression of the AKR1C3 gene.
Figure 6.
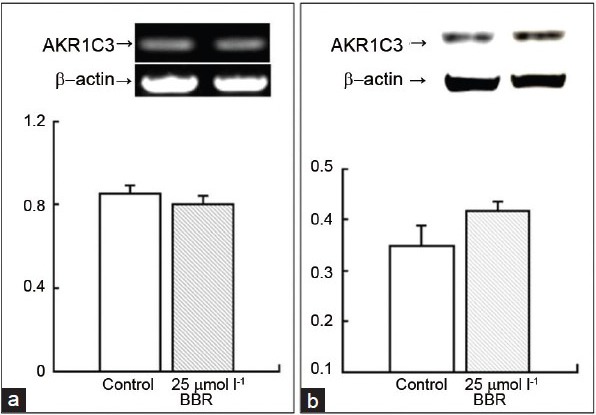
The influence of BBR on AKR1C3 in the level of mRNA and protein. (a) RT-PCR for AKR1C3 gene, the upper panel shows the mRNA bands for the control group and the BBR group, and the lower panel shows the bars for their quantification. (b) Western blot for AKR1C3 gene, the upper panel shows the protein bands for the control group and the BBR group, and the lower panel shows bars for their quantification.
In vitro inhibition of AKR1C3 enzyme activity
The aforementioned results demonstrated that AKR1C3 did not change the mRNA expression or the protein expression after a 48 h treatment with 25 μmol l−1 BBR. However, along with cell proliferation, the intracellular testosterone production significantly reduced. Therefore, we inferred that the inhibition of enzymatic activity by AKR1C3 was the main reason for the suppression of prostate cancer cell growth. Regarding the AKR1C3-catalyzed reduction of Δ4-adione to T, we examined the inhibition of BBR on AKR1C3 reductive enzyme activity by using Δ4-adione as a substrate and INN as a positive control. It is shown that both BBR and INN displayed potent inhibitory effects on the activity of recombinant human AKR1C3 (Figure 7a and 7b). The IC50 values for BBR was 4.08 μmol l−1 with a 95% Confidence Intervals of 3.47 μmol l−1 to 4.69 μmol l−1. The IC50 values for INN was 7.26 μmol l−1 with a 95% Confidence Intervals of 6.82 μmol l−1 to 7.70 μmol l−1. The IC50 value for BBR was much lower than that for INN implicating that BBR had higher efficacy in inhibiting the activity of AKR1C3.
Figure 7.
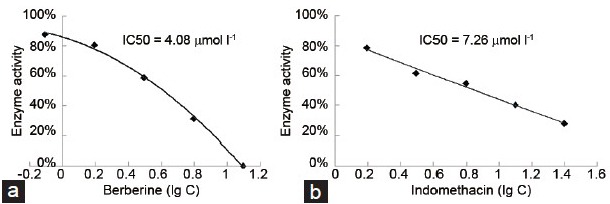
The enzyme inhibition curve of (a) BBR and (b) INN.
Molecular docking studies
To gain insight into interactions inside the enzyme binding sites of BBR, we used AutoDock 4.2 to observe the optical interactions of BBR (Figure 8 Ia–Ic) with the cavity center of AKR1C3. Two control drugs were involved in the analysis, INN (Figure 8 IIa–IIc) and EM1404 (Figure 8 IIIa–IIIc), which are potent AKR1C3 inhibitors. Our data showed that BBR could enter the steroid/inhibitor binding cavity of AKR1C3 and form p-p interactions with amino-acid residues Phe306 and Phe311 (Figure 8 Ic). To identify the binding forces contributing to the interaction between the inhibitors and AKR1C3, the estimated free energy of binding (FEB), which predicts the Van der Waals energy, electrostatic energy, hydrogen-bond energy, de solv energy, final total internal energy, torsional free energy, and unbound system energy, were calculated. As estimated by Discovery Studio (DS) 3.5 Visualizer software, the most stable conformation exhibited the lowest FEB. The binding energies of BBR, INN, and EM1404 were −9.63, −9.79, and −12.55 kcal mol−1, respectively (Table 1). These results provided theoretical support for and explanation of the effects of BBR on AKR1C3 activity.
Figure 8.
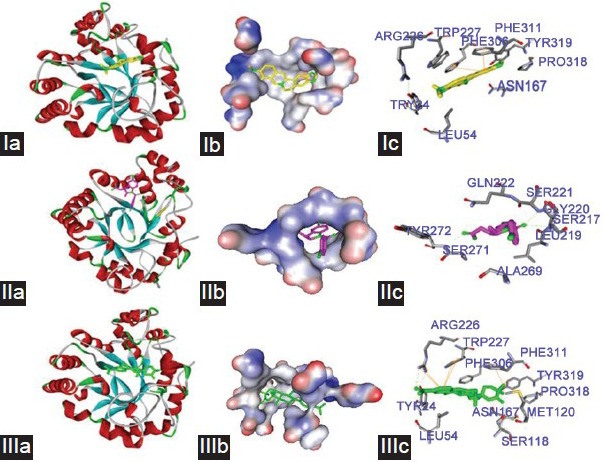
The interaction of BBR (I), INN (II) and EM1404 (III) with AKR1C3. BBR (yellow), INN (pink) and EM1404 (green). (a) The optimize graphic performance of ligands with AKR1C3. (b) The created surface around ligands in the active center of AKR1C3. (c) Ligand binding site atoms of AKR1C3. Hydrogen bonds and π-π interaction formed between BBR (I), INN (II) and EM1404 (III) with the active center of AKR1C3. Hydrogen bonds are shown by green dash lines. π-π interactions are shown by yellow solid line.
Table 1.
The binding energy of BBR, INN and EM1404 with AKR1C3

DISCUSSION
Recently researchers demonstrated that prostate cancer cells acquired ability of intratumoral steroidogenesis from cholesterol or adrenal androgens in the absence of blood androgens, which made prostate cancer evolve from an endocrine disease to an autocrine/paracrine one.3,5 AKR1C3 is a key steroidogenic enzyme to catalyze the conversion of low active androstenedione and androsterone hormone precursors to highly active testosterone and dihydrotestosterone in steroid synthesis pathway.11 In recent, accumulating amount of data reported that AKR1C3 is associated with the transformation from hormone-dependent prostate cancer to CRPC.11,15 This concept has led to the increasing interest in the development of AKR1C3 inhibitors. Until now, several AKR1C3 inhibitors, such as ASP9521and SN33638, were evaluated in multi-center phase I/II study in patients with metastatic CRPC or in high AKR1C3-expressing cell lines. The results suggested that a subpopulation of CRPC patients with high AKR1C3 might benefit from AKR1C3 inhibitor therapy.21,22
BBR, as an isoquinoline alkaloid, has high capacity and spectrum for various tumors. Recent studies have found that BBR displays potent antitumor activity for prostate cancer without toxicity.19,23 In this research, BBR exhibited a dose-dependent inhibition on the proliferation of 22Rv1 cells at 48 h without influencing the mRNA level or the protein level of AKR1C3, but significantly reducing the formation of total testosterone. In enzyme assays, BBR displayed potent inhibition on the activity of recombinant human AKR1C3 with lower IC50 value than that for INN, implicating that BBR had higher efficacy in inhibiting the activity of AKR1C3. Some studies had shown that CRPC cells were prone to generate significantly higher amounts of T, rather than DHT to promote prostate cancer growth.5,11,24 Therefore, AKR1C3 is regarded as a vital therapeutic target in the treatment of CRPC. In cell proliferation assay, BBR exhibited dose-dependent growth inhibition on high AKR1C3-expressing 22Rv1 cell, but INN had only a limited effect on the cell growth mediated by Δ4 -adione. After blocking AKR1C3, prostate cancer cells reverted to their overall androgen metabolic profile from the conversion Δ4 -adione to the 5a-dione, which bound with the mutated AR to stimulate androgen signaling pathway.16 Therefore, the desired therapeutic effect of AKR1C3 inhibitors could be weakening in this circumstance. In our previous study, we confirmed the BBR also induced the truncated AR splice variants degradation. Consequently, BBR could block cell proliferation either by intratumoral steroidogenesis or by AR antagonist. That's why the more potent effect was shown in BBR than that of INN.
In order to better understanding the molecular mechanism of BBR on AKR1C3, we used AutoDock Tools to study the molecular interaction between BBR and AKR1C3. According to the X-ray crystal structure, AKR1C3 consists of 323 amino acids, which form a (β/a) 8-barrel structure with a large, multi-cavity active site that exhibits flexibility at the level of individual side chains and entire loop regions on ligand binding.25 AKR1C3 catalytic sites were consist of five compartments, an oxyanion site (formed by Tyr55, His117, and NADP+), a steroid channel (Trp227 and Leu54), and three subpockets, SP1 (Ser118, Asn167, Phe306, Phe311, and Tyr319), SP2 (Trp86, Leu122, Ser129, and Phe311), and SP3 (Tyr24, Glu192, Ser221, and Tyr305).17 The difference among the AKR1C enzymes lies in the three subpockets. The SP1 pocket is expected to give more potent and selective AKR1C3 inhibitors.17,26 Structural differences at position 306 in SP3 and 311 in SP2 make the AKR1C3 differ from the other enzymes.17 Our data show that BBR could enter the steroid/inhibitor binding cavity of AKR1C3 and formed p-p interaction in the amino-acid residue Phe306 and Phe311. Although further study should be carried out to certificate this interaction, this molecular docking theoretically gave the supports and explanation on the effects of BBR on AKR1C3 enzymatic activity.
CONCLUSIONS
The present findings reveal that BBR could decrease intracellular androgen synthesis due to the suppression of AKR1C3 enzyme activity. The mechanisms of BBR on CRPC provide a basis on the design, research, and development of future AKR1C3 inhibitors using BBR as the lead compound.
AUTHOR CONTRIBUTIONS
JL conceived the study, coordinated the experimental design of the study. YTT and LJZ carried out the critical revision of results, analyzed the data and drafted the manuscript. YW carried out the computer analysis. DX carried out an in vitro pharmacology study. HTZ, YL, and XJZ helped to draft and to revise the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (81302206 and 81560422), the Development and Reform Commission of Jilin Province (2013C026-2), and the Young Scholars Program of Norman Bethune Health Science Center of Jilin University (2013201012), the Health and Family Planning Commission of Jiangxi Province (20143207) and the Natural Science Foundation of Jiangxi Province of China (20151BAB205016 and 20132BAB205008).
REFERENCES
- 1.Titus M, Tomer KB. Androgen quantitation in prostate cancer tissue using liquid chromatography tandem mass spectrometry. Methods Mol Biol. 2011;776:47–57. doi: 10.1007/978-1-61779-243-4_3. [DOI] [PubMed] [Google Scholar]
- 2.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 3.Mostaghel EA, Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab. 2008;22:243–58. doi: 10.1016/j.beem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 5.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pezaro CJ, Mukherji D, De Bono JS. Abiraterone acetate: redefining hormone treatment for advanced prostate cancer. Drug Discov Today. 2012;17:221–6. doi: 10.1016/j.drudis.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Rehman Y, Rosenberg JE. Abiraterone acetate: oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. Drug Des Devel Ther. 2012;6:13–8. doi: 10.2147/DDDT.S15850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 10.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, et al. Hormonal impact of the 17alpha-hydroxylase/C (17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–25. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013;137:136–49. doi: 10.1016/j.jsbmb.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura Y, Suzuki T, Nakabayashi M, Endoh M, Sakamoto K, et al. In situ androgen producing enzymes in human prostate cancer. Endocr Relat Cancer. 2005;12:101–7. doi: 10.1677/erc.1.00914. [DOI] [PubMed] [Google Scholar]
- 13.Pelletier G, Luu-The V, El-Alfy M, Li S, Labrie F. Immunoelectron microscopic localization of 3beta-hydroxysteroid dehydrogenase and type 5 17beta-hydroxysteroid dehydrogenase in the human prostate and mammary gland. J Mol Endocrinol. 2001;26:11–9. doi: 10.1677/jme.0.0260011. [DOI] [PubMed] [Google Scholar]
- 14.Lapouge G, Marcias G, Erdmann E, Kessler P, Cruchant M, et al. Specific properties of a C-terminal truncated androgen receptor detected in hormone refractory prostate cancer. Adv Exp Med Biol. 2008;617:529–34. doi: 10.1007/978-0-387-69080-3_53. [DOI] [PubMed] [Google Scholar]
- 15.Tian Y, Zhao L, Zhang H, Liu X, Zhao X, et al. AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn Pathol. 2014;9:42. doi: 10.1186/1746-1596-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrns MC, Mindnich R, Duan L, Penning TM. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5alpha-reductase inhibitor finasteride. J Steroid Biochem Mol Biol. 2012;130:7–15. doi: 10.1016/j.jsbmb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Liu Q, Liu Z, Li B, Sun Z, et al. Berberine, a genotoxic alkaloid, induces ATM-Chk1 mediated G2 arrest in prostate cancer cells. Mutat Res. 2012;734:20–9. doi: 10.1016/j.mrfmmm.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Cao B, Liu X, Fu X, Xiong Z, et al. Berberine suppresses androgen receptor signaling in prostate cancer. Mol Cancer Ther. 2011;10:1346–56. doi: 10.1158/1535-7163.MCT-10-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantena SK, Sharma SD, Katiyar SK. Berberine, a natural product, induces G1-phase cell cycle arrest and caspase-3-dependent apoptosis in human prostate carcinoma cells. Mol Cancer Ther. 2006;5:296–308. doi: 10.1158/1535-7163.MCT-05-0448. [DOI] [PubMed] [Google Scholar]
- 21.Loriot Y, Fizazi K, Jones RJ, Van den Brande J, Molife RL, et al. Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs. 2014;32:995–1004. doi: 10.1007/s10637-014-0101-x. [DOI] [PubMed] [Google Scholar]
- 22.Yin YD, Fu M, Brooke DG, Heinrich DM, Denny WA, et al. The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17beta-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front Oncol. 2014;4:159. doi: 10.3389/fonc.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi MS, Oh JH, Kim SM, Jung HY, Yoo HS, et al. Berberine inhibits p53-dependent cell growth through induction of apoptosis of prostate cancer cells. Int J Oncol. 2009;34:1221–30. [PubMed] [Google Scholar]
- 24.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 25.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3 alpha-hydroxysteroid dehydrogenase reveals a “push-pull” mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–48. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 26.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, et al. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–8. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]