

Abstract
Objective:
We prospectively screened a large European cohort of patients presenting with hyperCKemia and/or limb-girdle muscular weakness (LGMW) for acid α-glucosidase (GAA) deficiency by dried blood spot (DBS) investigation.
Methods:
DBS were collected from 3,076 consecutive adult patients from 7 German and British neuromuscular centers. All specimens were investigated for GAA deficiency by fluorometry. Samples with reduced enzyme activity were subsequently investigated for GAA gene mutations.
Results:
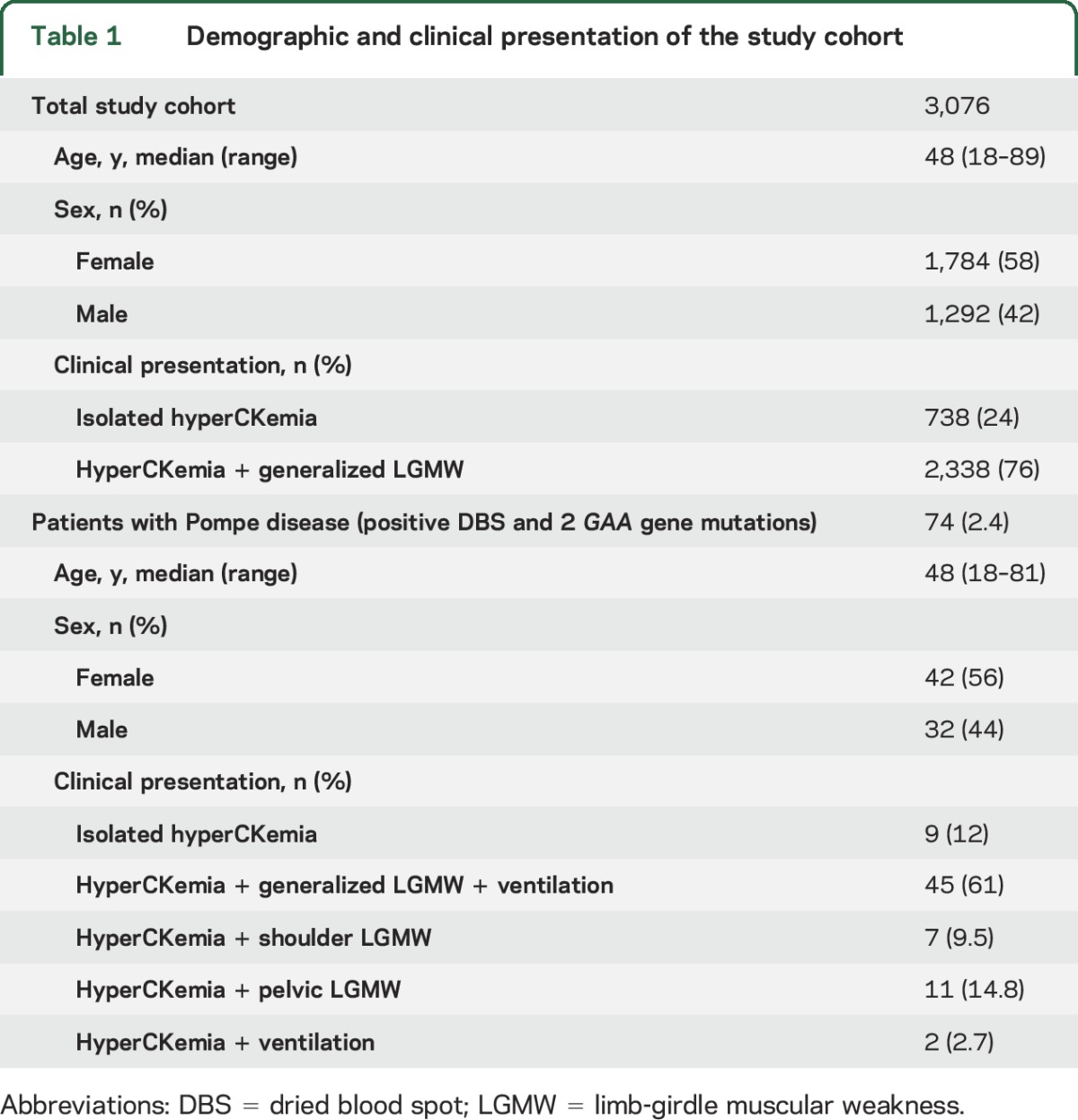
Of 3,076 patients with DBS samples, 232 patients (7.6%) showed low GAA enzyme activity. Of these 232 patients, 55 (24%) presented with isolated hyperCKemia and 176 (76%) with hyperCKemia and LGMW. With both features present, 94% of the patients showed a low enzymatic activity. Mutational analysis found GAA gene mutations in 74 patients (2.4%); herein 70 patients were heterozygote for the common GAA gene splice-site mutation c.-32-13T>G. The most common clinical presentation in the confirmed Pompe cohort was a limb-girdle phenotype (85.3%) combined with ventilatory insufficiency (61%). Isolated hyperCKemia was found in 12%, while 2.7 had hyperCKemia and ventilatory insufficiency only.
Conclusions:
In a large cohort of unselected adult patients with hyperCKemia and/or LGMW, we found a prevalence of late-onset Pompe disease of 2.4%. Therefore, targeted screening of such a population should be encouraged in clinical practice.
Pompe disease, also termed glycogen storage disease type II or acid maltase deficiency, is a multisystemic metabolic disease caused by deficiency of the lysosomal enzyme acid α-1,4-glucosidase (GAA).1 Late-onset Pompe disease has a variable age at onset and phenotype. In most patients, the presenting symptom is axial and proximal skeletal muscle weakness; however, morbidity and mortality are in particular related to the ventilatory insufficiency.2 The advent of enzyme replacement therapy has arguably increased the need for awareness of the condition. However, late-onset Pompe disease presents with a wide spectrum of symptoms including pauci-neuromuscular symptoms at onset, and the diagnosis can be easily overlooked.3 Delay in diagnosis has been reported to be on average 7 to 10 years.4
METHODS
The participants were prospectively identified through medical examination at 7 in- and outpatient neuromuscular clinics in Germany (Munich, Halle) and the United Kingdom (London, Newcastle, Oswestry, Oxford, Salford) between the years 2009 and 2014.
Inclusion criteria.
Consecutive patients with unclassified proximal and/or axial limb-girdle muscular weakness (LGMW) and/or unexplained sustained elevation of serum creatine kinase (CK) levels were included. We defined the LGMW as weakness of the proximal muscles in the arms and legs, including the muscles of the shoulders, upper arms, axial paravertebral area, pelvic area, and thighs.1–3 Repeated CK elevation as an inclusion criterion was defined as a minimum of 2× the upper limit of normal for women (<180 IU/L) and men (<200 IU/L).
Sampling and fluorometry.
A blood sample was collected from a peripheral vein in an EDTA-coated tube, and blood was immediately afterward spotted onto a filter paper (Whatman 903 Specimen Collection Paper). All samples were anonymized and analyzed at the Metabolic Laboratory, Institute of Clinical Chemistry, Hamburg University Medical Centre, Germany. A fluorometric assay was used for analysis.5
Statistics.
For descriptive statistical analyses, the statistics program SPSS for Windows version 23.0 (IBM Corp., Armonk, NY) was used.
Gene sequencing.
Sanger sequencing of the coding and exon flanking region of the GAA gene was performed if the DBS activity of GAA was below 0.9 nmol/punch × 21 hours in 2 independently taken DBS samples.
Standard protocol approvals, registrations, and patient consents.
Oral and written informed consent was obtained from all participants before blood sampling. The study was conducted in accordance with the Declaration of Helsinki. The study was approved by the ethics committee of the Ludwig Maximilian University of Munich, Germany (no. 201-09).
RESULTS
The cohort included 3,076 patients aged between 18 and 89 years with a median of 48 years of participation (58.2% women). The demographic and clinical presentation of the studied cohort is summarized in table 1. GAA activity results were within the normal range for 2,844 patients (92.2%). The average value of CK levels in all patients was 563 (SD 371) IU/L (normal range, 15,000 IU/L). DBS activity of GAA was below the cutoff of 0.9 nmol/punch × 21 hours in 232 patients with a median age of 40.7 years. On average, the DBS GAA activity with the specific inhibitor acrabose was 1.73 nmol/punch × 21 hours (median 1.22 nmol/punch × 21 hours) in nonaffected individuals. In contrast, all 74 patients with finally confirmed Pompe disease showed an average GAA activity with inhibitor of 0.18 nmol/punch × 21 hours (median 0.12 nmol/punch × 21 hours). Forty-two women (56%) and 32 men were finally diagnosed by mutational analysis as having late-onset Pompe disease. Sequencing of the GAA gene revealed 2 heterozygous known pathogenic mutations, including the common GAA gene splice-site mutation c.-32-13T>G on one allele in 70 patients, confirming the diagnosis. One hundred fifty-eight possible carriers of Pompe disease were also identified by borderline GAA enzyme activity; however, a second DBS test had normal GAA enzyme activities in 123 patients. An alternative diagnosis was finally established in 52 of the 123 patients (recessive limb-girdle dystrophies [n = 32], Becker muscular dystrophies [n = 4], myotonic dystrophy type 2 [n = 8], myositis [n = 6], limb-girdle myasthenic syndrome [n = 1], spinal muscular atrophy type 3 [n = 1]). In 95 patients, the borderline GAA enzyme activity and diagnosis remained unsolved. In 24 of these 95 patients, a second DBS test could not be performed. Eleven patients were found to be heterozygous for the common splice-site GAA gene mutation c.-32-13T>G without detection of a second GAA gene mutation.
Table 1.
Demographic and clinical presentation of the study cohort
DISCUSSION
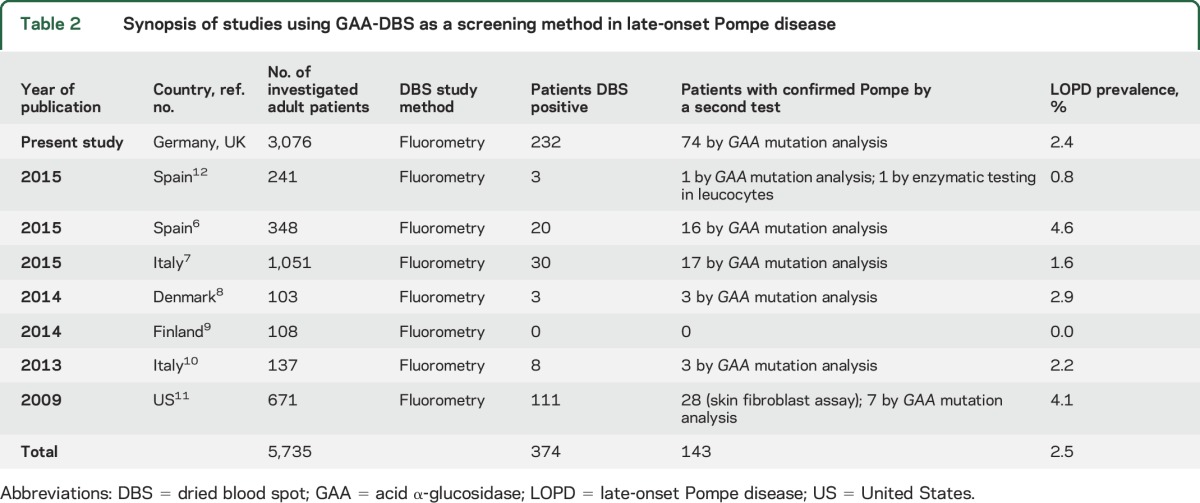
This is the largest prospective cohort screening for late-onset Pompe disease in a population of unclassified muscle diseases so far. The screening efficacy of DBS has been described in newborn screening and other smaller focused studies in Pompe disease (table 2).6–12 All 74 of our newly diagnosed patients with Pompe disease revealed the clinical key elements of late-onset Pompe disease (table 1). In our study, the main finding is the prevalence of adult Pompe disease of 2.4% in patients with unclassified LGMW and/or hyperCKemia. This finding is in accordance with smaller published studies, with a combined prevalence of 2.5%.6–12 Although using similar fluorometric assays, all 8 DBS screening studies show a slight difference in the prevalence. These differences may reflect country-specific differences in the frequency of a rare disease. In the Black American, Dutch, and Taiwanese populations, there is known to be a higher prevalence of Pompe disease compared to other countries worldwide.1,4 Deficient DBS GAA activity cannot always be confirmed on analysis of a second sample. Known reasons include blood draw after transfusion, incorrect DBS spotting and sampling, and environmental circumstances such as high temperature during transport. Even in this study of experienced centers, errors in spotting the blood or environmental conditions cannot be completely excluded. However, the internal control of the DBS assay by measurement of neutral maltase activity was performed in all assays and was acceptable in the first sample for our patients. There is still a chance of a false-positive result; therefore, we recommend that DBS GAA deficiency be confirmed using a second tissue, e.g., fibroblasts, muscle, or the gold standard of GAA Sanger gene sequencing. Classically, muscle biopsy in late-onset Pompe disease shows autophagic vacuolar myopathic changes, glycogen accumulation, and acid phosphatase–positive inclusions. The latter may be seen without obvious glycogen accumulation. However, it is well recognized by neuromuscular specialists that glycogen accumulation and acid phosphatase inclusions may be absent, and if so, that should not preclude DBS analysis. Arguably, DBS could be performed before biopsy whenever there is a clinical suspicion of Pompe disease.
Table 2.
Synopsis of studies using GAA-DBS as a screening method in late-onset Pompe disease
Given the relative rarity of late-onset Pompe disease, the relatively nonspecific clinical features of proximal weakness and elevated CK, and the frequent absence of diagnostic muscle biopsy findings, even experienced neuromuscular clinicians may not consider the diagnosis.
On the basis of our findings, we recommend that a DBS test be undertaken early in the diagnostic workup of patients with unclassified LGMW or persistent hyperCKemia to exclude late-onset Pompe disease. Key clinical elements that should alert clinicians to perform a DBS screening for late-onset Pompe disease are (1) axial, pelvic, and respiratory muscle involvement, and (2) persistent CK elevation.
Finally, there is growing evidence to suggest that early treatment of late-onset Pompe disease with enzyme replacement therapy, before there is extensive, possibly irreversible muscle damage, is more efficacious than later treatment, emphasizing the importance of early diagnosis.1,7,11,13
ACKNOWLEDGMENT
The authors thank all participants and the patient support groups for their ongoing help and commitment, and Drs. P. Schneiderat and J. Schessl for contributing patients to this study.
GLOSSARY
- CK
creatine kinase
- DBS
dried blood spot
- GAA
acid α-glucosidase
- LGMW
limb-girdle muscular weakness
AUTHOR CONTRIBUTIONS
Z. Lukacs and B. Schoser: design of study, analysis, acquisition and interpretation of all data, and drafting of the manuscript. T.A. Willis, T. Evangelista, M. Roberts, M. Guglieri, R. Quinlivan, D. Hilton-Jones, V. Straub: acquisition and interpretation of data from the UK and critical revision of the manuscript. P. Nieves Cobos, S. Wenninger, S. Zierz, B. Schlotter-Weigel, M.C. Walter, P. Reilich, T. Klopstock, M. Deschauer, W. Müller-Felber: acquisition of data in Germany and critical revision of the manuscript.
STUDY FUNDING
This study was supported in part by an unrestricted research grant from Genzyme, a Sanofi company, to Dr. Lukacs, Hamburg, Germany.
DISCLOSURE
Z. Lukacs has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. P. Nieves Cobos, S. Wenninger, T. Willis, and T. Evangelista report no disclosures relevant to the manuscript. M. Roberts has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. M. Guglieri reports no disclosures relevant to the manuscript. R. Quinlivan has received honoraria from Genzyme and research funding and honoraria from PTC bio. D. Hilton-Jones, S. Zierz, B. Schlotter-Weigel, M. Walter, and P. Reilich report no disclosures relevant to the manuscript. T. Klopstock has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. M. Deschauer has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. V. Straub has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. Dr. Straub is a member of the Genzyme Pompe Disease Global Advisory Board. W. Müller-Felber has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. B. Schoser has received research support, honoraria, and travel funding from Genzyme, a Sanofi company, during the past 5 years. Dr. Schoser is a member of the Genzyme Pompe Disease Global Advisory Board. Dr. Schoser received honoraria and travel funding from BioMarin Pharmaceutical and Audentes Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet 2008;372:1342–1353. [DOI] [PubMed] [Google Scholar]
- 2.Güngör D, de Vries JM, Hop WC, et al. Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy. Orphanet J Rare Dis 2011;6:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schüller A, Wenninger S, Strigl-Pill N, Schoser B. Toward deconstructing the phenotype of late-onset Pompe disease. Am J Med Genet C Semin Med Genet 2012;160C:80–88. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Amartino HM, Lindberg C, et al. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A 2013;161A:2431–2443. [DOI] [PubMed] [Google Scholar]
- 5.Lukacs Z, Nieves CP, Mengel E, et al. Diagnostic efficacy of the fluorometric determination of enzyme activity for Pompe disease from dried blood specimens compared with lymphocytes: possibility for newborn screening. J Inherit Metab Dis 2010;33:43–50. [DOI] [PubMed] [Google Scholar]
- 6.Gutiérrez-Rivas E, Bautista J, Vílchez JJ, et al. Targeted screening for the detection of Pompe disease in patients with unclassified limb-girdle muscular dystrophy or asymptomatic hyperCKemia using dried blood: a Spanish cohort. Neuromuscul Disord 2015;25:548–553. [DOI] [PubMed] [Google Scholar]
- 7.Musumeci O, la Marca G, Spada M, et al. LOPED Study: looking for an early diagnosis in a late-onset Pompe disease high-risk population. J Neurol Neurosurg Psychiatry 2016;87:5–11. [DOI] [PubMed] [Google Scholar]
- 8.Palmio J, Auranen M, Kiuru-Enari S, et al. Screening for late-onset Pompe disease in Finland. Neuromuscul Disord 2014;24:982–985. [DOI] [PubMed] [Google Scholar]
- 9.Spada M, Porta F, Vercelli L, et al. Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 2013;109:171–173. [DOI] [PubMed] [Google Scholar]
- 10.Preisler N, Lukacs Z, Vinge L, et al. Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 2013;110;287–289. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein JL, Young SP, Changela M, et al. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 2009;40:32–36. [DOI] [PubMed] [Google Scholar]
- 12.Pérez-López J, Selva-O'Callaghan A, Grau-Junyent JM, et al. Delayed diagnosis of late-onset Pompe disease in patients with myopathies of unknown origin and/or hyperCKemia. Mol Genet Metab 2015;114:580–583. [DOI] [PubMed] [Google Scholar]
- 13.Schoser B, Toscano A. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013;260:951–959. [DOI] [PubMed] [Google Scholar]