

Abstract
Objective:
To describe the neurologic and neuroimaging manifestations associated with Cantú syndrome.
Methods:
We evaluated 10 patients with genetically confirmed Cantú syndrome. All adult patients, and pediatric patients who were able to cooperate and complete the studies, underwent neuroimaging, including vascular imaging. A salient neurologic history and examination was obtained for all patients.
Results:
We observed diffusely dilated and tortuous cerebral blood vessels in all patients who underwent vascular imaging. White matter changes were observed in all patients who completed an MRI brain study. Two patients had a persistent trigeminal artery. One patient had an occluded right middle cerebral artery. One patient had transient white matter changes suggestive of posterior reversible encephalopathic syndrome. Four patients had migraines with one patient having complicated migraines. Seizures were seen in early life but infrequent. The majority of patients had mild developmental delays and one patient had a diagnosis of autism.
Conclusions:
Cantú syndrome is associated with various neurologic manifestations, particularly cerebrovascular findings including dilated and tortuous cerebral vessels, white matter changes, and persistent fetal circulation. Involvement of the KATP SUR2/Kir6.1 subtype potentially plays an important role in the neurologic manifestations of Cantú syndrome.
Cantú syndrome is a rare disorder characterized by congenital hypertrichosis, distinctive facial features, osteochondrodysplasia, and multiple cardiovascular abnormalities. Mutations in the genes ABCC9 (SUR2, NM_020297) and KCNJ8 (Kir6.1 NM_004982), both encoding proteins involved in ATP-sensitive potassium (KATP) channels, have been identified as causes for the syndrome.1–4
Ion channel mutations or channelopathies have been implicated in many neurologic disorders including migraines, epilepsy, neuromuscular disorders, and movement disorders.5 Few studies, however, have examined the neurologic manifestations of Cantú syndrome. One case series of 9 patients reported variable neurologic manifestations including macrocephaly, developmental delay, and behavioral and mood disturbances.6 Additionally, Cantú syndrome is associated with significant cardiovascular and systemic vascular anomalies, including cardiomegaly, patent ductus arteriosus, and pericardial effusions.7,8 These findings raise the potential for cerebrovascular manifestations. A prior case report of a single patient with Cantú syndrome demonstrated cerebral vascular anomalies with tortuous cerebral arteries and veins, as well as diminutive and absent venous sinuses and veins.2
In this study, we describe neurologic and neuroimaging manifestations of Cantú syndrome in a series of 10 patients.
METHODS
Ten patients participated in the study through attendance at a special Cantú syndrome research clinic at Washington University, St. Louis, Missouri. All patients were diagnosed by a clinical geneticist based on clinical presentation/phenotype followed by confirmatory genetic testing except one patient who was initially identified by whole-exome sequencing and subsequently referred to the clinic. All patients had a confirmed mutation for the disorder. A salient history and focused neurologic examination performed by both an adult and pediatric neurologist were completed for the patients able to attend the clinic. Neuroimaging studies including vascular imaging were obtained in all adult patients and in pediatric patients who could cooperate and complete the studies. Contrast-enhanced MRI sequences were obtained in all adult patients. Prior neuroimaging studies were also reviewed if available. MRI examinations (1.5T Siemens Magnetom Avanto; Munich, Germany) were done with conventional sequences that included fast spin-echo T1-weighted, fast spin-echo T2-weighted, T2-weighted fluid-attenuated inversion recovery, susceptibility-weighted, diffusion-weighted, and contrast-enhanced T1-weighted imaging. Contrast images were obtained after the administration of an IV gadolinium–DTPA contrast agent (0.1 mmol/kg). The apparent diffusion coefficient map was automatically processed on the magnetic resonance (MR) scanner. MR angiography (MRA) and MR venography (MRV) of the head were done without contrast, utilizing a 3D time-of-flight technique. Evaluation of the head and neck vessels was done with a 3D Flash dynamic contrast-enhanced sequence using the same bolus of contrast. All neuroimages were reviewed and interpreted by a neuroradiologist.
Standard protocol approvals, registrations, and patient consents.
We received approval from The Washington University Human Studies Committee prior to initiation of the study. Written informed consent was obtained from each participant or, if under 18, parents before enrollment in the study.
RESULTS
The table provides a summary of the patient characteristics and neurologic manifestations. Ages ranged from 18 months to 48 years. Four patients were from one family. Half of the patients were female. All patients had a missense mutation in ABCC9.
Table.
Patient characteristics, clinical information, and neuroradiographic findings
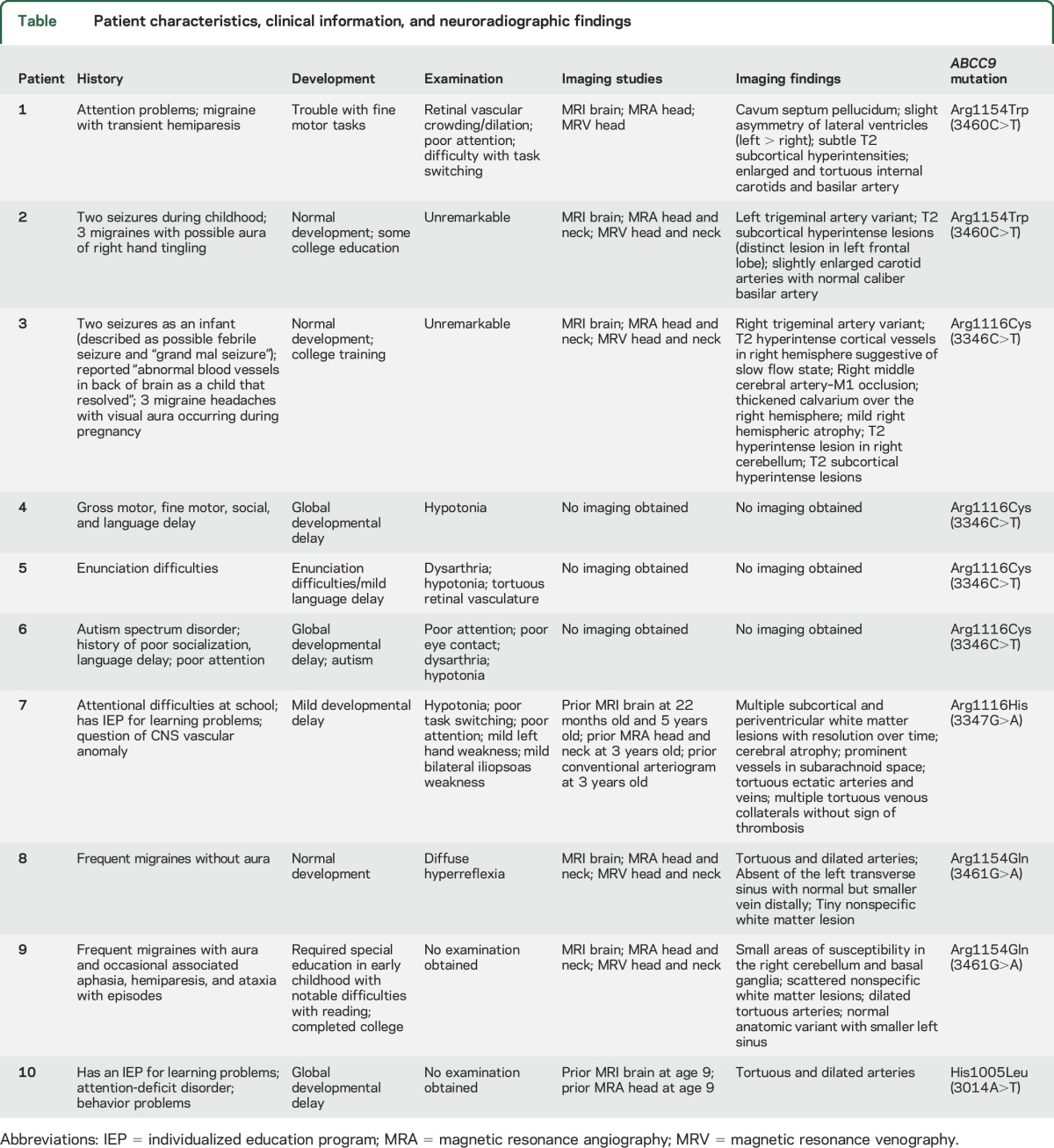
Patient 1.
Patient 1 is a 13-year-old boy with an ABCC9 missense mutation, c.3460C>T, p.R1154W. His mother reported problems with attention and concentration along with difficulty with fine motor tasks early in life. A few months after presentation at the Cantú clinic, he was diagnosed with a complicated migraine. The episode consisted of right hemiplegia, right-side numbness, and speech difficulties lasting 30–40 minutes, associated with a severe headache lasting 15 hours. He was evaluated in an emergency department and a CT head without contrast and an MRI brain without contrast were reportedly negative for an acute process.
Mental status was notable for poor concentration requiring significant redirection throughout the visit. The remainder of his neurologic examination was unremarkable. Undilated funduscopic examination was notable for retinal vascular dilation and crowding.
An MRI brain at the time of the study identified subtle T2 subcortical hyperintensities, slight asymmetry of the lateral ventricles, and a cavum septum pellucidum. An MRA/MRV of the head showed enlarged and tortuous internal carotid and basilar arteries (figure, A).
Figure. Neuroimaging manifestations of Cantú syndrome.

(A) Magnetic resonance angiography (MRA) of the head demonstrates dilated and tortuous arteries. (B) MRA of the head shows a left persistent trigeminal artery. (C) MRA of the head shows a right middle cerebral artery M1 division occlusion. (D) MRI T1-weighted without contrast shows mild right hemispheric atrophy. (E) MRA of the head demonstrates an abundance of distal collateral vessels. (F) Cerebral arteriogram shows an abundance of distal collateral vessels. (G) MRI brain T2-weighted fluid-attenuated inversion recovery (FLAIR) shows subcortical white matter changes. (H) MRI brain T2-weighted FLAIR shows white matter lesion in the right cerebellum. (I) Magnetic resonance venography of the head demonstrates aplasia of the left transverse sinus. (J) MRI brain T2-weighted FLAIR shows subcortical white matter lesions predominantly in the posterior circulation with (K) subsequent improvement/resolution of the white matter lesions on follow-up MRI brain T2-weighted FLAIR.
Patient 2.
Patient 2 is a 19-year-old woman with an ABCC9 missense mutation, c3460C>T, p.R1154W. She has had normal development and has completed some college education. She had 2 generalized tonic-clonic seizures as a child. She noted 3 migraines in her life associated with an aura described as right hand tingling.
Neurologic examination and undilated funduscopic examination were unremarkable.
An MRI brain at the time of the study revealed multiple T2 subcortical hyperintensities. An MRA/MRV of the head and neck showed enlarged and tortuous carotid arteries, normal caliber vertebral and basilar arteries, and a persistent left trigeminal artery (figure, B).
Patients 3–6.
Patients 3–6 are a members of the same immediate family, all with the ABCC9 missense mutation, c.3346C>T, p.R1116C.
Patient 3 is a 33-year-old woman and the mother of patients 4–6. She has had normal development and has completed some college education. She had 2 seizures as a child, one described as a febrile seizure and the other as a generalized tonic-clonic seizure. She has had 3 migraines in her life associated with visual aura and all occurring during her pregnancies.
Neurologic examination and undilated funduscopic examination were unremarkable.
An MRI brain at the time of the study showed multiple subcortical T2 hyperintensities with a distinct hyperintensity in the right cerebellum (figure, H). An MRA/MRV of the head and neck showed a right middle cerebral artery (MCA) M1 segment occlusion (figure, C). There were pronounced T2 hyperintense cortical veins in the right hemisphere suggestive of a slow flow state along with mild right hemispheric atrophy on MRI of the brain (figure, D), associated with the right MCA occlusion. There was no history of prior stroke but she recalls being told she had abnormal blood vessels in the back of her brain as a child that had reportedly resolved. The carotid arteries were tortuous and dilated. The vertebral basilar arteries were atretic due to a persistent right trigeminal artery.
Patient 4 is an 18-month-old girl, the youngest child of patient 3. She has had global delayed development.
Neurologic examination was notable for diffuse hypotonia. She was not able to cooperate with an undilated funduscopic examination. She was unable to tolerate an MRI study.
Patient 5 is a 4-year-old boy, the middle child of patient 3. His mother reported that he has had mild language delay described as trouble with enunciation.
Neurologic examination was notable for mild dysarthria and mild diffuse hypotonia. Undilated funduscopic examination revealed tortuous retinal vasculature.
He was unable to tolerate an MRI study.
Patient 6 is a 7-year-old boy, the oldest child of patient 3. His mother reported global developmental delay particularly with language and socialization skills with autistic features. Per report, he recently completed formal neuropsychiatric evaluation and was diagnosed with autism by his local pediatrician.
Neurologic examination was notable for poor concentration, poor eye contact, mild dysarthria, and mild diffuse hypotonia. He was not able to cooperate with an undilated funduscopic examination.
He was unable to tolerate an MRI study.
Patient 7.
Patient 7 is an 8-year-old boy with an ABCC9 missense mutation, c.3347G>A, p.R1116H. His parents reported global developmental delay and learning disabilities requiring an individualized education program at school.
Neurologic examination was notable for poor concentration, requiring redirection throughout the visit, trouble with task switching, diffuse hypotonia, mild weakness with left hand grip, and mild weakness with bilateral hip flexors. The patient was not able to cooperate with an undilated funduscopic examination.
An MRI brain study obtained at 22 months of age because of decreased responsiveness demonstrated multiple subcortical and periventricular T2 hyperintensities predominantly in the posterior region (figure, J). A serial MRI of the brain at 3 years of age demonstrated decrease/resolution of the T2 hyperintensities in the posterior region (figure, K). Stable cerebral atrophy and prominent tortuous vessels in the subarachnoid space were noted on both MRI brain studies. An MRA/MRV of the head and neck at 5 years of age showed tortuous, dilated, ectatic arteries with normal dural sinuses and enlarged cortical veins (figure, E). A conventional arteriogram at 5 years of age redemonstrated tortuous arteries as well as multiple tortuous venous collaterals (figure, F). The patient was unable to tolerate an MRI study.
Patient 8.
Patient 8 is a 19-year-old woman with an ABCC9 missense mutation, c.3461G>A, p.R1154Q. She has had normal intellectual development and is enrolled in college. She has had frequent migraine headaches since early adolescence, characterized by photophobia and nausea, without associated aura or focal neurologic symptoms.
Neurologic examination was notable for diffuse hyperreflexia. Undilated funduscopic examination was unremarkable.
An MRI of the brain at the time of the study revealed a single small left frontal subcortical T2 hyperintensity. An MRA/MRV of the head and neck showed enlarged and tortuous carotid arteries and vertebral, basilar, and cerebral vessels. Additionally, aplasia of the left transverse sinus was noted on MRV (figure, I).
Patient 9.
Patient 9, the mother of patient 8, is a 48-year-old woman with an ABCC9 missense mutation, c.3461G>A, p.R1154Q. She reported reading difficulty requiring special education in early childhood, but has since completed a college degree and works as an accountant. She has had frequent and severe migraine headaches with photophobia, phonophobia, and nausea, as well as visual aura and associated aphasia, hemiparesis, and ataxia. These attacks have required intermittent hospitalization. She also described intermittent transient neurologic symptoms not associated with headache, including unilateral weakness and paresthesias, which last for several hours at a time.
An MRI of the brain at the time of the study was notable for several small frontal and occipital subcortical T2 hyperintensities (figure, G), as well as 2 areas of increased susceptibility in the left basal ganglia and right cerebellum, indicative of prior hemorrhage or calcification. MRA of the head and neck revealed diffusely tortuous dilated arteries. MRV of the head was notable for a diminutive left transverse sinus.
She was unable to attend the multidisciplinary Cantú clinic and followed up on a later date; therefore, we were unable to obtain a neurologic examination.
Patient 10.
Patient 10 is a 14-year-old boy with an ABCC9 missense mutation, c.3014A>T, p.H1005L. This mutation has not been described previously in the literature. His parents reported global developmental delay and learning disabilities requiring an individualized education program at school, physical therapy, occupational therapy, and speech therapy. He also has had behavioral problems and attention-deficit disorder. He was a poor feeder and had hypotonia as an infant.
An MRI of the brain at the time of the study was unremarkable. An MRA of the head revealed diffusely tortuous and dilated cerebral arteries.
The patient was unable to attend the multidisciplinary Cantú clinic and followed up on a later date; therefore, we were unable to obtain a neurologic examination.
DISCUSSION
KATP channels are widely expressed in the human body and provide a unique link between metabolic demands and electric potentials at the cellular level.9 They are composed of pore-forming Kir6 channel subunits and regulatory SUR subunits.9 In particular, mutations in the genes encoding the protein subtypes Kir6.1 and SUR2 have been identified as causes of Cantú syndrome.1,2 Recent studies indicate that Cantú syndrome results from a gain of function mutation in the KATP channels generated by these subunits.1,3,10
In our study, we observed numerous cerebrovascular changes associated with Cantú syndrome. The predominant KATP channel subtype found in vascular smooth muscle consists of Kir6.1 and SUR2B (both implicated in Cantú syndrome1,2) although other subtypes are likely expressed in various vascular beds.8 Overactivation of vascular smooth muscle KATP channels results in vasodilation.10 Consistent with chronically dilated vessels, we observed diffusely dilated and tortuous cerebral blood vessels in all patients who underwent vascular imaging (figure, A illustrates this finding). This finding aligns with the prior observations made in one patient with a KCNJ8 mutation.2 Unexpectedly, however, patient 3 had an occluded right middle cerebral artery (figure, C). She had no history of a prior stroke and her neurologic examination was nonfocal. There was associated mild right hemispheric atrophy (figure, D). Given the absence of radiographic evidence of cerebral injury, we postulate the occlusion was likely a slow, progressive lesion, allowing for the development of compensatory cerebral blood flow.
Subcortical white matter changes were observed in all patients who had an MRI of the brain (figure, G and H). KATP channels have been theorized to play a role in cerebral ischemia.11 Additionally, mice in which one copy of KCNJ8 (Kir6.1) had been deleted demonstrated worsened cerebral injury compared to controls under ischemic conditions, suggesting KATP channels might play a protective role in ischemic injury.12 Although reduced expression of KATP channels seems to result in worsening ischemia, the corollary of overactivation of KATP channels being neuroprotective in cerebral ischemia may not necessarily be the case, particularly with the significant white matter changes observed in our cohort. The role of KATP channels is likely complex and dynamic with significant variability in expression of various subtypes across cerebrovascular beds.8 Conceivably, white matter changes may be related to the absence of cerebrovascular reserve. Because the arteries and arterioles are chronically dilated, it is possible that the arterioles regulating blood flow lack the capacity for compensatory vasoconstriction in the setting of persistent hypotension seen in Cantú syndrome. Kir6.1 gain-of-function mouse models resulted in a phenotype with hypotension similar to what is seen in patients with Cantú syndrome.10 Indeed, an abundance of distal collaterals was observed in patient 7, possibly to permit compensatory flow in the setting of persistent hypotension (see figure, E and F).
Interestingly, patient 7 had transient, predominantly posterior, white matter changes on MRI, at 22 months of age (figure, J). Repeat MRI at 3 years of age (figure, K) demonstrated improvement in some of the white matter changes. The transient white matter changes appear similar to findings observed in posterior reversible encephalopathic syndrome. KATP channels in vascular smooth muscle potentially play a role in cerebral vascular autoregulation, coupling metabolic demands to vascular tone. Again, normal cerebral blood flow autoregulation may be impeded in Cantú syndrome because of overactive KATP channels limiting appropriate vasoconstriction.
Two of the patients had a persistent primitive trigeminal artery (see figure, B for an example). In utero, the trigeminal artery connects the developing internal carotid artery with the developing posterior circulation.13 The incidence of a persistent trigeminal artery in the general population is low, estimated to be 0.1%–0.6%, making our finding unlikely to be incidental. Additionally, 2 patients had aplasia of the left transverse sinus (figure, I); however, the significance of this finding is unclear since variations within the venous system are common in the general population.14 Patent ductus arteriosus has also been described frequently in Cantú syndrome15 and 4 of our 10 patients had persistent patent ductus arteriosus in the first years of life that required intervention for closure. Collectively, these findings suggest that persistent fetal vasculature may be a common finding in Cantú syndrome, and that overactivity of Kir6.1/SUR2-dependent KATP channel activity may preclude a necessary constriction for vessel elimination.
Four adult patients and one of the children had a history of migraines, with and without aura. The severity and frequency varied across patients. Two patients had transient focal neurologic deficits associated with the migraines. Prior pharmacologic studies using synthetic KATP channel openers include headache as an adverse side effect in clinical studies consistent with KATP channel opening being involved in migraine.16 Again, underlying etiologies are unclear, but this does suggest that antagonists of the Kir6.1 and SUR2B subunits may be potential pharmacologic targets for migraine treatment.17 Additionally, given the vascular anomalies and white matter changes observed in Cantú syndrome it may be prudent to obtain neuroimaging, particularly vascular imaging, in patients with Cantú syndrome who have migraines.
Two adult patients had infrequent seizures as children. Detailed history regarding the circumstances and any particular provocations resulting in seizures is limited. None of the patients required long-term antiepileptic drugs. KATP channels consisting of Kir6.1 and SUR1 are found in excitatory glutamate synaptic terminals in the hippocampus and gene silencing of Kir6.1 in mice increased the susceptibility of kainic-induced seizures, which model human temporal lobe epilepsy.18
Similar to the developmental pattern observed in a prior case series,6 several of the individuals in our cohort had a history of early motor and language developmental delays as well as parent-reported attention problems. One patient in our study had a diagnosis of autism. Despite early developmental delays, however, it appears that most patients with Cantú syndrome retain normal and independent intellectual function by adulthood.
Cantú syndrome is associated with various neurologic manifestations, particularly cerebrovascular findings, including dilated and tortuous cerebral vessels, white matter changes, and persistent fetal circulation. Involvement of the KATP SUR2/Kir6.1 subtype within vascular smooth muscle cells potentially plays an important role in the neurologic manifestations of Cantú syndrome. Additional studies on the pathophysiology and long-term neurologic outcomes in Cantú syndrome are needed.
ACKNOWLEDGMENT
The authors thank the patients and their families who participated and helped make this study possible.
GLOSSARY
- KATP
ATP-sensitive potassium
- MCA
middle cerebral artery
- MR
magnetic resonance
- MRA
magnetic resonance angiography
- MRV
magnetic resonance venography
AUTHOR CONTRIBUTIONS
Dr. Christopher R. Leon Guerrero is responsible for manuscript drafting, acquisition of the clinical data, and evaluating and interpreting the data. Dr. Sheel Pathak is responsible for manuscript drafting, acquisition of the clinical data, and evaluating and interpreting the data. Dr. Dorothy K. Grange is responsible for the study concept and design, revising the manuscript for intellectual content, funding, and organizing/supervising the clinical data. Dr. Gautam Singh is responsible for analysis and interpretation of the data and revising the manuscript for intellectual content. Dr. Colin G. Nichols is responsible for analysis and interpretation of the data and revising the manuscript for intellectual content. Dr. Jin-Moo Lee is responsible for the study concept and design, analysis and interpretation of the data, and revising the manuscript for intellectual content. Dr. Katie D. Vo is responsible for the study concept and design, analysis and interpretation of the data, acquisition and interpretation of the radiographic data, and revising the manuscript for intellectual content.
STUDY FUNDING
Supported by a grant (to C.G.N.) from the Children's Discovery Institute at Washington University and by a CIMED Pilot and feasibility award (CIMED-14-03) to D.K.G.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Harakalova M, van Harssel JJ, Terhal PA, et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet 2012;44:793–796. [DOI] [PubMed] [Google Scholar]
- 2.Brownstein CA, Towne MC, Luquette LJ, et al. Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities: support for the role of K(ATP) channels in this condition. Eur J Med Genet 2013;56:678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper PE, Reutter H, Woelfle J, et al. Cantú syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat 2014;35:809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Bon BW, Gilissen C, Grange DK, et al. Cantú syndrome is caused by mutations in ABCC9. Am J Hum Genet 2012;90:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernard G, Shevell MI. Channelopathies: a review. Pediatr Neurol 2008;38:73–85. [DOI] [PubMed] [Google Scholar]
- 6.Scurr I, Wilson L, Lees M, et al. Cantú syndrome: report of nine new cases and expansion of the clinical phenotype. Am J Med Genet A 2011;155A:508–518. [DOI] [PubMed] [Google Scholar]
- 7.Grange DK, Lorch SM, Cole PL, Singh GK. Cantú syndrome in a woman and her two daughters: further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. Am J Med Genet A 2006;140:1673–1680. [DOI] [PubMed] [Google Scholar]
- 8.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res 2013;112:1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 2006;440:470–476. [DOI] [PubMed] [Google Scholar]
- 10.Li A, Knutsen RH, Zhang H, et al. Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J Am Heart Assoc 2013;2:e000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun HS, Feng ZP. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol Sin 2013;34:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong YF, Wang LX, Huang X, et al. Kir6.1 knockdown aggravates cerebral ischemia/reperfusion-induced neural injury in mice. CNS Neurosci Ther 2013;19:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meckel S, Spittau B, McAuliffe W. The persistent trigeminal artery: development, imaging anatomy, variants, and associated vascular pathologies. Neuroradiology 2013;55:5–16. [DOI] [PubMed] [Google Scholar]
- 14.Alper F, Kantarci M, Dane S, Gumustekin K, Onbas O, Durur I. Importance of anatomical asymmetries of transverse sinuses: an MR venographic study. Cerebrovasc Dis 2004;18:236–239. [DOI] [PubMed] [Google Scholar]
- 15.Grange DK, Nichols CG, Singh GK. Cantú syndrome and related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al. eds. GeneReviews®. Seattle: University of Washington; 2014:1993–2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK246980/. Accessed January 2016. [PubMed] [Google Scholar]
- 16.Ploug KB, Amrutkar DV, Baun M, et al. K(ATP) channel openers in the trigeminovascular system. Cephalalgia 2012;32:55–65. [DOI] [PubMed] [Google Scholar]
- 17.Ploug KB, Baun M, Hay-Schmidt A, Olesen J, Jansen-Olesen I. Presence and vascular pharmacology of KATP channel subtypes in rat central and peripheral tissues. Eur J Pharmacol 2010;637:109–117. [DOI] [PubMed] [Google Scholar]
- 18.Soundarapandian MM, Wu D, Zhong X, et al. Expression of functional Kir6.1 channels regulates glutamate release at CA3 synapses in generation of epileptic form of seizures. J Neurochem 2007;103:1982–1988. [DOI] [PubMed] [Google Scholar]