

Abstract
This Letter describes the chemical optimization of a novel series of M4 positive allosteric modulators (PAMs) based on a 5,6-dimethyl-4-(piperidin-1-yl)thieno[2,3-d]pyrimidine core, identified from an MLPCN functional high-throughput screen. The HTS hit was potent and selective, but not CNS penetrant. Potency was maintained, while CNS penetration was improved (rat brain:plasma Kp = 0.74), within the original core after several rounds of optimization; however, the thieno[2,3-d]pyrimidine core was subject to extensive oxidative metabolism. Ultimately, we identified a 6-fluoroquinazoline core replacement that afforded good M4 PAM potency, muscarinic receptor subtype selectivity and CNS penetration (rat brain:plasma Kp > 10). Moreover, this campaign provided fundamentally distinct M4 PAM chemotypes, greatly expanding the available structural diversity for this exciting CNS target.
Keywords: M4, Muscarinic acetylcholine receptor, Positive allosteric modulator (PAM), Schizophrenia, Structure–Activity Relationship (SAR)
Positive allosteric modulators (PAMs) of the muscarinic acetylcholine receptor subtype 4 (M4) have garnered a great deal of attention as a novel therapeutic approach for the treatment of multiple symptom domains of schizophrenia,1–6 Huntington’s disease7 and Alzheimer’s disease.8 However, the diversity and breadth of small molecule M4 PAMs is quite limited, and challenged by many of the most concerning issues with allosteric modulators (non-obvious, subtle structural modifications that engender steep SAR, species differences in affinity/cooperativity and subtype selectivity, solubility, and/or P-gp-mediated efflux).9–12 From the beginning, Lilly’s LY2033298 (1) demonstrated that major species’ disconnects in activity were possible with M4 PAMs;1,2 however, second generation M4 PAMs, such as VU0152100 (2, also know as ML108),3,4 were similarly potent on human and rat M4, and displayed antipsychotic-like activity in rats by potentiation of receptor activation by endogenous acetylcholine (Fig. 1).3,4,13 Structurally related M4 PAMs were also reported (VU0010010,14 ML253,15 ML17316), which reverted back to steep SAR and/or species differences in potency. VU0448088 (3, ML293) was optimized from an HTS hit, but again proved to be a human-preferring M4 PAM with steep, intractable SAR.17,18 Recently, we disclosed the pyridazine congener VU0467154 (4), which displayed robust in vivo efficacy across a broad range of rodent antipsychotic and cognition models, further increasing interest in the target.18 However, all of the known chemotypes of M4 PAMs (1–4) posed significant challenges en route to a clinical candidate. In this Letter, we detail the chemical optimization, SAR, DMPK profile and in vivo efficacy of a fundamentally new M4 PAM chemotype.
Figure 1.

Structures of representative M4 PAMs 1–4, highlighting the limited diversity of known chemotypes.
In order to identify a new M4 PAM chemotype, we performed a new functional M4 HTS in 1536-well assay format against the ~360,000 compound MLSMR collection, and four structurally novel M4 PAMs were identified.19 Of the four, VU0473619 (5) represented an exciting lead (Fig. 2), as it was similarly potent on both human and rat M4 (EC50s of 410 nM and 160 nM, respectively, i.e., little or no apparent species difference), and was inactive on both human M1 and M5 (built-in HTS counterscreen). For a lead, the DMPK profile was acceptable; however, 5 was not CNS penetrant, likely due to the primary sulfonamide moiety that engendered a cLogP of 1.2. Thus, the optimization effort would focus on replacing this detrimental functional group, and, in addition, assess the 5,6-dimethyl moieties as potential CYP450 metabolic liabilities as well as the amide linker as another potentially CNS-limiting functional group. Upon resynthesis, the potency of 5 for human M4 confirmed, but as a weaker hM4 PAM (EC50 = 1.20 μM, pEC50 = 5.92 ± 0.10, 91 ± 4%).
Figure 2.

Structure and key biological data for the M4 PAM HTS hit VU0473619 (5), and three areas to address in the lead optimization campaign. Potency values represent means from at least three independent determinations in a functional Ca2+ mobilization assay with rat or human M4/Gqi5-CHO cells performed in the presence of an ~EC20 fixed concentration of acetylcholine.
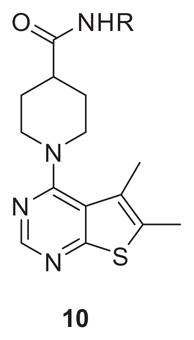
The chemistry to produce amide analogs of 5 was straightforward (Scheme 1), and the initial optimization focused on replacing the sulfonamide. Commercially available 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 6 was subjected to a standard SNAr reaction with piperidine 7, to afford 8 in 90% yield. Saponification delivered 9, which then underwent a HATU-mediated coupling with diverse amines to afford analogs 10 or 11 in yields ranging from 22% to 90%.
Scheme 1.

Reagents and conditions: (a) DIEA, NMP, MW, 200 °C, 15 min, 90%; (b) LiOH, aqueous MeOH, rt, 95%; (c) HNR1R2, HATU, DCE/DMF (9:1), rt, 22–90%.
Table 1 highlights select SAR for the first generation of analogs 10 employing primary amines/anilines. Efficacy (% ACh Max) proved to be relatively low across all of the analogs 10, but we were pleased to see that the primary sulfonamide of 5 could be replaced with other moieties such as a sulfone (10e) or the more lipophilic halogenated anilines 10g and 10h while retaining selectivity versus M1 and M5 (data not shown). However, SAR was steep, efficacy was uniformly low, and physiochemical and DMPK properties remained poor.
Table 1.
Structures and activities for M4 PAM analogs 10
| |||
|---|---|---|---|
| Compd | R | hM4 EC50 a (μM) [% ACh Max ± SEM] | hM4 pEC50 (±SEM) |
| 10a | H | >10 [25 | >5 |
| 10b |
|
2.3 [67 ± 1%] | 5.63 ± 1.1 |
| 10c |
|
0.39 [56 ± 6%] | 6.41 ± 0.01 |
| 10d |
|
0.57 [70 ± 3%] | 6.24 ± 0.13 |
| 10e |

|
0.85 [62 ± 6%] | 6.07 ± 0.11 |
| 10f |

|
6.2 [40 ± 8%] | 5.21 ± 0.20 |
| 10g |

|
1.6 [55 ± 5%] | 5.80 ± 0.18 |
| 10h |

|
3.8 [69 ± 5%] | 5.42 ± 0.09 |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n = 3) independent experiments performed in triplicate.
We then elected to evaluate tertiary amides, where we found greater success in terms of M4 potency, efficacy and disposition. As shown in Table 2, many analogs 11 displayed submicromolar potency and improvements in efficacy. The thiomorpholine congener 11e was considerably more potent and efficacious than the analogous morpholine derivative 11d. Further exploration of pyrrolidine amides 11a and 11b provided encouraging results. In in vitro DMPK assays, PAM 11a displayed good free fraction in both rat (fu = 0.04) and human (fu = 0.06) plasma and importantly proved to be CNS penetrant (rat brain:plasma Kp = 0.45, Kp,uu = 0.45). However, in intrinsic clearance studies, 11a was highly metabolized with predicted hepatic clearances near the organ blood flow rate (predicted CLhep = 67 and 16 mL/min/kg for rat and human respectively). The fluorinated congener, 11b, showed increased free fraction in both rat (fu = 0.12) and human (fu = 0.05) plasma, but CNS penetration decreased (rat brain:plasma Kp = 0.17, Kp,uu = 0.12) and intrinsic clearance remained high (predicted CLhep = 68 and 14 mL/min/kg for rat and human, respectively).
Table 2.
Structures and activities for M4 PAM analogs 11

| |||
|---|---|---|---|
| Compd | NR1R2 | hM4 EC50 a (μM) [% ACh Max ± SEM] | hM4 pEC50 (±SEM) |
| 11a |

|
1.1 [68 ± 3%] | 5.94 ± 0.11 |
| 11b |

|
0.97 [79 ± 10%] | 6.01 ± 0.03 |
| 11c |

|
1.9 [60 ± 5%] | 5.73 ± 0.21 |
| 11d |

|
1.7 [65 ± 4%] | 5.77 ± 0.10 |
| 11e |

|
0.82 [75 ± 7%] | 6.09 ± 0.27 |
| 11f |

|
0.96 [69 ± 5%] | 6.02 ± 0.06 |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n = 3) independent experiments performed in triplicate.
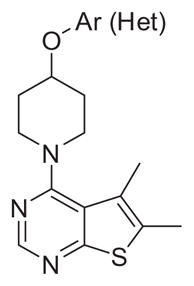
These data suggested that efforts needed to focus on replacement of the amide linker as well as the 5,6-dimethylthieno[2,3-d]pyrimidine core. We first elected to replace the amide linker with ether linkages (Scheme 2), which could be readily accessed. Here, 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 6 was subjected to a standard SNAr reaction with piperidine 12 to afford 13 in 70–95% yield. A subsequent Mitsunobu reaction installed the aryl/heteroaryl ether moiety, producing analogs 14. These ether analogs, devoid of the amide linker, displayed high CNS distribution (Kps > 1), but variable unbound CNS distribution Kp,uu. Of these, 14b (VU6002703) stood out as a potent M4 PAM (hM4 EC50 = 600 nM, 65% ACh Max) but exhibited high intrinsic clearance in vitro (hepatic microsomes) with predicted hepatic clearances near organ blood flow rates in both human and rat (CLhep = 20 and 68 mL/min/kg, respectively), as did all analogs 14. Clearly, a metabolic instability issue rested within the 5,6-dimethylthieno[2,3-d]pyrimidine core. Therefore, we performed metabolite identification (MetID) experiments in rat and human hepatic microsomes (Fig. 3) with 14b, and found hydroxylation of the 5,6-dimethyl moiety was the major pathway of metabolism in rat, while heteroatom (likely the thiophene sulfur) oxidation was the major metabolic route in human. Tying back the 5,6-dimethyl moiety into five- and six-membered unsaturated rings was tolerated for potency and CNS penetration, but had no impact on lowering in vitro clearance (data not shown). Therefore, we needed to identify an alternate core (see Table 3).
Scheme 2.

Reagents and conditions: (a) DIEA, NMP, MW, 200 °C, 15 min, 70–95%; (b) ArOH/Het-OH, PPh3, DtBAD, THF, rt, 55–86%.
Figure 3.

Metabolism of 14b (VU6002703) in rat and human hepatic microsomes. All biotransformation pathways shown are NADPH-dependent.
Table 3.
Structures and activities for M4 PAM analogs 14
| ||||
|---|---|---|---|---|
| Compd | NR1R2 | hM4 EC50 a (μM) [% ACh Max ± SEM] | hM4 pEC50 (±SEM) | Rat Kp (Kp,uu)b |
| 14a |

|
4.4 [60 ± 5%] | 5.36 ± 0.09 | 1.77 (0.65) |
| 14b |

|
0.60 [65 ± 4%] | 6.22 ± 0.04 | 1.16 (0.74) |
| 14c |

|
1.8 [68 ± 4%] | 5.76 ± 0.03 | 2.80 (0.34) |
| 14d |

|
4.3 [64 ± 4%] | 5.37 ± 0.11 | 2.56 (0.17) |
| 14e |
|
2.3 [74 ± 5%] | 5.64 ± 0.09 | 1.25 (1.38) |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n = 3) independent experiments performed in triplicate.
Total and calculated unbound brain:plasma partition coefficients determined at 0.25 h post-administration of an IV cassette dose (0.20–0.25 mg/kg) to male, SD rat (n = 1); in conjunction with in vitro rat plasma protein and brain homogenate binding assay data.
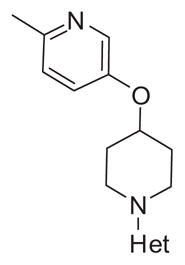
Thus, we maintained the 2-methyl-5-(piperidin-4-yloxy)pyridine moiety in 14b and surveyed a variety of alternative heteroaryl and heterobiaryl ring systems to replace the 5,6-dimethylthieno [2,3-d]pyrimidine core. New analogs 17 were readily obtained by a Mitsunobu reaction, Boc deprotection and SNAr reaction (Scheme 3) in good overall yields. SAR for analogs 17 was steep, but substituted quinazoline derivatives 17a and 17b, a quinoline congener 17c, and a pyrrolotriazine, 17d, were active (M4 PAM EC50s 600 nM to 1.2 μM) and highly CNS penetrant (Kps 0.94–21.1). Intrinsic clearance remained high (predicted CLhep near hepatic blood flow in both human and rat) for all analogs 17 except 17b (VU6003130). 17b was the most potent human M4 PAM in this new series (human M4 EC50 = 600 nM, 62%) with moderate activity at rat M4 (rat EC50 = 950 nM, 57%) and selective versus M1–3,5 (EC50s > 10 μM). Moreover, 17b had a moderate to high free fraction in rat (fu = 0.02) and human (fu = 0.16) plasma as well as in rat brain homogenate (fu = 0.03). In addition, 17b was highly CNS penetrant (rat brain:plasma Kp = 11.1, Kp,uu > 10) and was the only analog to show a notable improvement in metabolic stability in vitro (predicted CLhep = 12 and 50 mL/min/kg in human and rat, respectively), moving it into the moderate clearance range. Thus, this optimization effort provided two fundamentally novel, potent and selective M4 PAMs, 14b (VU6002703) and 17b (VU6003130), with excellent CNS distribution (see Table 4).
Scheme 3.

Reagents and conditions: (a) 6-methylpyridin-3-ol, PPh3, DtBAD, THF, rt, 88%; (b) Het-Cl, DIEA, NMP, MW, 200 °C, 15 min, 62–94%.
Table 4.
Structures and activities for M4 PAM analogs 17
| ||||
|---|---|---|---|---|
| Compd | NR1R2 | hM4 hEC50 a (μM) [% ACh Max ± SEM] | hM4 pEC50 (±SEM) | Rat Kp (Kp,uu)b |
| 17a |

|
1.2 [49 ± 7%] | 5.93 ± 0.08 | 0.94 (0.62) |
| 17b |

|
0.6 [62 ± 7%] | 6.24 ± 0.07 | 11.1 (>10) |
| 17c |

|
0.69 [64 ± 5%] | 6.16 ± 0.03 | 21.4 (18.2) |
| 17d |

|
1.1 [58 ± 3%] | 5.98 ± 0.17 | 1.42 (1.95) |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n = 3) independent experiments performed in triplicate.
Total and calculated unbound brain:plasma partition coefficients determined at 0.25 h post-administration of an IV cassette dose (0.20–0.25 mg/kg) to male, SD rat (n = 1); in conjunction with in vitro rat plasma protein and brain homogenate binding assay data.
Before further advancement of 14b and 17b, we also evaluated replacement of the ether linkage in 14b with an amine linker, as in 18 (Fig. 4). The majority of analogs were inactive, but once the alkyl chain reached 3-carbons in length, a ‘molecular switch’ was noted (rare amongst muscarinic allosteric modulators20–22 and the first report for M4), wherein the ligands became M4 antagonists. For example, 19 (human M4 IC50 = 2.4 μM, 9.3% ACh min) and 20 (human M4 IC50 = 1.5 μM, 8.7% ACh min) were robust M4 antagonists. Analogs 19 and 20 had attractive DMPK profiles (fus > 0.10 across species, Kps ~ 0.2 and acceptable CYP450 inhibition profiles); however, both were similarly potent at the other muscarinic receptors (M1–3,5), suggesting binding had switched from an allosteric site to the orthosteric site. While not the desired outcome, this finding was interesting and will enable computational/docking strategies moving forward.
Figure 4.

Generic structure of amine-linked analogs 18 and specific analogs 19 and 20 that underwent a ‘molecular switch’ into pan-orthosteric mAChR antagonists.
In summary, an HTS campaign identified a fundamentally new M4 PAM chemotype, but with unfavorable disposition and no CNS penetration. Iterative parallel synthesis developed SAR and afforded two novel M4 PAMs, 14b (VU6002703) and 17b (VU6003130), with excellent CNS distribution and enhanced M4 PAM potency relative to the HTS hit. Importantly, this effort also identified a ‘molecular switch’ that modulated the mode of pharmacology from PAM to orthosteric antagonist. Further study and optimization within this series is ongoing, and will be reported in due course.
Acknowledgments
We thank the NIH for funding via the NIH Roadmap Initiative 1X01 MH077607 (C.M.N.), the Molecular Libraries Probe Center Network (U54MH084512 to P.S.H. and U54MH084659 to C.W.L.) We also thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
References and notes
- 1.Chan WY, McKinize DL, Bose S, Mitchell SN, Witkins JM, Thompson RC, Christopoulos A, Birdsall NJ, Bymaster FP, Felder CC. Proc Natl Acad Sci USA. 2008;105:10978. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leach K, Loiancono RE, Felder CC, McKinize DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. Neuropsychopharmacology. 2010;35:855. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady A, Jones CK, Bridges TM, Kennedy PJ, Thompson AD, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey J, Conn PJ, Lindsley CWJ. Pharm Exp Ther. 2008;327:941. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byun NE, Grannan M, Bubser M, Barry RL, Thompson A, Rosanelli J, Gowrishnakar R, Kelm ND, Damon S, Bridges TM, Melancon BJ, Tarr JC, Brogan JT, Avison MJ, Deutch AY, Wess J, Wood MR, Lindsley CW, Gore JC, Conn PJ, Jones CK. Neuropsychopharmacology. 2014;39:1578. doi: 10.1038/npp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrell M, Roth BL. Neuropsychopharmacology. 2010;35:851. doi: 10.1038/npp.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones CK, Byun N, Bubser M. Neuropsychopharmacology. 2012;37:16. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pancani T, Foster DJ, Bichell T, Bradley E, Bridges TM, Klar R, Daniels JS, Jones CK, Bowman AB, Lindsley CW, Xiang Z, Conn PJ. Proc Natl Acad Sci USA. 2015;112:14078. doi: 10.1073/pnas.1512812112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen W, Plotkin JL, Francardo V, Ko WKD, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, Conn PJ, Greengrad P, Bezard E, Cenci MA, Surmeier DJ. Neuron. 2015;88:762. doi: 10.1016/j.neuron.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conn PJ, Christopolous A, Lindsley CW. Nat Rev Drug Disc. 2009;8:41. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Nat Rev Drug Disc. 2014;13:692. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huynh T, Valant C, Crosby IT, Sexton PM, Christopoulos A, Capuano B. ACS Chem Neurosci. 2015;6:838. doi: 10.1021/acschemneuro.5b00035. [DOI] [PubMed] [Google Scholar]
- 13.Pacani T, Bolarinwa C, Smith Y, Lindsley CW, Conn PJ, Xiang Z. ACS Chem Neurosci. 2014;5:318. doi: 10.1021/cn500003z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. Nat Chem Biol. 2008;4:42. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- 15.Le U, Melancon BJ, Bridges TM, Utley TJ, Lamsal A, Vinson PN, Sheffler DJ, Jones CK, Morrison R, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2013;23:346. doi: 10.1016/j.bmcl.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy JP, Bridges TM, Gentry PR, Brogan JT, Brady AE, Shirey JK, Jones CK, Conn PJ, Lindsley CW. Chem Med Chem. 2009;4:1600. doi: 10.1002/cmdc.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salovich JM, Sheffler DJ, Vinson PN, Lamsal A, Utley TJ, Blobaum AL, Bridges TM, Le U, Jones CK, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2012;22:5084. doi: 10.1016/j.bmcl.2012.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bubser M, Bridges TM, Thorbeck DD, Gould RW, Grannan M, Noetzel MJ, Niswender CM, Daniels JS, Melancon BJ, Tarr JC, Wess J, Duggan ME, Brandon NJ, Dunlop J, Wood MW, Wood MR, Lindsley CW, Conn PJ, Jones CK. ACS Chem Neurosci. 2014;5:920. doi: 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith E, Chase P, Niswender CM, Conn PJ, Lindsley CW, Madoux F, Acosta M, Scampavia L, Spicer T, Hodder PJ. Biomol Screening. 2015;20:858. doi: 10.1177/1087057115581770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Digby GJ, Utley TJ, Lamsal A, Sevel C, Sheffler DJ, Lebois EP, Bridges TM, Wood MR, Niswender CM, Lindsley CW, Conn PJ. ACS Chem Neurosci. 2012;3:1025. doi: 10.1021/cn300103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheffler DJ, Sevel C, Le U, Lovell KM, Tarr JC, Cho HP, Digby GJ, Niswender CM, Conn PJ, Hopkins CR, Wood MR, Lindsley CW. Bioorg Med Chem Lett. 2013;23:223. doi: 10.1016/j.bmcl.2012.10.132. [DOI] [PMC free article] [PubMed] [Google Scholar]