

ABSTRACT
The infiltration of myeloid cells helps tumors to overcome immune surveillance and imparts resistance to cancer immunotherapy. Thus, strategies to modulate the effects of these immune cells may offer a potential therapeutic benefit. We report here that tasquinimod, a novel immunotherapy which targets S100A9 signaling, reduces the immunosuppressive properties of myeloid cells in preclinical models of bladder cancer (BCa). As single anticancer agent, tasquinimod treatment was effective in preventing early stage tumor growth, but did not achieve a clear antitumor effect in advanced tumors. Investigations of this response revealed that tasquinimod induces an increase in the expression of a negative regulator of T cell activation, Programmed-death-ligand 1 (PD-L1). This markedly weakens its antitumor immunity, yet provokes an “inflamed” milieu rendering tumors more prone to T cell-mediated immune attack by PD-L1 blockade. Interestingly, the combination of tasquinimod with an Anti-PD-L1 antibody enhanced the antitumor immune response in bladder tumors. This combination synergistically modulated tumor-infiltrating myeloid cells, thereby strongly affecting proliferation and activation of effector T cells. Together, our data provide insight into the rational combination of therapies that activate both innate and adaptive immune system, such as the association of S100A9-targeting agents with immune checkpoints inhibitors, to improve the response to cancer immunotherapeutic agents in BCa.
KEYWORDS: Bladder cancer, immunotherapy, myeloid cells, PD-L1, treatment combination
Abbreviations
- APC
Antigen-presenting cells
- DAMP
Damage-associated molecular pattern
- EMEM
Eagle's minimum essential medium
- FACS
Fluorescence-activated cell sorting
- FDA
Food and Drug Administration
- FFPE
Formalin fixed paraffin embedded
- MDSCs
Myeloid-derived suppressor cells
- MFI
Median fluorescence intensity
- MRI
Magnetic resonance imaging
- PD-1
Programmed death 1
- PD-L1
Programmed-death-ligand 1
- PMA
Phorbol 12-Myristate 13-Acetate
- RPMI
Roswell Park Memorial Institute medium
- TAMs
Tumor-associated macrophages
- TILs
Tumor-infiltrating lymphocytes
- TMA
Tissue microarray
- TME
Tumor microenvironment.
Introduction
BCa is the most common urinary tract cancer with an estimated incidence of 386,000 cases and 150,000 deaths per year worldwide.1 Significant progress has been made in the last decades in understanding the biology of BCa. Overactivation of the PI3K/AKT/mTOR and MAPK pathways as well as epigenetic alterations are frequently found in bladder tumor cells.2 In addition to changes in the tumor cells themselves, the tumor microenvironment (TME) plays a major role in promoting tumor development and metastasis.3 Several cell populations of the TME are implicated in tumor growth and progression such as endothelial cells, pericytes, fibroblasts, regulatory T cells (Tregs) and myeloid cells.4 The myeloid lineage cells are a heterogeneous population of bone-marrow derived cells, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which are actively recruited to the TME.
TAMs play a key role in promoting tumor cell proliferation, angiogenesis and repression of adaptive immunity.5 Two distinct subsets of TAMs have been proposed. Type 1 macrophages (M1) express multiple pro-inflammatory factors and cytokines, such as IL-12 and iNOS and exert antitumor immunity. In contrast, type 2 macrophages (M2) express a wide variety of anti-inflammatory molecules, such as IL-10, TGF-β, and Arginase-1. The M2 phenotype predominates in most human tumors and provides an immunosuppressive microenvironment that fosters tumor growth.6
MDSCs are a heterogeneous immature cell population that increases during inflammation and cancer, and suppresses T-cell activation.7 S100A9 regulates the accumulation of MDSCs, leading to tumor-promoting immunosuppressive functions.8 Mice lacking S100A9 rejected implanted EL4 tumors, whereas STAT-3 inducible upregulation of S100A9 increases the accumulation of tumor-associated MDSCs.9 Administration of wild-type MDSCs to S100A9 null-mice reversed this effect. Moreover, a recent study showed that S100A9 may be useful as a molecular imaging marker to monitor MDSCs and TAMs activity in primary tumor lesions.10 S100A9 imaging revealed a strong correlation with tumor growth and metastasis formation.
In addition to myeloid cells, which negatively regulate T cell function, a large number of immune checkpoints such as PD-1/PD-L1, CTLA-4, Tim-3 or LAG-3 are implicated in tumor-induced immunosuppression.11 In particular, the PD-1/PD-L1 axis attenuates antitumor immunity via several mechanisms such as T cell anergy, exhaustion and apoptosis.12 PD-L1 is expressed on multiple lymphoid cells (B cells, T cells), myeloid cells (MDSCs, TAMs) as well as on tumor cells in a wide variety of tumor types, including melanoma,13 non-small cell lung,14 and bladder carcinomas.15 Interestingly, patients with superficial BCa showed lower PD-L1 expression than those with invasive BCa.16 Moreover, blockade of the PD-1/PD-L1 axis was shown to induce a potent antitumor immune response in preclinical mouse models as well as in the clinic.17,18 Based on the efficacy of Anti-PD-L1 directed therapy, the FDA granted a Breakthrough Therapy Designation for the use of Anti-PD-L1 (MPDL3280A) in metastatic BCa in 2014.
Tasquinimod is a small molecule with a quinoline-3-carboxamide structure that binds to S100A9 and blocks its interaction with TLR4, RAGE and EMMPRIN expressed on different subsets of myeloid cells.19,20 Tasquinimod has been shown to exert immunomodulatory, anti-angiogenic and anti-metastatic properties in preclinical studies.21,22
In this study, we screened first multiple human tumors for S100A9 expression. Among all tumor types, BCa appears to express the highest amounts of S100A9. Then, we tested the activity of tasquinimod in preclinical models of BCa. We hypothesized that tasquinimod induces a tumor inflammatory state that could enhance PD-L1 expression in the TME which in turn may reduce tasquinimod-induced antitumor response. We therefore investigated whether the association of tasquinimod with Anti-PD-L1 may enhance the antitumor immunity. This study also explored the mechanisms of reciprocal communication between tumor-infiltrating myeloid cells and T cells.
Results
S100A9 is highly expressed in human BCa
It is well known that tasquinimod inhibits in a dose-dependent way the interaction between S100A9 and TLR4 or RAGE,19 and reduces TNFα release upon LPS challenge in a S100A9-dependent model in vivo.24 Recent data have also shown that tasquinimod significantly improved the survival of DP42 tumor-bearing mice but lost its antitumor activity in S100A9 knock-out mice model.25 These data all together confirm that S100A9 is a pharmacological target of tasquinimod. This protein appears to be critical for tumor growth and progression.23 However, little is known about S100A9 expression and distribution in human cancer tissues. Here, we performed a comparative study of S100A9 gene expression across multiple tumor types, which aimed to identify potential therapeutic indications for tasquinimod. S100A9 mRNA was variable among all tumor types analyzed and heterogeneous among individual data sets, reflecting the differences in the number of patients and the related clinical parameters (Fig. 1A). Nevertheless, S100A9 mRNA was detected in all tumor samples. Very low S100A9 mRNA expression (ΔCt<0.6) was observed in gastrointestinal stromal tumors (GIST). Low to medium expression (0.6<ΔCt<17) was noted in several tumor types, such as thyroid, testicular and renal cancers. Interestingly, the highest mRNA expression of S100A9 (ΔCt>17) was observed in esophageal and BCa compared to other tumor tissues. In this report, we focused our study on the understanding of the role of S100A9 in BCa.
Figure 1.

S100A9 is expressed in human tumors. (A) Fraction of samples with low (ΔCt<0.6), medium (0.6<ΔCt<17) or high (ΔCt>17) mRNA expression levels normalized to Hmbs across 17 different human tumor types. (B) Percentage of bladder samples (n = 14) with weak, moderate or strong immunohistochemistry staining for S100A9 in paraffin-embedded tumors (C) Representative images from urinary BCa showing variable S100A9 expression exemplifying (C1) weak (W) or (C2) moderate (M) staining intensities in tumor cells. Original magnification X50, inset X200. Arrow: stromal cells showing strong staining. Scale bars: 100 µm. (D) Representative images showing S100A9 strong staining in stromal cells of MBT-2 and AY-27 tumors. Original magnification X200.
First, we examined whether S100A9 protein was predominately expressed in the tumor cells and/or the stromal compartment of the human bladder tumors. Weak to moderate expression of S100A9 protein in tumor cells was observed in 93% of the analyzed tumors (Fig. 1B and C). However, strong S100A9 staining was always observed in the tumor stroma (Fig. 1C). Therefore, these data gave rise to the question whether stromal S100A9 may be a critical factor for the growth and the progression of BCa and whether bladder tumors could be targeted by pharmacological inhibitors of S100A9 signaling.
To this end, we investigated the ability of tasquinimod to exert an antitumor activity in preclinical models of BCa using two different animal models: the MBT-2 mouse model and the AY-27 rat model. Both primary tumors express S100A9 protein exclusively in the stromal compartment (Fig. 1D).
Tasquinimod prevents the growth of bladder tumors
Previous investigations revealed that the human and murine S100A9 share a higher degree of functional homology than of sequence similarity.26 In addition, the binding of tasquinimod to S100A9 does not appear to be species specific.24 Thus, the dosage regimens of tasquinimod treatment was based on the pharmacokinetics parameters (bioavailability) and the tolerated doses in animals. Previous studies have shown that tasquinimod at 30 mg/kg did not induce any organ/systemic toxicity or weight loss in mice (data not shown). However, a dose level between 0.3 and 3 mg/kg/d is recommended in rats.
Tasquinimod treatment at the dose of 2 mg/kg prevented intravesical AY-27 tumor growth in rats (Fig. 2A and B) and induced a reduction of 60% in tumor weight (Fig. 2D) as compared to the control group. In contrast, cisplatin, a current standard of care treatment for muscle-invasive human BCa, showed only very moderate activity in this model (Fig. 2C and D).
Figure 2.

Early treatment with tasquinimod prevents tumor growth in two preclinical models of BCa. (A–D). AY-27 tumor cells (106) were injected orthotopically in the bladder of female rats. Mice were left (A) untreated (Control) or treated with (B) tasquinimod 2 mg/kg (oral gavage, twice daily), or with (C) cisplatin 2 mg/kg (Intraperitoneal injection, once per week) at day 4 post-tumor cell inoculation. Each curve corresponds to the tumor growth of a single tumor monitored by MRI measurements at 4, 31, 41 and 48 d post-tumor cell inoculation (n = 10 animals per group). (D) Weight of AY-27 tumors left untreated or treated with tasquinimod or cisplatin at the end of the experiment (day 48) (One-way ANOVA; ***p < 0.001). (E) MBT-2 tumor cells (106) were injected subcutaneously into C3H/HeNRj mice. Treatment with 4 doses of tasquinimod: 0.1–1–10 and 30 mg/kg was initiated the next day following tumor cell injection. MBT-2 tumor growth for each dose of tasquinimod treatment as compared to control. Fold change of mRNA expression of different inflammatory genes in (F) AY-27 and (G) MBT-2 treated tumors relative to their respective control set to 1. Data are mean ± SEM (n = 10 mice). Asterisks denote statistical significance (One-way ANOVA; *p < 0.05; **p < 0.005; ***p < 0.001).
The activity of tasquinimod in the MBT-2 model was also assessed with oral administration of tasquinimod at 0.1, 1, 10 and 30 mg/kg twice daily in C3H/HeNRj mice which possess a normal TLR-4 response (Fig. 2E). Tasquinimod at the doses of 0.1 and 1 mg/kg was not sufficiently effective to inhibit tumor growth. In contrast, tasquinimod prevented MBT-2 tumor growth in a dose dependent-manner at 10 and 30 mg/kg. These data obtained from two different models suggest that S100A9-targeting agents like tasquinimod have potential activity against BCa.
We also found that tasquinimod was effective in preventing MBT-2 tumor growth in TLR4-defective C3H/HeJ mice (Fig. S1). This potentially suggests that the antitumor activity of tasquinimod was not dependent on TLR4 signaling but rather to S100A9 interaction with RAGE or EMMPRIN in BCa model.
Tasquinimod reprograms the immunosuppressive properties of the BCa microenvironment
To investigate the mechanism by which tasquinimod induces the antitumor response in vivo, we performed gene expression profiling in AY-27 tumors treated with tasquinimod at the dose of 2 mg/kg (Fig. 2F), and in MBT-2 tumors treated with different doses of tasquinimod (Fig. 2G). Interestingly, in both models, tasquinimod induced a significant increase in the expression of different markers of type 1 macrophages (M1), such as Nos2, Cxcl5, Cxcl9 and Cxcl11 (Fig. 2F and G).
IL-12 is a pro-inflammatory cytokine that is known to be secreted by antigen-presenting cells (APC) in response to pathogens.27 IL-12 induces T-bet and controls the differentiation of naive T cells into Th1 cells.28 It is also a potent inducer of IFNγ production. Interestingly, Il-12b, T-bet and Ifng mRNAs were found to be increased in AY-27 tumors treated with tasquinimod (Fig. 2F). We also examined the expression of Serpinb2 that encodes a protease inhibitor whose expression has been shown to correlate positively with increased survival of patients with breast cancer or pancreatic carcinomas.29 A dose-dependent increase in Il-12b and Serpinb2 was observed in MBT-2 tumors treated with tasquinimod (Fig. 2G). Moreover, an increase in the expression of different pro-inflammatory cytokines, IFNγ, IL-1α, IL-1β and TNF-α, was observed in AY-27 tumors treated with tasquinimod as compared to control and/or cisplatin (Fig. S2C–F). Similarly, the expression of other pro-inflammatory cytokines such as MIG, IP-10 and LIX was upregulated in MBT-2-treated tumors at the highest dose used for tasquinimod 30 mg/kg as compared to control (Fig. S3). Taken together, these data showed that tasquinimod induced an increase in pro-inflammatory cytokines and inflammatory related genes in the TME, which were consistent with impaired bladder tumor growth.
Tasquinimod modulates immunoregulatory myeloid cells
The profile of the different immune cells that infiltrated primary tumors after tasquinimod treatment was also investigated. We chose the dose of 30 mg/kg that showed the greatest antitumor activity in the MBT-2 model (Fig. 2E). We found that tasquinimod neither changed the percentage of CD4+ (Fig. S4A), CD8+ (Fig. S4B) tumor-infiltrating-lymphocytes (TILs), nor NK cells (Fig. S4E). In addition, the percentage of tumor-infiltrating myeloid cells CD11b+ (Fig. 3B), macrophages CD11b+ F4/80+ (Fig. 3C) or MDSCs (Fig. S4D) were not modified by tasquinimod treatment. The percentage of tumor-infiltrating CD8+ cells was also unchanged in AY-27 tumors treated with tasquinimod (Fig. S2G). However, a decrease in the expression of CD206+, a M2 TAM phenotype marker, gated on CD11b+ F4/80+ (Fig. 3D) was observed in MBT-2 tumors treated with tasquinimod. To conduct a detailed phenotypic and molecular analysis of myeloid cells exposed to tasquinimod treatment, we isolated CD11b+ (purity more than 95%) from tumors and evaluated the expression of different markers of alternatively (M2) or classically activated (M1) macrophages based on the classification reported in several studies.5,6,30 Isolated CD11b+ cells derived from tumors treated with tasquinimod had decreased expression of M2-associated genes such as F13a1, Tgfb1, Fn1, Mrc1/CD206, Ccl2, Ccl7, Arginase-1 and Lgals1, whereas expression of M1-associated genes including Nos2, Tnf, Cxcl9, Cxcl11 and Serpinb2 was increased (Fig. 3E). Together, these data indicated that tasquinimod was able to re-educate tumor-infiltrating myeloid cells toward a M1 phenotype, which was associated with a marked antitumor response. Our data strongly suggest that tasquinimod induced the activation of the innate immune system within the TME.
Figure 3.

Tasquinimod treatment modulates the immunosuppressive activity of tumor-infiltrating myeloid cells (A) MBT-2 tumor growth treated with the most effective dose of tasquinimod in this model: 30 mg/kg (oral gavage, twice daily) using 10 mice per group. Treatment was initiated at day 1 post-tumor cell inoculation (Student test; **p < 0.005; ***p < 0.001). (B) Quantitative data of the percentage of (B) tumor infiltrating myeloid cells (CD11b+), (C) macrophages (CD11b+ F4/80+) and (D) M2 macrophages (CD11b+ F4/80+ CD206+) at day 20. Representative gating strategy is shown in the upper figure. Quantitative data were pooled from two independent experiments in the lowest figure. Each experiment was conducted with five mice per group using cytometric analysis (Student test; *p < 0.05). (E) CD11b+ cells were sorted from MBT-2 tumors treated or non-treated with tasquinimod at 30 mg/kg for 20 d using BD FACSAria II. mRNA levels are normalized by cyclophilin-A mRNA level (delta CT method). Data are expressed relative to their respective control set to 1. Fold change of gene expression profiling for M2 (gray bars) or M1 markers (black bars) of TAMs is indicated. Data are mean ± SEM. Asterisks denote statistical significance using student test (*p < 0.05; **p < 0.005; ***p < 0.001).
Expression of PD-L1 is increased in tumor tissue following tasquinimod treatment
We also investigated whether tasquinimod was able to inhibit tumor progression on established tumors when given at a later time point after tumor implantation. To this end, animals were treated when MBT-2 tumors reached a tumor volume ranging between 50 and 100 mm3(Fig. 4A and B). In this setting, surprisingly, tasquinimod (30 mg/kg) lost its ability to inhibit tumor growth. Despite the immune stimulatory effects of tasquinimod that were still maintained (Table S1), an optimal activation of the adaptive immune response to eradicate primary tumors seems to be compromised. We hypothesized that this resistance to tasquinimod treatment may be due to the induction of T-cell inhibitory pathways, such as the PD-1/PD-L1 axis. Indeed, the mRNA expression of PD-L1 was found to be increased in MBT-2 tumors treated with tasquinimod (Table S1). In addition, we observed an increase in the expression of PD-L1 gated on CD11b+ cells, including monocytic MDSCs, derived from MBT-2 tumors (Fig. 4C and D; Fig. S5). The expression level of PD-1 was not changed as a result of tasquinimod treatment (Fig. S2B and S4C). These data suggest that the alterations in the PD-L1 expression may be responsible for the lack of tumor growth inhibition in established MBT-2 tumors exposed to tasquinimod treatment. Our findings identified elevated PD-L1 expression on myeloid cells as a potential resistance mechanism by which tumors escape the effects of tasquinimod treatment. These findings indicate that the use of a combined treatment regimen including tasquinimod and PD-L1/PD-1 axis blockade may overcome this resistance.
Figure 4.
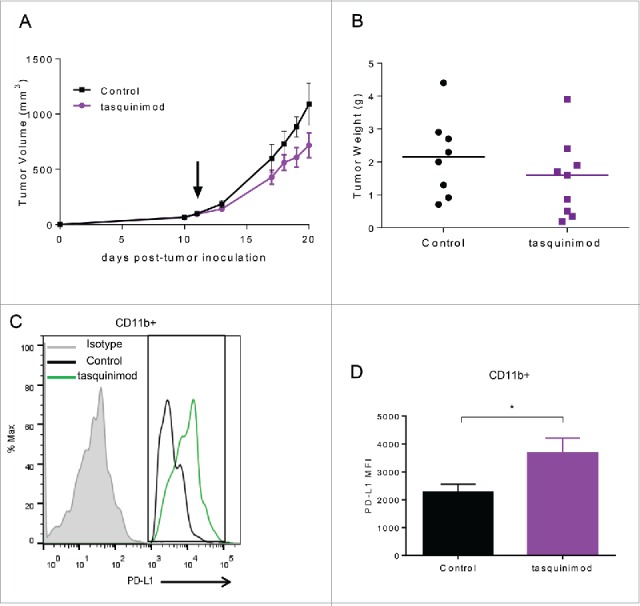
Treatment with tasquinimod had no effect on established MBT-2 tumor growth and induced an alteration in the profile of PD-1/PD-L1 axis. (A) Growth curves and (B) tumor weight of MBT-2 tumors treated with tasquinimod at 30 mg/kg (oral gavage, twice daily) after randomization at day 11 post tumor-cell inoculation (n = 12). Mice were sacrificed. Tumors were harvested, digested and then subjected to surface staining. (C) PD-L1 expression on myeloid cells treated with vehicle (control) or tasquinimod 30 mg/kg at day 15. (D) Quantitative data of the Median Fluorescence Intensity of PD-L1 gated on infiltrating myeloid cells CD11b+ (*p < 0.05; Mann–Whitney test, n = 5 mice per group).
Treatment of combined tasquinimod/Anti-PD-L1 enhances antitumor effects in BCa
As observed with tasquinimod treatment in the MBT-2 tumor model, anti-PD-L1 prevented tumor development when given as a single agent on day 1 after tumor cell inoculation (Fig. S6A). However, the antitumor activity of Anti-PD-L1 alone was lost in treating established tumors, potentially due to the increase in the tumor burden (Fig. 5B and Fig. S6B). Therefore, we investigated whether combining a modulator of myeloid cell functions and an immune checkpoint inhibitor may enhance the antitumor response (Fig. 5A). The results showed that mice treated with tasquinimod in combination with Anti-PD-L1 exhibited a significant slow-down in tumor growth (Fig. 5B; Control 413 ± 51 mm3; Anti-PD-L1 325 ± 52 mm3; tasquinimod 343 ± 67; combination 129 ± 15 mm3) and tumor weight (Fig. 5C) as compared to single treatments or control group. We demonstrated that the combination was superior to monotherapy with either agent in exerting an antitumor response.
Figure 5.
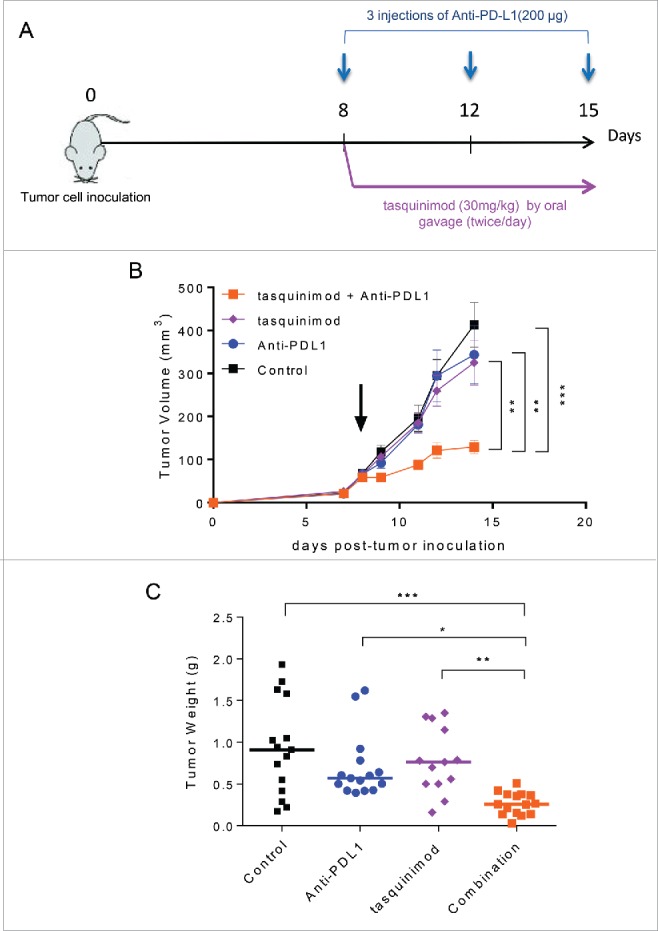
Combination of tasquinimod with Anti-PD-L1 therapy synergistically reduces tumor growth. (A) Study design: Subcutaneous MBT-2 tumors were allowed to grow until reaching an average size ranging between 50 and 100 mm3 (day 8). Mice (n = 16) were treated with IgG2B (control), Anti-PD-L1, tasquinimod or the combination of tasquinimod + Anti-PD-L1. (B) Tumor growth curves represent serial caliper measurements. Error bars indicate mean ± SEM (One-way ANOVA; **p < 0.005, ***p < 0.001). Tumor weights at the endpoint (day 15) are shown in (C) (Kruskal–Wallis test; *p < 0.05, **p < 0.005, ***p < 0.001). The experiments were repeated at least four times. Results from one representative experiment are shown.
Combination of treatments increases the activation of T cells
We next examined whether combined treatments could activate adaptive immune responses. We analyzed the percentage of TILs and their ability to release effector cytokines. The combination of tasquinimod with Anti-PD-L1 induced a 2.7-fold increase in CD8+ TILs (Fig. 6A). In parallel, a significant increase in the percentage of lymphocytic cells producing granzyme B was also observed in the combination treatment group compared to control (Fig. 6B). We also analyzed the cytokine expression profile in tumors exposed to treatment for 7 d (Fig. 6C). IL-7 and IL-15, both belonging to IL-2 superfamily, have been reported to increase the survival and cytotoxic effects of T cells to a greater extent than IL-2.31 Strikingly, strong increases in the production of IL-7, IL-15 and IL-12 were found in the tumors treated with the combination of tasquinimod and Anti-PD-L1 as compared to control (Fig. 6C). These cytokines were not significantly increased in the single treatment groups.
Figure 6.
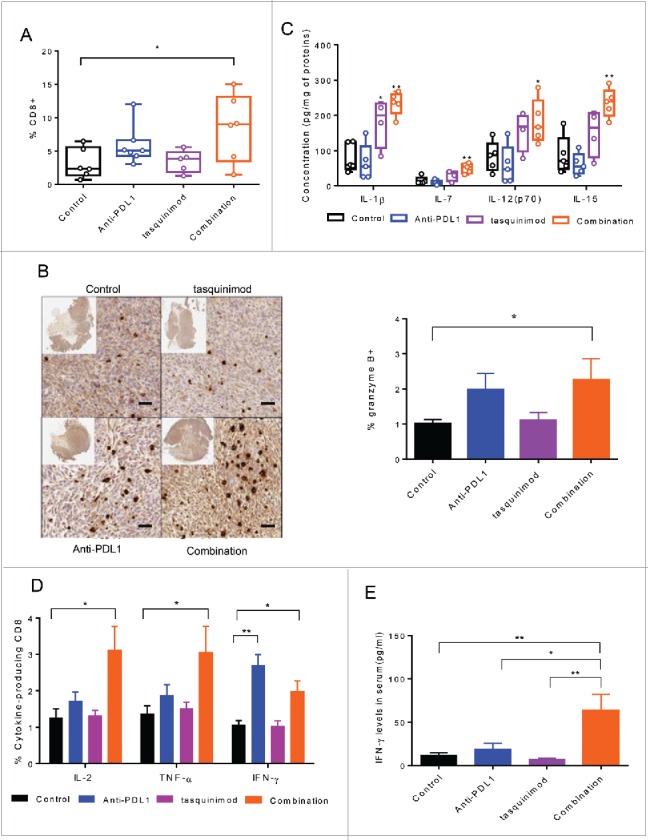
Combination of tasquinimod with Anti-PD-L1 increases cytotoxic T cell activity. (A) Quantitative data of the percentage of tumor-infiltrating CD8+ cells on day 15 after treatment (n = 6). (B) Left panel: Representative images showing immunostaining for granzyme B (brown staining) in tumors from control or treated groups. Original magnification: X200, inset: tumor overview. Scale bar: 50 µm. Right panel: Quantification of granzyme B positive cells on tumor sections expressed as a percentage of total cells using an antibody against granzyme B (Kruskal–Wallis test; *p = 0.0326). (C) The concentration levels (pg/mL) of the following cytokines: IL-1β, IL-7, IL-12(p70) and IL-15 in tumor lysate from each group were quantified using Luminex Technology (n = 5). (D) Splenocytes (n = 5) from each group were stimulated with PMA/ionomycin in the presence of Brefeldin A. IL-2, TNF-α and IFNγ production was examined by intracellular staining. Representative data (means ± SEM) showed the percentage of the different cytokines gated on CD8+ analyzed by flow cytometry. Asterisks denote statistical significance using one-way ANOVA (*p < 0.05; **p < 0.005). (E) Bars represented IFNγ concentrations in the serum of 10 mice from each group of treatment. p values were calculated based on Kruskal–Wallis test between the different groups (*p < 0.05; **p < 0.005).
To further investigate the immune responses that were induced by the combination of tasquinimod with Anti-PD-L1 in the MBT-2 tumor model, we isolated splenocytes from tumor-bearing mice and subjected them to stimulation with PMA/ionomycin for 4 h. An increase in the intracellular expression of IL-2, IFNγ and TNF-α gated on CD8+ was found in the combination group as compared to control (Fig. 6D). CD8+ producing IFNγ was also increased in the tumors treated with Anti-PD-L1 alone. In addition, high amounts of IFNγ into the serum of mice treated with the combination therapies were found as compared to single agents or to the control group (Fig. 6E). These data all together indicated that the combination of tasquinimod with Anti-PD-L1 treatment activated the adaptive immune system to exert a cytotoxic immune response.
Activation of both innate and adaptive immune cells is required to induce a potent immune response
To further understand the mechanism that underlies the observed increase in CD8+ cells producing cytotoxic cytokines in the combination group, we hypothesized that myeloid cells derived from treated tumors may directly interact with T cells and affect their function. To this end, we isolated myeloid cells CD11b+ from tumors and put them in culture with T cells derived from the spleen of naive mice.
The immune modulation of CD11b+ derived from established tumors treated with tasquinimod has limited ability to increase the proliferation of stimulated CD8+ T cells ex vivo (Fig. 7A and B). In addition, blockade of PD-L1 in CD11b+ derived from Anti-PD-L1 treated tumors had also moderate ability to activate T cells ex vivo. Importantly, combining modulators of both myeloid cells and T-cell inhibitory functions strongly increased the percentage of proliferating CD8+ cells. This was accompanied by a potent secretion of IFNγ into the supernatant as compared to control, tasquinimod alone or Anti-PD-L1 alone (Fig. 7C). In summary, we found that the combination of tasquinimod with Anti-PD-L1 in MBT-2 tumors modulates immunosuppressive myeloid cells affecting CD8+ T cell proliferation and production of IFNγ. These data further corroborate the synergistic interplay between myeloid cells and T cells and suggest therapeutic antitumor interventions aimed at modulating the communication between cell populations of both the innate and adaptive immune system.
Figure 7.

Combining a modulator of infiltrating-myeloid cells and an inhibitor of PD-1/PD-L1 axis increases T cell proliferation and T cell producing IFNγ. Myeloid cells CD11b+ were isolated from tumors using BD FACSAria II (BD Biosciences). T cells were isolated from spleen of naive mice using mouse pan T cell isolation Kit (Miltenyei). CFSE-labeled T cells were stimulated with CD3/CD28 beads ratio 1:1 (Life Technologies). Stimulated T cells were cultured with CD11b+ (at a ratio CD11b:T cells of 1:1) and incubated for 72 h at 37°C. (A) Representative histograms obtained by FACS analysis showing the fluorescence intensity of CFSE-T cells gated on CD8+. (B) The percentage of proliferating CD8+ cells from the different treated groups is shown. (C) IFNγ secretion in the supernatant of the co-culture is measured 72 h following incubation at 37°C using Luminex Technology. Experiments were repeated twice (Kruskal–Wallis test, *p < 0.05).
Discussion
In this report, we found that S100A9 was highly expressed in human BCa. We also observed the differential abundance of S100A9 expression in bladder tumor cells, whereas stromal cells always expressed high levels of S100A9. Elevated S100A9 expression in tumor stroma has been previously reported to be correlated with increased tumor cell invasiveness, macrophages recruitment, interleukin-6 production and a shorter survival in patients.32,33 Here, we investigated the role of stromal S100A9, including the myeloid compartment, in BCa progression. To this end, we used two preclinical models: MBT-2 and AY-27, both expressing S100A9 protein exclusively in the tumor stroma. We found that tasquinimod, a S100A9-targeting small molecule, prevented tumor growth in both models. The antitumor effects of tasquinimod may be explained by its ability to re-educate the TME, and particularly the myeloid cell compartment, toward a pro-inflammatory antitumor milieu. These observations fully corroborated with earlier investigations showing that tasquinimod was able to modulate tumor-infiltrating macrophages in mouse models of prostate cancer and melanoma.34
Surprisingly, tasquinimod treatment alone was shown effective in suppressing early stage tumor growth, but having minimal antitumor effect in rejecting established late stage tumors. These data raised the question whether tasquinimod may primarily prevent the early establishment of an immunosuppressive TME but once in place, the immunoregulatory functions of this environment could not be reversed by treatment. Nonetheless, we found that tasquinimod was also able to induce inflammatory stimuli in an established tumor milieu.
It is well know that tumors with a broad pro-inflammatory chemokine profile, as seen with tasquinimod treatment, are indicative of innate immune activation.35 Similarly, traditional chemotherapy or radiotherapy also triggers innate immune activation through a process involving immunogenic cell death.36-39 However, these tumors can escape immune surveillance and become resistant to therapeutic interventions through the activation of T cell-inhibitory pathways.35 Indeed, previous reports have shown that the PD-1/PD-L1 axis might be a key mechanism of acquired radioresistance in tumors.40,41 In agreement with these findings, we found that once bladder tumors progress, tasquinimod alone was not sufficient to achieve a complete activation of the immune system to eliminate primary tumors. One potential explanation for the observed resistance to tasquinimod treatment was the reported upregulation of PD-L1 in myeloid cells, potentially induced by the tumor inflammatory state related to tasquinimod treatment. The high expression of PD-L1 limits an efficacious immune response and thus promotes tumor relapse. Here, we demonstrated that tasquinimod increased the protein expression of IL-1β in tumors (Fig. 6C) which may in turn upregulate PD-L1 expression. This leads to an “inflamed” phenotype rendering tumors more sensitive to T-cell-mediated killing induced by the inhibition of the PD-1/PD-L1 axis.
The prevention of tumor growth by tasquinimod was also accompanied by an increase in the expression levels of PD-L1 (Fig. 3E; Fig. S2A). Here, we have shown that tasquinimod modulates the innate immune system. In early stage tumors, the competing kinetics between the tumor growth and the innate immune responses was in favor of a reduction in the tumor burden following tasquinimod treatment. However, this immune activation by tasquinimod was not sufficient to eradicate advanced tumors.
Multiple clinical trials with immune checkpoints inhibitors, such as antibodies against PD-1 or PD-L1, are currently ongoing in several types of cancer, including BCa.42-44 Early results with antibodies targeting PD-L1 have shown promise as potential therapeutics in this setting.43 Nevertheless, some patients do not respond to Anti-PD-L1 therapy.
In a phase II clinical study, tasquinimod improved progression free survival in patients with minimally symptomatic metastatic castration-resistant prostate cancer.45 In a pivotal phase III clinical study, tasquinimod reduced the risk of radiographic cancer progression or death (rPFS HR = 0.69; 95% CI: 0.60–0.80) in patients with metastatic castration-resistant prostate cancer who had not received chemotherapy. However, tasquinimod did not extend overall survival (HR = 1.09; 95% CI: 0.94–1.28).46
It is important to note that therapeutic immune interventions with single agents modulating innate immune system appear to be limited because of the plasticity of innate cells within the TME.47,48 Thus, approaches that involve the combination of innate immune system activation with immune checkpoints inhibitors may be capable of generating a more potent antitumor immunity and an increase in the efficacy of either treatment alone.
Our results demonstrated that tasquinimod/Anti-PD-L1 treatment elicited a synergistic tumor growth inhibition along with a potent antitumor immune response with the increase in the expression of IL-7 and IL-15 in the tumors of treated animals. Previously published data have shown that tumor-specific T cells activated and expanded with IL-7/IL-15 ex vivo and transferred back into tumor-bearing mice induced tumor regression.49 Both cytokines have been shown to promote tumor immunity by enhancing the function of effector immune cells. In addition, advances in adoptive cell therapy have relied on the use of such cytokines to create an optimal in vitro stimulation and expansion of effector T cells.50 Indeed, we found that the increased expression of IL-7 and IL-15 in tumors treated with combined tasquinimod/anti-PD-L1 was associated with an upregulation in the production of granzyme B. Granzyme B is known to be released by both cytotoxic T cells and NK cells. However, the density of NK cells was very low in MBT-2 tumors (Fig. S4E). This potentially suggests that the majority of this serine protease is produced by CD8+ cells and reflects an enhancement in their killing abilities in the TME. Interestingly, the increase in the infiltration of cytotoxic T cells was only seen in the combination group. In addition, after combined tasquinimod therapy and PD-L1 blockade, the production of pro-inflammatory cytokines IFNγ, TNF-α and IL-2 by CD8 T cells from spleen were greatly increased. High IFNγ levels were also detected in the serum of mice treated with the combination of treatments. All these data indicate that tasquinimod synergizes with Anti-PD-L1 to induce a potent antitumor immune response mainly through a cytotoxic T-cell-dependent mechanism. However, further analysis, such as the depletion of CD8+ cells in the combination group, may conclusively demonstrate their involvement.
The immunomodulation of myeloid cell functions by tasquinimod was not sufficient to mediate T cell producing IFNγ ex vivo, probably due to the upregulation of PD-L1 on myeloid cells which dampens the effector T cell response. In addition, blocking PD-L1 receptors on myeloid cells alone induced a limited increase in the proliferation of CD8+ T cells since their suppressive functions, including their ability to produce immunosuppressive molecules such as Lgals1, Tgfb1 and Il-10 were not modulated by anti-PD-L1 treatment (Fig. S7). Both molecules, tasquinimod and Anti-PD-L1, were crucial to induce a strong activation of T cell expansion and production of IFNγ ex vivo and also to increase the infiltration of cytotoxic T cells in tumors in vivo. Moreover tasquinimod alone at the dose of 30 mg/kg was not able to increase the median survival of mice (27 d) compared with 29 d for control mice (Fig. S8). However, despite the ability of Anti-PD1 treatment to increase the median survival of mice to 43 d, the combination of tasquinimod with Anti-PD1 was superior to either agent used alone.
Our data are in agreement with preliminary clinical results of Anti-PD-L1 (MPDL3280A) in urothelial BCa where a low signature of myeloid associated markers correlated with a better response to Anti-PD-L1 therapy, suggesting a potential role of myeloid cell biology in resistance to this type of therapy.51,52 These data highlight the importance of profiling tumors for rationally designing combination therapies based on the expression levels of myeloid markers and also taking into consideration the presence of T cells and PD-L1 expression as reported by Teng and colleagues.53 Here, we demonstrated that in tumors with high levels of immunosuppressive myeloid markers, a full immune engagement of the myeloid cells to induce T cell activation requires (i) an innate immune modulation, (ii) an inflamed tumor milieu leading to an increase in PD-L1 expression and (iii) an adaptive immune stimulation to release the brakes using PD-1/PD-L1 inhibitors.
In summary, the combination of tasquinimod with Anti-PD-L1 antibody synergizes to promote tumor regression and modulation of the TME in animal models of BCa. A combination of therapeutic strategies to improve innate immune system activation and T cell trafficking into the TME was found to be much more effective than either agent alone in this tumor type. Additional therapeutic strategies for combination of drugs that target the innate immune system, such as tasquinimod, and vaccination or T cell transfer should also be considered to increase the number and potency of tumor specific T cells, before blocking T-cell inhibitory pathways. All these combination strategies may be necessary to achieve clinical benefit in BCa patients.
Materials and methods
Cell lines
MBT-2 was purchased from the JCRB Cell Bank and cultured in EMEM (Life Technologies) supplemented with 10% fetal bovine serum. AY-27 was provided by Oncodesign (Dijon, France) and maintained in RPMI 1640 supplemented with 10% fetal bovine serum. The cell lines were free of mycoplasma contamination. No other authentication assay was performed.
In vivo experiments
6 to 7 week old male C3H/HeNRj mice were purchased from JANVIER Labs. 5 to 6 weeks old female Fischer 344 (F344/IcoCrl) rats were obtained from CHARLES RIVER.
MBT-2 cells (1 × 106) were injected subcutaneously into the flanks of mice and resulting tumors were allowed to grow for 21 d. Tumors were measured by caliper and tumor volume (mm3) was calculated using the formula = (Width)2 × Length/2.
Animals were treated with different doses of tasquinimod (0, 1, 1, 10 and 30 mg/kg) by oral gavage at a volume of 10 mL/kg twice a day for 21 d. To block PD-L1, 200 µg of Anti-PD-L1 (10F.9G2; BioXCell) or its isotype control (LTF-2; BioXCell) was administered in a volume of 100 µL by intraperitoneal route to mice every 3 d for a total of three to four injections for each experiment.
Procedures for the intravesical AY-27 cancer model were performed by Oncodesign (Dijon, France). Tumor cells (1 × 106) were injected orthotopically into the internal face of the bladder wall. Treatment with tasquinimod was initiated on day 4 at a dose of 2 mg/kg twice a day for 28 consecutive days. Cisplatin (CDDP, EBEVE) was given at 2 mg/kg once a day every 7 d starting day 4 following tumor cell inoculation. All the magnetic resonance images (MRIs) were performed using ParaVision® (Bruker Biospin).
All procedures using animals were validated by the Animal Care and Use Committee of Oncodesign (Oncomet) and IPSEN (C2EA), and were authorized by the French Ministry of Research.
FACS analysis
Single cell suspensions were prepared from tumors by incubating the tumors cross-cut into small pieces in 8 mg/mL Collagenase IV (Life Technologies) and 0.1% DNase (Sigma-Aldrich) for 45 min at 37°C. Cells were blocked with Fc-blockers (2.4G2), and then stained with different antibodies against CD11b (M1/70), F4/80 (clone BM8), CD206 (C068C2), Ly6C (AL-21), Ly6G (1A8), CD4+ (RM4-5), CD3ϵ (145-2C11), NK-1.1 (PK136) and CD8a (53-6.7) purchased from BD Biosciences, eBioscience and BioLegend. For cytokine staining, cells were stimulated in vitro with Leukocyte Activation Cocktail for 4 h in the presence of GolgiPlug™ (BD Biosciences), permeabilized and fixed using BD Cytofix/Cytoperm™ (BD Biosciences), then stained with anti-IL-2 (JES6-5H4), anti-TNF-α (MP6-XT22), and anti-IFNγ (XMG1.2) antibodies purchased from eBioscience. Flow cytometric analysis was performed with a BD Fortessa X-20 (BD Biosciences). Data were analyzed using FlowJo software (Tree Star Inc.). CD11b+ sorting was run on a BD FACSAria™ II (BD Biosciences) with the support of the Curie Institute core Facility (Orsay, France) and the final purity reached was more than 95%. Alternatively, CD11b+ cells were separated using MACS® microbeads (Miltenyi). This procedure yielded predominantly CD11b+ cells with purity greater than 80% as assessed by FACS analysis.
Ex vivo T cell proliferation assay
T cells (1 × 105) were isolated from the spleen of naive mice using a Pan T cell isolation kit (Miltenyi). T cells were labeled with CellTrace™ CFSE Cell Proliferation Kit (Life Technologies) and activated by Dynabeads® Mouse T-Activator CD3/CD28 (Life Technologies) at a bead-to-cell ratio of 1:1. Isolated CD11b+ cells (1 × 105) from tumors were added to labeled T cells at a ratio CD11b:T cells of 1:1 and were incubated in culture for 72 h.
Cytokine induction assay
Splenocytes (1 × 106) were stimulated with a mix of PMA and ionomycin in the presence of GolgiPlug™ for 4 h (BD Biosciences). Cells were harvested and stained for surface markers, then permeabilized, fixed and stained for intracellular cytokines with anti-IL-2 (JES6-5H4), anti-TNF-α (MP6-XT22), and anti-IFNγ (XMG1.2) antibodies.
Immunochemistry
S100A9 staining was performed on FFPE tissue sections from human tissue microarray (TMA) consisting of multiple cancer tissues (cancer survey, Origen and Top 4 multi tumor from Asterand) or BCa tumors (FFPE TMA, #BLC241 and urinary bladder carcinoma section #HuCAT416, Usbiomax). The tumor sections were incubated with an antibody against S100A9 (1:5000; Abcam #ab92507) after antigen retrieval in a low pH solution (Dako) and peroxidase/diaminobenzidine reaction. Staining intensity was assessed semi-quantitatively.
Animal tumors were sampled, cut in two pieces and either embedded in OCT compound or immersion-fixed in formalin for 24 h and embedded in paraffin. FFPE sections (5 µm) were incubated with granzyme B (1:100; Abcam #ab4059), S100A9 (1:1000; R&D systems #AF2065/ Abcam #ab62227) or CD8+ (1:200; AbD Serotec #MCA48R) antibody after antigen retrieval in low pH solution (Dako). Staining was revealed by peroxidase/diaminobenzidine reaction. Image analysis was performed on slide scans using Halo software (Indica labs). Granzyme B Stained cells were counted and were reported in relation to the total number of cells in the tumor section.
Cytokine determination by Multiplex assay
Cytokines were extracted from a 1 mm thick section of frozen OCT-compound (VWR, France) embedded tumors. After three washes in PBS, the pellet was resuspended in PBS + Protease Inhibitor Cocktail (Roche) and ground by ceramic beads in a homogenizer (Fastprep®, MP Biomedicals). The different samples were assayed for protein concentration. Cytokines were measured using Multiplex immuno-assay kits (Merck-Millipore) according to the manufacturer's instructions. Signal detection was performed on Luminex 200 (Luminex), and the Median Fluorescence Intensity (MFI) was recorded.
Quantitative Real-time PCR (qRT-PCR)
Cancer Survey cDNA Array was purchased from Origene and comprised 381 cDNA (2–3 ng/well) from 17 human tissues types either from normal or disease area.
RNA extraction of murine CD11b+ was performed using the PicoPure® RNA Isolation Kit (Life Technologies). RNA in tumors was isolated from 100 µm thick cryosections of OCT-embedded tumors using Trizol Reagent (Life Technologies). cDNAs were prepared using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies) following the manufacturer's instructions. cDNA from CD11b+ isolated cells was pre-amplified (14 cycles) using the TaqMan PreAmp Master Mix (Life Technologies). Real time PCR (q-PCR) was performed with a two-step PCR protocol (95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min) using Taqman gene expression (Life Technologies). The probes that were used are documented in Table S2. Hmbs was used as “housekeeping” gene whose expression was correlated to other housekeeping quantified genes (e.g. Cyclophilin A). Expression levels were calculated as normalized ΔCt expression values between target gene and “housekeeping” genes.
Statistics
Data were analyzed using the Prism 6.0 software (GraphPad Software) and validated by a biostatistician. Experiments were repeated two to four times as required. Normal data distribution was evaluated using the Shapiro–Wilk test. In this case, the p values were assessed by either Student's t test or by analysis of variance (ANOVA). For other data distributions, a Mann–Whitney or Kruskal–Wallis test was used. A p value less than 0.05 was considered statistically significant (*p <0.05; **p < 0.01; ***p < 0.001).
Supplementary Material
Disclosure of potential conflicts of interest
All authors are employees of Ipsen.
Acknowledgments
We would like to thank Oncodesign (Dijon) for performing in vivo experiments for AY-27 tumor model. We also acknowledge Dr. Tarig Bashir from Ipsen for his critical review and helpful comments.
Author Contributions
Conception and design: J. Nakhlé (all studies), F. Meyer-Losic (design of animal experiments and collaboration with Oncodesign for AY-27 tumor model). Development of methodology: J. Nakhlé (all studies). Acquisition of data: J. Nakhlé (all studies), V. Pierron (gene expression profiling), AL. Bauchet (analyzed human TMAs and immunohistochemistry), P. Plas (ran Luminex experiments for cytokine analysis). Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Nakhlé (all studies), A. Thiongane (pharmacodynamics of tasquinimod) and Jean-Luc Blachon (statistical analysis; Ipsen). Writing review and/or revision of the manuscript: J. Nakhlé and F. Schmidlin. All the authors read and approved the final manuscript. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Emilie Lebraud and Marie Daugan (both Ipsen). Study supervision: J. Nakhlé.
Funding
This work was financially supported by Ipsen.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/ 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2.Carneiro BA, Meeks JJ, Kuzel TM, Scaranti M, Abdulkadir SA, Giles FJ. Emerging therapeutic targets in bladder cancer. Cancer Treat Rev 2015; 41:170-8; PMID:25498841; http://dx.doi.org/ 10.1016/j.ctrv.2014.11.003 [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 4.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/ 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787-95; PMID:22378047; http://dx.doi.org/ 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De PM, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277-86; PMID:23518347; http://dx.doi.org/ 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 7.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markowitz J, Carson WE. Review of S100A9 biology and its role in cancer. Biochim Biophys Acta 2013; 1835:100-9; PMID:23123827; http://dx.doi.org/ 10.1016/j.bbcan.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T et al.. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 2008; 205:2235-49; PMID:18809714; http://dx.doi.org/ 10.1084/jem.20080132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker A, Hokamp NG, Zenker S, Flores-Borja F, Barzcyk K, Varga G, Roth J, Geyer C, Heindel W, Bremer C et al.. Optical in vivo imaging of the alarmin s100a9 in tumor lesions allows for estimation of the individual malignant potential by evaluation of tumor-host cell interaction. J Nucl Med 2015; 56:450-6; PMID:25678492; http://dx.doi.org/ 10.2967/jnumed.114.146688 [DOI] [PubMed] [Google Scholar]
- 11.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348:74-80; PMID:25838376; http://dx.doi.org/ 10.1126/science.aaa6204 [DOI] [PubMed] [Google Scholar]
- 12.Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol 2014; 193:3835-41; PMID:25281753; http://dx.doi.org/ 10.4049/jimmunol.1401572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massi D, Brusa D, Merelli B, Ciano M, Audrito V, Serra S, Buonincontri R, Baroni G, Nassini R, Minocci D et al.. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann Oncol 2014; 25:2433-42; PMID:25223485; http://dx.doi.org/ 10.1093/annonc/mdu452 [DOI] [PubMed] [Google Scholar]
- 14.Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, Herbst RS, Gettinger SN, Chen L, Rimm DL. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest 2014; 94:107-16; PMID:24217091; http://dx.doi.org/ 10.1038/labinvest.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inman BA, Sebo TJ, Frigola X, Dong H, Bergstralh EJ, Frank I, Fradet Y, Lacombe L, Kwon ED. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer 2007; 109:1499-505; PMID:17340590; http://dx.doi.org/ 10.1002/cncr.22588 [DOI] [PubMed] [Google Scholar]
- 16.Wang YU, Liu AN, Zhao SH. Association between B7-H1 expression and bladder cancer: a meta-analysis. Genet Mol Res 2015; 14:1277-86; PMID:25730066; http://dx.doi.org/ 10.4238/2015.February.13.6. [DOI] [PubMed] [Google Scholar]
- 17.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450-61; PMID:25858804; http://dx.doi.org/ 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol 2015; 42:363-77; PMID:25965355; http://dx.doi.org/ 10.1053/j.seminoncol.2015.02.015 [DOI] [PubMed] [Google Scholar]
- 19.Källberg E, Vogl T, Liberg D, Olsson A, Björk P, Wikström P, Bergh A, Roth J, Ivars F, Leanderson T. S100A9 interaction with TLR4 promotes tumor growth. PLoS One 2012; 7:e34207; PMID:22470535; http://dx.doi.org/ 10.1371/journal.pone.0034207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hibino T, Sakaguchi M, Miyamoto S, Yamamoto M, Motoyama A, Hosoi J, Shimokata T, Ito T, Tsuboi R, Huh NH. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res 2013; 73:172-83; PMID:23135911; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3843 [DOI] [PubMed] [Google Scholar]
- 21.Isaacs JT, Pili R, Qian DZ, Dalrymple SL, Garrison JB, Kyprianou N, Björk A, Olsson A, Leanderson T. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 2006; 66:1768-78; PMID:16955399; http://dx.doi.org/ 10.1002/pros.20509 [DOI] [PubMed] [Google Scholar]
- 22.Gupta N, Al UO, Shen L, Pili R. Mechanism of action and clinical activity of tasquinimod in castrate-resistant prostate cancer. Onco Targets Ther 2014; 7:223-34; PMID:24600234; http://dx.doi.org/ 10.2147/OTT.S53524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leanderson T, Ivars F. S100A9 and tumor growth. Oncoimmunology 2012; 1:1404-5; PMID:23243608; http://dx.doi.org/ 10.4161/onci.21027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Björk P, Björk A, Vogl T, Stenström M, Liberg D, Olsson A, Roth J, Ivars F, Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol 2009; 7:e97; PMID:19402754; http://dx.doi.org/ 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramachandran IR, Lin C, hase T, Gabrilovich DI, Nefedova Y. A Novel Agent Tasquinimod Demonstrates a Potent Anti-Tumor Activity in Pre-Clinical Models of Multiple Myeloma. Blood 2014; 124 (21); 5729-5729 [Google Scholar]
- 26.Nacken W, Sopalla C, Propper C, Sorg C, Kerkhoff C. Biochemical characterization of the murine S100A9 (MRP14) protein suggests that it is functionally equivalent to its human counterpart despite its low degree of sequence homology. Eur J Biochem 2000; 267:560-5; PMID:10632726; http://dx.doi.org/ 10.1046/j.1432-1327.2000.01040.x [DOI] [PubMed] [Google Scholar]
- 27.Steding CE, Wu ST, Zhang Y, Jeng MH, Elzey BD, Kao C. The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology 2011; 133:221-38; PMID:21453419; http://dx.doi.org/ 10.1111/j.1365-2567.2011.03429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol 2013; 13:777-89; PMID:24113868; http://dx.doi.org/ 10.1038/nri3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croucher DR, Saunders DN, Lobov S, Ranson M. Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat Rev Cancer 2008; 8:535-45; PMID:18548086; http://dx.doi.org/ 10.1038/nrc2400 [DOI] [PubMed] [Google Scholar]
- 30.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V et al.. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19:1264-72; PMID:24056773; http://dx.doi.org/ 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sim GC, Radvanyi L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev 2014; 25:377-90; PMID:25200249; http://dx.doi.org/ 10.1016/j.cytogfr.2014.07.018 [DOI] [PubMed] [Google Scholar]
- 32.Tidehag V, Hammarsten P, Egevad L, Granfors T, Stattin P, Leanderson T, Wikström P, Josefsson A, Hägglöf C, Bergh A. High density of S100A9 positive inflammatory cells in prostate cancer stroma is associated with poor outcome. Eur J Cancer 2014; 50:1829-35; PMID:24726733; http://dx.doi.org/ 10.1016/j.ejca.2014.03.278. [DOI] [PubMed] [Google Scholar]
- 33.Fang WY, Chen YW, Hsiao JR, Liu CS, Kuo YZ, Wang YC, Chang KC, Tsai ST, Chang MZ, Lin SH et al.. Elevated S100A9 expression in tumor stroma functions as an early recurrence marker for early-stage oral cancer patients through increased tumor cell invasion, angiogenesis, macrophage recruitment and interleukin-6 production. Oncotarget 2015; 6:28401-24; PMID:26315114; http://dx.doi.org/ 10.18632/oncotarget.4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen L, Sundstedt A, Ciesielski M, Miles KM, Celander M, Adelaiye R, Orillion A, Ciamporcero E, Ramakrishnan S, Ellis L et al.. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol Res 2015; 3:136-48; PMID:25370534; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014-22; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology 2013; 2:e26536; PMID:24404424; http://dx.doi.org/ 10.4161/onci.26536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vacchelli E, Aranda F, Eggermont A, Galon J, Sautès-Fridman C, Cremer I, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014; 3:e27878; PMID:24800173; http://dx.doi.org/ 10.4161/onci.27878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ 2014; 21:39-49; PMID:23832118; http://dx.doi.org/ 10.1038/cdd.2013.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngiow SF, McArthur GA, Smyth MJ. Radiotherapy complements immune checkpoint blockade. Cancer Cell 2015; 27:437-8; PMID:25873170; http://dx.doi.org/ 10.1016/j.ccell.2015.03.015 [DOI] [PubMed] [Google Scholar]
- 40.Deng L, Liang H, Burnette B, Weicheslbaum RR, Fu YX. Radiation and anti-PD-L1 antibody combinatorial therapy induces T cell-mediated depletion of myeloid-derived suppressor cells and tumor regression. Oncoimmunology 2014; 3:e28499; PMID:25050217; http://dx.doi.org/ 10.4161/onci.28499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014; 124:687-95; PMID:24382348; http://dx.doi.org/ 10.1172/JCI67313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eggermont AM, Maio M, Robert C. Immune Checkpoint Inhibitors in Melanoma Provide the Cornerstones for Curative Therapies. Semin Oncol 2015; 42:429-35; PMID:25965361; http://dx.doi.org/ 10.1053/j.seminoncol.2015.02.010 [DOI] [PubMed] [Google Scholar]
- 43.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL et al.. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014; 515:558-62; PMID:25428503; http://dx.doi.org/ 10.1038/nature13904 [DOI] [PubMed] [Google Scholar]
- 44.Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buqué A, Senovilla L, Baracco EE, Bloy N, Castoldi F, Abastado JP, Agostinis P et al.. Classification of current anticancer immunotherapies. Oncotarget 2014; 5:12472-508; PMID:25537519; http://dx.doi.org/ 10.18632/oncotarget.2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pili R, Häggman M, Stadler WM, Gingrich JR, Assikis VJ, Björk A, Nordle O, Forsberg G, Carducci MA, Armstrong AJ. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol 2011; 29:4022-8; PMID:21931019; http://dx.doi.org/ 10.1200/JCO.2011.35.6295 [DOI] [PubMed] [Google Scholar]
- 46.Carducci MA, Armstrong AJ, Pili R, Ng S, Huddart R, Agarwal N, Khvorostenko D, Lyulko O, Brize A, Vogelzang NJ et al.. A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Tasquinimod (TASQ) in Men with Metastatic Castrate Resistant Prostate Cancer (mCRPC). Abstract presented at the 18th ECCO - 40th ESMO European Cancer Congress, 25–29 September Vienna, Austria 2015. [Google Scholar]
- 47.Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer 2013; 13:365-76; PMID:23535846; http://dx.doi.org/ 10.1038/nrc3498 [DOI] [PubMed] [Google Scholar]
- 48.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014; 74:5057-69; PMID:25082815; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim HR, Hwang KA, Park SH, Kang I. IL-7 and IL-15: biology and roles in T-Cell immunity in health and disease. Crit Rev Immunol 2008; 28:325-39; PMID:19166383; http://dx.doi.org/ 10.1615/CritRevImmunol.v28.i4.40 [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011; 3:3856-93; PMID:24213115; http://dx.doi.org/ 10.3390/cancers3043856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao Y, Rabe C, Kowanetz M, Powles T, Vogelzang NJ, Petrylak DP, Loriot Y, Denker M, Nakamura R, Wu QJ et al.. Myeloid cell biology and inhibition of anti-tumor immune responses by MPDL3280A in urothelial bladder cancer. J ImmunoTher Cancer 2014; S3-131; http://dx.doi.org/ 10.1186/2051-1426-2-S3-P131 [DOI] [Google Scholar]
- 52.Zhu Y, Hawkins WG, DeNardo DG. Regramming myeloid responses to improve cancer immunotherapy. Oncoimmunology 2015; 4:e974399; PMID:26155432; http://dx.doi.org/ 10.4161/2162402X.2014.974399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res 2015; 75:2139-45; PMID:25977340; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.