

Abstract
Scope
High glycemic load diets have been associated with increased breast cancer risk in population-based studies, but the evidence is mixed. This investigation determined whether diets differing in glycemic load affected the carcinogenic process using a preclinical model.
Methods and results
Human diets, formulated to differ 2-fold in glycemic load, were evaluated in the 1-methyl-nitrosourea-induced (37.5 mg/kg) mammary carcinogenesis model. Cancer incidence (23.3 vs 50.0%, P = 0.032 ), multiplicity, (0.40 vs 1.03, P = 0.030) and burden, (0.62 vs 1.19 g/rat, P = 0.037) were reduced in the low versus high glycemic load diets, respectively. However, the low glycemic protective effect was attenuated when two purified diets that differed in resistant starch and simulated the glycemic effects of the human diets were fed. Protection was associated with alterations in markers of cell growth regulation.
Conclusion
Our findings show that human low or high glycemic load dietary patterns differentially affect the carcinogenic response in a non-diabetic rodent model for breast cancer. However, factors that are associated with these patterns, in addition to dietary carbohydrate availability, appear to account for the differences observed.
Keywords: glycemic load, cell growth regulation, mammary carcinogenesis, resistant starch
Graphical Abstract
Human diets, formulated to differ 2-fold in glycemic load, were evaluated in the 1-methyl-nitrosourea-induced (37.5 mg/kg) mammary carcinogenesis model. Cancer incidence (23.3 vs 50.0%, P = 0.032 ), multiplicity, (0.40 vs 1.03, P = 0.030) and burden, (0.62 vs 1.19 g/rat, P = 0.037) were reduced in the low versus high glycemic load diets, respectively. However, the low glycemic protective effect was attenuated when two purified diets that differed in resistant starch and simulated the glycemic effects of the human diets were fed. Protection was associated with alterations in markers of cell growth regulation.
Introduction
A dominant focus of investigations of the role of diet in affecting breast cancer risk has centered on the amount and type of fat ingested [1, 2]. However, increasing attention is being given to the potential role of carbohydrates [3]. That interest is not limited to the evaluation of dietary fiber’s contribution to carbohydrate quality, but also to the glycemic response that a food invokes and that is quantified as glycemic index [4]. In addition, the effect of total amount of carbohydrate in a meal on postprandial glycemic response can be quantified and is referred to as dietary glycemic load [5]. With advances in methodology, it has become possible to characterize the patterns of foods consumed as either high or low glycemic load patterns [6].
High glycemic load dietary patterns are associated with persistent increases in circulating levels of glucose and insulin, the impact of which is determined in part by the metabolic state of an individual [4]. For example, the therapeutic benefits of low glycemic load diets for glucose and insulin regulation in obesity-associated insulin resistance and in diabetes are well recognized [7, 8]. Thus, given the evidence of increased risk for breast cancer in diabetics [9], the potential role of different glycemic load dietary patterns in affecting risk for breast cancer has been investigated and mixed evidence has emerged [10–17].
One approach to investigating glycemic load has been to develop a specific menu and recipe based dietary pattern of calculated glycemic load and to conduct highly controlled metabolic human feeding studies as reported in [18]. The use of this approach created an opportunity for the study reported herein. For years there have been concerns about the relevance of animal models for cancer to the human disease [19–26]. There also has been considerable debate about the limitations imposed by using the gold standard approach of purified diets in rodent models to study the effects of diet and nutrition on the carcinogenic process because human populations do not eat purified diets [27–30]. While such discussions have identified both strengths and weaknesses of animal models for cancer and the various dietary formulations that can be fed to rodents, our study was designed to address a simple question: does feeding defined diets that were fed to a population of human participants in a randomized controlled format [31–34], provide any insights when the same human diets are fed to rats in a well characterized model for breast cancer with established relevance to the disease process in women? This paper reports the effects of two dietary glycemic load patterns on the carcinogenic response and on host systemic and cell autonomous processes considered to play a role in human breast carcinogenesis.
Materials and methods
Experiment 1 Evaluation of human diets differing in glycemic load
Female Sprague Dawley rats were obtained from Taconic Farms, Germantown, NY at 19 days of age (DOA) and injected with 37.5 mg of 1-methyl-1-nitrosourea/kg body weight, intraperitoneally, at 20 DOA as previously described [35]. All animals were fed purified AIN-93G diet for 1 week following carcinogen injection and were then randomized to one of two dietary groups (n = 30 rats/diet group) and fed either the human high or low glycemic load diets that were formulated to differ by a factor of 2 (Supplementary Tables S1 and S2). The menus are described in supplemental information section of [33]. While consideration was given to feeding a purified diet in this experiment, it was decided that it would be more informative to construct purified diets that mirrored the macronutrient content of the human diet (Experiment 2).
Preparation and characterization of human diets: Each day of a 7-day menu was prepared in the same metabolic facility used in the clinical study and was homogenized in a Hobart blender [31]. All 7 days of homogenized food for each diet arm were combined and further processed in a paddle mixer until a homogenous mixture was obtained. That mixture was then divided into 50 g portions and immediately frozen. Portions were labeled in a manner that blinded the investigators to whether the portions came from the high or low glycemic load diet. Proximate analysis of both diets in the form fed to the rats was performed by a commercial laboratory (Warren Analytical, Greeley, CO). The glycemic response to each diet was determined using a previously reported procedure [36].
Experiment 2 Evaluation of purified diets differing in glycemic load
Female Sprague Dawley rats were obtained from Taconic Farms, Germantown, NY at 19 DOA and injected with 37.5 mg of 1-methyl-1-nitrosourea/kg body weight, intraperitoneally, at 20 DOA as previously described [35]. All animals were fed purified AIN-93G diet for 1 week post carcinogen injection and were then randomized to one of two dietary groups (n = 50 rats/diet group) and fed either the low or high glycemic load purified diet formulated using corn starch that differed in its content of resistant starch (detailed formulation, Supplementary Table S2).
Procedures common to both experiments
Rats were fed 2 meals per day (08:00–11:00 and 14:00–17:00), 7 days per week until the end of the study; food consumption was ad libitum during each meal. All rats were weighed weekly and palpated twice per week for detection of mammary tumors beginning at 21 days post-carcinogen until study termination. Both experiments were terminated 8 weeks post carcinogen based on our previous work [35]. All animals were group housed (3/cage) in an AAALAC accredited vivarium. The animal room was maintained at 22 ± 1°C with 50% relative humidity and a 12-h light/12-h dark cycle. The work reported was reviewed and approved by Colorado State University Animal Care and Use Committee and conducted according to that committee’s guidelines.
Necropsy and Sample Collection
Following an overnight fast, rats were killed over a 3-hour time interval via inhalation of gaseous carbon dioxide. The sequence in which rats were euthanized was stratified across groups so as to minimize the likelihood that order effects would masquerade as treatment-associated effects. Detected tumors were excised, weighed, and cut into two parts; one section was fixed in formalin for histological classification and the remainder of each tumor was snap frozen in liquid nitrogen. All tumors were histopathologically classified according to established criteria [37] and only mammary carcinomas were used in tabulating the carcinogenic response and for molecular assessments. Because a statistically significant effect on the carcinogenic response was observed only in Experiment 1, additional analyses were limited to that experiment.
Analyses of plasma
Plasma from the animals in the carcinogenesis experiment was isolated by centrifugation at 1000 × g for 10 min at room temperature and then stored at −80C until it was analyzed. Plasma was subjected to the assessment of the following molecules: insulin-like growth factor 1 (IGF-1), IGF binding protein 3 (IGFBP-3), adiponectin, insulin, and leptin using commercially available ELISA assays (Diagnostic Systems Laboratory, Webster, TX; Cayman Chemicals, Ann Arbor, MI; and Millipore, Billerica, MA). Glucose in plasma was determined enzymatically using a commercially available kit (Pointe Scientific, Inc., Canton, MI.). Assays were performed according to manufacturer’s instructions.
Statistical Analyses of the carcinogenesis experiments and plasma analytes
Experiment 1 was powered on cancer incidence with the assumed incidence in the high glycemic load group of 70% and with a predicted reduction of 50% in the low glycemic load group based on the epidemiological literature [10–17].; the power was 80% with an n = 30/group. However, the actual power based on results was approximately 50%, primarily because the cancer incidence in the high glycemic load (referent) group only reached 50%. In Experiment 2, the n was increased to 50 rats/per group based on Experiment 1 results. With n = 50 rats/group, 80% power existed to detect the differences observed in Experiment 1.
Effects on cancer incidence were evaluated by Pearson chi-square analysis and cancer latency was evaluated by survival analysis [38]. Differences between groups in cancer multiplicity (number of mammary carcinomas/rat) and in cancer mass were evaluated by the Kruskal Wallis test. Differences between groups in body weight and plasma molecules were analyzed by ANOVA. All analyses were performed using Systat statistical analysis software version 12 (Systat Software, San Jose, CA).
Proteomic analysis
Western blotting was used in a manner analogous to the use of protein array technology. Primary antibodies used in this study were anti-cyclin-D1, anti-p27Kip1 from Thermo Fisher Scientific (Kalamazoo, MI); anti-Bax and anti-Bcl-2 from BD Biosciences (San Diego, CA); anti Caspase 3, anti-pRB/RB, anti-pAMPK, anti-AMPK, anti-pAkt, anti-Akt, anti-pmTOR, anti-mTOR, anti-pP70S6, anti-P70S6, anti- pTSC2, anti-XIAP, anti-rabbit immunoglobulin-horseradish peroxidase-conjugated secondary antibody and LumiGLO reagent with peroxide were purchased from Cell Signaling Technology (Beverly, MA); anti-TSC2, anti-p21Cip1, anti-VEGF and anti-mouse immunoglobulin-horseradish peroxidase-conjugated secondary antibody were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); mouse anti-β-Actin primary antibody was obtained from Sigma Aldrich (St. Louis, MO). For analysis, tissue was homogenized in lysis buffer [40 mM Tris-HCl (pH 7.5), 1% Triton X-100, 0.25 M sucrose, 3 mM EGTA, 3 mM EDTA, 50 μM β-mercaptoethanol, 1 mM phenyl-methylsulfonyl fluoride, and complete protease inhibitor cocktail (Calbiochem, San Diego, CA)]. The lysates were centrifuged at 7500 × g for 10 min at 4 °C and supernatant fractions collected and stored at −80 °C. Supernatant protein concentrations were determined by the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Western blotting was performed as described previously. Briefly, 40 μg of protein lysate per sample was subjected to 8–16% sodium dodecyl sulfate-polyacrylamide gradient gel electrophoresis (SDS-PAGE) after being denatured by boiling with SDS sample buffer [63 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue] for 5 min. After electrophoresis, proteins were transferred to a nitrocellulose membrane and thereafter cut into strips at the appropriate molecular weights for each target protein. The amounts of target proteins were determined using specific primary antibodies, followed by treatment with the appropriate peroxidase-conjugated secondary antibodies and visualized by LumiGLO reagent western blotting detection system. The chemiluminescence signal was captured using a ChemiDoc densitometer (Bio-Rad) that was equipped with a CCD camera having a resolution of 1300 × 1030. Quantity One software (Bio-Rad) was used in the analysis. The Quantity One software has a warning algorithm that notifies the user if pixel density is approaching saturation so that all signals used for analysis are in the linear range. All Western blot signals were within a range where the signal was linearly related to the mass of protein and actin-normalized scanning density data were used for analysis.
Data evaluation
Data from plasma and Western blot analyses were evaluated via unsupervised and supervised multivariate techniques per our previously published approach [39–42]. Briefly, principal components analysis (PCA), an unsupervised clustering technique was first applied to each data set [43]. The PCA model can be written: X = Xbar +TP′ + E where X is the matrix of measured variables, Xbar is a vector of means (all 0 when the data are centered), T is a matrix of scores that summarize the X variables, P is a matrix of loadings, and E is a matrix of residuals. PCA analysis was followed by evaluation of the same data sets using orthogonal projections to latent structures for discriminant analysis (OPLD-DA), a supervised class-based method [39–41, 44]. The OPLS-DA model can be written: X = TpPp′ + ToPo′ + E where the interpretation of equation 2 is similar to that for the PCA model; however, an additional rotation has been applied using the class information to partition TP′ into a predictive, TpPp′, and an orthogonal, ToPo′, component. The number of predictive and orthogonal components in the models was determined by cross-validation.
Visualization of PCA and OPLS-DA
Scatter plots of the first two score vectors for the PCA models were drawn, along with 95% confidence ellipses based on Hotelling’s multivariate T2, to identify outliers that might bias the results. For OPLS-DA, class separation was shown in several ways. Biplots were used to co-chart scores and loadings from the OPLS-DA model for their simultaneous display and interpretation. Thus this plot displays similarities and dissimilarities among observations (each carcinoma assessed) and permits interpretation of the observations in terms of the variables (each target protein that was measured). Observations situated near variables are high in those variables and are low in variables situated opposite. Observations close to the plot origin have average properties. Variables close to the plot origin do not contribute to the formation of the scores in question, i.e. they are poorly described by the model components. S-plots were constructed to identify influential proteins in the separation of treatment groups. S-plots based on the first principal component show reliability (modeled correlation) plotted against feature magnitude (loadings or modeled covariance). If protein concentrations have variation in correlation and covariance between classes, this plot will assume an S-shape, with heavily influential features separating from other features at the upper right and lower left tails of the feature cloud within the model space [40, 41]. Variable importance in the projections (VIP) plots were also computed. VIP expresses the influence on Y (class assignment, i.e., low or high glycemic load diet) of every variable xk. The VIP for each variable is expressed with its respective 95% confidence interval. VIP can be compared with one another. VIP greater than 1 are considered most relevant to accounting for class separation; whereas, VIP values less than 0.5 are considered unimportant. VIP scores between 0.5 and 1.0 are considered to be in a gray area relative to interpreting their importance to class separation. All multivariate analyses were done using SIMCA-P+ v.12.0.1 (Umetrics, Umea, Sweden).
Results
Low glycemic load diet protects against mammary carcinogenesis
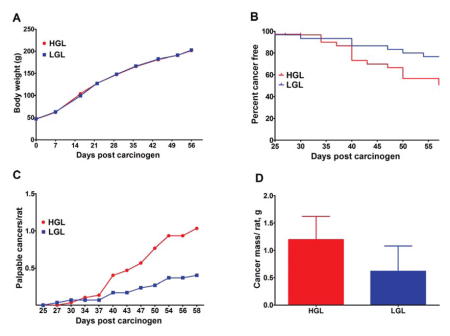
The effect of human diets that differed in glycemic load was initially determined (Experiment 1). Growth rates of the rats fed the low or high human glycemic load diets were similar (Figure 1A). Cancer incidence was reduced (23.3 vs 50.0%, P = 0.032, Odds ratio = 0.30, 95% CI = 0.10 to 0.92), and cancer latency prolonged (Figure 1B, Hazard ratio = 0.37, 95% CI = 0.16 to 0.88, P = 0.024) in rats fed the low glycemic diet. Cancer multiplicity over time (Figure 1C), cancer multiplicity at necropsy, (0.40, 95% CI = 0.05 to 0.75, vs 1.03, 95% CI = 0.49 to 1.58, P = 0.030), and cancer burden (Figure 1D: 0.62, 95% CI = −0.32 to 1.56, vs 1.19, 95% CI = 0.32–2.07, P = 0.037), were also reduced in the low versus high glycemic diet group.
Fig. 1.

Effects of high or low glycemic load human diets on body weight gain and the carcinogenic response. (A) change in group mean body weight over time. (B) time to event curves; cancer latency was delayed by the LGL diet (P = 0.024); final cancer incidence was reduced by 50% in LGL versus HGL (P = 0.032). (C) mean number of palpable cancers per rat over time; final cancer multiplicity was reduced by 40% in LGL versus HGL, (P = 0.030). (D) cancer mass per rat (cancer burden) was reduced by 52% in LGL versus HGL (P = 0.037). HGL = high glycemic load diet; LGL = low glycemic load diet.
Human dietary patterns that differ in glycemic load also vary in many other respects, which conceivably could account for the mixed findings observed in population-based studies [10–17]. Therefore, we constructed purified diets that had a similar macronutrient content to the 7-day human cycle menu (Supplementary Table S1), but that differed in resistant starch which is a means of manipulating the glycemic load of the diet formulation. The purified diets were calculated to have a 2-fold difference in glycemic load, the same as the human diets. The effect of these purified diets on the carcinogenic response was determined (Experiment 2). Growth rates of the rats fed these diets were similar (Figure 2A). However, the effect of glycemic load on the carcinogenic response was greatly attenuated when the purified diets were fed (Figure 2B–D). Specifically, in the low glycemic diet group, cancer incidence was lower (49.0% vs 62.7%, P = 0.165, odds ratio = 0.5, 95% CI = 0.26 to 1.26), and cancer latency was prolonged (Figure 2B, hazard ratio = 0.66, 95% CI = 0.38 to 1.13, P = 0.131), but these differences were not statistically significant. Similarly, neither cancer multiplicity over time (Figure 2C), cancer multiplicity at necropsy, 1.18, 95% CI = 0.73 to 1.63, vs 1.14, 95% CI = 0.81 to 1.46, P = 0.479), nor cancer burden (Figure 2D, 1.75, 95% CI = 0.72 to 2.77, vs 2.10, 95% CI = 1.03 to 3.17, P = 0.251), were significantly affected in rats fed purified low versus high glycemic load diets.
Fig. 2.

Effects of high or low glycemic load purified rodents diets on body weight gain and the carcinogenic response. (A) change in group mean body weight over time. Time to event curves; cancer latency was unaffected by the LGL diet (P = 0.13). (B) final cancer incidence was not significantly affected in LGL versus HGL (P = 0.23). (C) ave. number of palpable cancers per rat over time; final cancer multiplicity was not significantly affected in LGL versus HGL (P = 0.048). (D) cancer mass per rat (cancer burden) was unaffected in LGL versus HGL (P = 0.25). HGL = high glycemic load diet; LGL = low glycemic load diet.
Glycemic response does not account for the differences in carcinogenic response observed between human and purified rodent diets
We reasoned that since the glycemic load of the human diets and purified rodent diets were based on calculated values, the glycemic response to these diets might differ in the rat and thus account for the differences observed in the carcinogenic response. The glycemic response to the same amount of carbohydrate (1 g/kg body weight) from each diet, human and rodent, was determined by gavaging a slurry of each diet to fasted rats. The area under the curve analysis showed that the patterns of glycemic response were similar (Figure 3) between the human and rodent diets. Factorial analysis of variance of the area under the curve values for each diet (n = 8 rats per diet group) indicated that the low versus high glycemic load diets differed (P = 0.009), but the differences between the human and purified rodent diets were not statistically significant (P = 0.705). Thus, we judge that it is unlikely that the small differences that were apparent in Figure 3 between human diets and the purified rodent diets account for the marked differences that were observed in the carcinogenic response (Figures 1 and 2).
Fig. 3.

Effect of diets varying in available carbohydrate content on glycemic response in the rat. The glycemic response to diets prepared for human consumption whose calculated glycemic load was 244 (HGL) and 117 (LGL). (A) when fasted rats were gavaged (1 g carbohydrate/kg body weight) with the composite human diets, prepared as described in the methods section, the mean area under the curve, using the analysis of incremental area, was 105 (95% CI = 26.6 to 183.6) and 18 (95% CI = 42.0 to 28.7) for HGL and LGL, respectively. The glycemic response for purified rodent diets (Supplementary Table S2). (B) the mean area under the curve, using the incremental area method of analysis, was 72 (95% CI = 47.2 to 96.3) and 19 (95% CI = −37.4 to 76.2) for HGL and LGL, respectively. Factorial analysis of variance of the area under the curve values for each diet (n = 8 per diet group) indicated that the low versus high glycemic load diets differed (P = 0.009), but the differences between the human and purified rodent diets were not statistically significant (P = 0.705). HGL = High glycemic load diet; LGL = low glycemic load diet.
Circulating analytes as indicators of cancer risk
Since only the human diets showed a differential effect of glycemic load on the carcinogenic process, we focused our analyses on tissue from Experiment 1. The plasma obtained from each animal (n = 30/diet group) was evaluated for the same analytes evaluated in the clinical studies that were conducted using these diets [31–34]. Differences were observed in concentrations of fasting plasma glucose, insulin, IGF-1, hsCRP, or IL-6, leptin and adiponectin in the low versus high glycemic load group, but none of those differences were statistically significant (Table 1). A similar response was observed in the clinical studies of the same low and high glycemic load diets (Supplementary Table S3); however, unlike those clinical studies of non obese women who were clinically free from cancer, we had data for cancer endpoints for each animal (Experiment 1). Therefore, plasma data were subjected to unsupervised and supervised analyses to determine whether plasma analytes in aggregate were predictive of dietary group assignment or the presence or absence of cancer. Unsupervised PCA revealed poor separation either between diet groups or between cancer bearing versus cancer free rats (Data not shown). However, when these data were subjected to supervised OPLS-DA, classification accuracy for diet group was 83% and for cancer status was 78%. Based on variable importance analyses (VIP), the most important plasma analytes for distinguishing between diet group or cancer bearing and cancer free rats were plasma insulin and adiponectin.
Table 1.
Effect of dietary glycemic load on plasma analytes
| Plasma analyte | Low glycemic load diet | High glycemic load diet | P-value |
|---|---|---|---|
| Glucose (mmol/L) | 8.1 ± 1.1 | 8.2 ± 1.9 | 0.734 |
| Insulin (pmol/L) | 161 ± 24 | 174 ± 28 | 0.078 |
| Insulin like growth factor-1 (ug/L) | 978 ± 202 | 1035 ± 149 | 0.215 |
| hsC-reactive protein (mg/L) | 300 ± 42 | 315 ± 51 | 0.239 |
| Interleukin-6 (ng/L) | 56.9 ± 9.8 | 61.2 ± 17.9 | 0.254 |
| Adiponectin (ug/L) | 14.2 ± 2.4 | 11.8 ± 1.1 | 0.001 |
| Leptin (ug/L) | 1.2 ± 0.3 | 1.4 ± 0.3 | 0.011 |
Values are means ± SD, n=30. Data were evaluated by ANOVA. P-values shown are unadjusted for the number of endpoints assessed. For statistical significance P<0.007 is required (P=0.05/7 (the number of endpoints measured)
Plasma analytes predict activity of downstream targets in mammary carcinoma
Insulin and adiponectin have been reported to mediate pro- or anti-carcinogenic effects, respectively, through their downstream targets, protein kinase B (Akt) and AMP activated protein kinase (AMPK). Therefore, we used regression analysis to determine the relationship between plasma insulin and activated AKT (r2= 0.31, p=0.04) and between adiponectin and activated AMPK (r2= 0.17, p=0.15) in mammary carcinomas. Both analyses are consistent with established canonical relationships and pointed to the potential involvement of mTOR network signaling in accounting differential responses to the human diets. To investigate this possibility, a more detailed proteomic analysis was undertaken.
Proteomic analyses indicate dissimilarity in cell growth regulation in carcinomas occurring in low versus high glycemic load animals
We reasoned that if glycemic response was driving the carcinogenic response via modulation of mTOR network activity mediated by insulin and adiponectin that differences in regulation of multiple nodes in that signaling cascade would be observed. To evaluate this hypothesis, Western blot data for 28 proteins were mean centered, pareto-scaled, and evaluated using unsupervised PCA (Figure 4A). There was partial separation of carcinoma by diet group; therefore, the same data set were subjected to supervised OPLS-DA in order to permit separation of variation due to diet treatment from unrelated variation and to noise (Figure 4B). The cross-validated predictive ability of this 7 component model, Q2(Y), was 54.5% with 72.9% of total variance, R2(X), explained, and with 100% classification accuracy. The biplot for this model (Figure 4C) shows not only the scores plot depicting the separation among carcinoma in rats fed low versus high glycemic load diets, but also the proteins most influential in accounting for the observed separation. It is noteworthy that while there is distinct separation associated with glycemic load of the diet (x-axis), there is little clustering along the y-axis (the orthogonal component associated with variation unrelated to glycemic load, and presumably associated with tumor heterogeneity. The effects related to glycemic load were quantified via S-plot analysis (Figure 4D) and variable importance for projection analysis (VIP, Figure 4E). Supplementary Figure S1 shows the graphic presentation of arbitrary units of optical density for each Western blot. Only limited evidence was found that implicated mTOR related signaling. Rather, low glycemic load was associated with higher concentrations of proteins involved in inhibition of cell cycle (P21 and P271) and induction of apoptosis; whereas, carcinomas in the high glycemic load diet had higher concentrations of a protein associated with progression through the G1/S cell cycle checkpoint (cyclin D1), inhibition of caspase activity (XIAP), and angiogenesis (VEGF).
Fig. 4.

Effects of high or low glycemic load human diets on patterns of protein expression in mammary carcinomas. Multivariate discriminant analysis was used to determine whether Western blot data for 26 proteins assessed in mammary carcinomas could distinguish among treat groups. (A) To visualize inherent clustering patterns, the scatter plot represents unsupervised analysis through PCA. Incomplete separation of treatment groups is observed. Model fit: R2X(cum)= 0.747, with 5 components. (B) To determine contributing sources of variation, the scatter plot represents supervised analysis of the 2-class OPLS-DA model, which rotates the model plane to maximize separation due to class assignment. Complete separation of the 2 classes was observed. Model fit: R2Y(cum) = 0.984, Q2Y(cum) = 0.963. (C) To determine the proteins responsible for class separation multivariate analysis was used to construct a biplot that identified influential proteins responsible for the separation between classes. (D) An S-plot was constructed by plotting modeled correlation in the first predictive principal component against modeled correlation from the first predictive component. Upper right and lower left regions of S-plots contain candidate proteins with both high reliability and high magnitude discriminatory proteins (n = 28). (E) To determine the statistical reliability of the proteins shown in (4-D), jack-knifed confidence intervals (JKCI) were created on the magnitude of covariance in the first component for the 28 proteins and sorted in ascending order based on expression in the low glycemic load group; proteins with JKCI including 0 were considered not to account for separation.
Discussion
It is relatively uncommon for defined human diets to be fed in clinical trials and such studies are only feasible over short periods of time and at institutions in which metabolic study facilities are available. Hence, it is virtually impossible for effects of specific menu- and recipe-defined diets to be determined on cancer endpoints in human populations [31–34]. Thus, the possibility of evaluating specific human diets on cancer endpoints in animal models for cancer, with a linkage between the human trial and the rodent experiment being provided by comparison of effects of diet on plasma biomarkers, has significant potential to hasten advances in the field of diet, nutrition and cancer. Cancer incidence was reduced by 50%, cancer multiplicity by 40%, and tumor burden by 52% in the low versus high glycemic load human diet groups (Figure 1). These are meaningful differences in the animal model and if such differences were found in human populations they would be considered clinically important. Moreover, the evaluation of blood from all animals in Experiment 1 showed that as in the clinical trial, in normal weight individuals, the feeding of the low or high glycemic load diets had non-significant effects on biomarkers of glucose homeostasis and chronic inflammation and that the diet group related trends in these markers were in general in the same direction in the preclinical and clinical studies [31, 33].
One advantage of preclinical models, such as the rapid emergence model for breast used in this study, is that many experiments can be conducted in a relatively short timeframe, making it feasible to identify protective elements of an intervention [35]. Thus, in the experiment in which purified rodent diets were fed (Experiment 2), we designed two diets to determine if the effects of the human diets on the carcinogenic response could be directly attributed to differences in resistant starch, a dietary factor that significantly affects the glycemic response to a meal [45, 46]. We considered this experiment highly relevant to the human diet experiment (Experiment 1) in that the glycemic response between the two purified diets was similar to that observed between the two human diets (Figure 3). The purified diet experiment (Figure 2) shows that resistant starch per se has little effect on the carcinogenic response. This finding indicates that other components of the human diet, in addition to carbohydrate availability, are likely to account for protection against cancer. Consistent with this observation, there was only limited support that mTOR network activity, of which insulin mediated signaling is a component, was playing a dominant role in explaining the impact of the human diets on the carcinogenic response. Rather, supervised cluster analyses provided evidence that growth regulation reflected by indicators of cell cycle regulation, apoptosis, and angiogenesis were modulated in a manner consistent with the smaller tumor masses detected in the low glycemic load group (Figure 4). This finding demonstrates that diet affects biological processes cited as hallmarks of cancer [47]. Of particular interest was the suppression of XIAP protein expression in the low glycemic tumors, suggestive of differential effects of the human diets on unfolded protein responses that are involved in a number of metabolic diseases including cancer [48].
Strengths and Limitations
Feeding human diets to rodents has precedent, although the approach has been infrequently used in investigations of diet, nutrition and cancer [49–51]. The preparation of human diets in a manner that could be fed to rodents provided the same glycemic response in the rat as was predicted by calculation based on glycemic index and glycemic load data determined in human participants (Figure 3). There are also weakness to this approach which include the fact that the human diets fed to rodents were homogenized, thus eliminating variables that might exist due to differences in food matrices or eating behaviors. Another limitation is that the same blended human diet was fed to rats each day without the daily variations in food consumption that occur when a cycle menu is followed by human participants.
Implications
There is currently a movement toward integrating preclinical and clinical studies in the cancer chemotherapy field, an approach referred to as co-clinical investigation [52, 53]. The study reported herein represents an extension of that paradigm to enhance progress in the field of diet, nutrition, and cancer. This study informs a range of contemporary questions in this field including whether human dietary patterns can realistically be expected to alter a genetically driven disease process such as cancer, and whether experiments conducted in rodents fed purified diets are sufficient for detecting differences in cancer outcomes of dietary patterns actually consumed in real life circumstances by human populations. Our findings indicate not only that human dietary patterns can alter the carcinogenic process, but also that purified rodent diets designed to model the differences in glycemic response to the human diets fail to render the same effects on the carcinogenic response. Our study illustrates that human diets can be assessed for effects on the carcinogenic process in animal models for cancer. This approach has merit considering that dietary effects may be obscured in studies of human populations due to difficulties in measuring specific dietary patterns using standard dietary assessment tools such as food frequency questionnaires. The approach also addresses the inability to simulate all aspects of a human dietary pattern when rodent studies are limited to purified diet formulations.
Summary
This study shows that dietary glycemic load patterns impact the carcinogenic process but that more work is needed to identify the characteristics of different glycemic load dietary patterns that account for the observed effects. The combinatorial use of both clinical approaches and preclinical models affords an opportunity to unmask the beneficial effects of diet and nutrition in the prevention and control of cancer.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Cancer Institute (R01-CA52626, U54-CA116847).
The authors thank Zongjian Zhu and Weiqin Jiang for their excellent technical assistance.
Footnotes
Conflict of Interest Statement: None declared.
Authors’ Contributions
HJT, JNM, ESN, MLN, JWL, YS participated in the design or implementation of the study. HJT, JNM, MLN, JWL, AMT participated in data evaluation and interpretation. All authors participated in the preparation of the manuscript.
References
- 1.Thomson CA. Diet and breast cancer: understanding risks and benefits. Nutr Clin Pract. 2012;27:636–650. doi: 10.1177/0884533612454302. [DOI] [PubMed] [Google Scholar]
- 2.Michels KB, Mohllajee AP, Roset-Bahmanyar E, Beehler GP, Moysich KB. Diet and breast cancer: a review of the prospective observational studies. Cancer. 2007;109:2712–2749. doi: 10.1002/cncr.22654. [DOI] [PubMed] [Google Scholar]
- 3.Aune D, Chan DS, Greenwood DC, Vieira AR, et al. Dietary fiber and breast cancer risk: a systematic review and meta-analysis of prospective studies. Ann Oncol. 2012;23:1394–1402. doi: 10.1093/annonc/mdr589. [DOI] [PubMed] [Google Scholar]
- 4.Brand-Miller J, McMillan-Price J, Steinbeck K, Caterson I. Dietary glycemic index: health implications. J Am Coll Nutr. 2009;28(Suppl):446S–449S. doi: 10.1080/07315724.2009.10718110. [DOI] [PubMed] [Google Scholar]
- 5.Atkinson FS, Foster-Powell K, Brand-Miller JC. International tables of glycemic index and glycemic load values: 2008. Diabetes Care. 2008;31:2281–2283. doi: 10.2337/dc08-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuhouser ML, Tinker LF, Thomson C, Caan B, et al. Development of a glycemic index database for food frequency questionnaires used in epidemiologic studies. J Nutr. 2006;136:1604–1609. doi: 10.1093/jn/136.6.1604. [DOI] [PubMed] [Google Scholar]
- 7.Marsh K, Barclay A, Colagiuri S, Brand-Miller J. Glycemic index and glycemic load of carbohydrates in the diabetes diet. Curr Diab Rep. 2011;11:120–127. doi: 10.1007/s11892-010-0173-8. [DOI] [PubMed] [Google Scholar]
- 8.Thomas D, Elliott EJ. Low glycaemic index, or low glycaemic load, diets for diabetes mellitus. Cochrane Database Syst Rev. 2009:CD006296. doi: 10.1002/14651858.CD006296.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Bruijn KM, Arends LR, Hansen BE, Leeflang S, et al. Systematic review and meta-analysis of the association between diabetes mellitus and incidence and mortality in breast and colorectal cancer. Br J Surg. 2013;100:1421–1429. doi: 10.1002/bjs.9229. [DOI] [PubMed] [Google Scholar]
- 10.Romieu I, Ferrari P, Rinaldi S, Slimani N, et al. Dietary glycemic index and glycemic load and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) Am J Clin Nutr. 2012;96:345–355. doi: 10.3945/ajcn.111.026724. [DOI] [PubMed] [Google Scholar]
- 11.Sieri S, Pala V, Brighenti F, Agnoli C, et al. High glycemic diet and breast cancer occurrence in the Italian EPIC cohort. Nutr Metab Cardiovasc Dis. 2013;23:628–634. doi: 10.1016/j.numecd.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Shikany JM, Redden DT, Neuhouser ML, Chlebowski RT, et al. Dietary glycemic load, glycemic index, and carbohydrate and risk of breast cancer in the Women’s Health Initiative. Nutr Cancer. 2011;63:899–907. doi: 10.1080/01635581.2011.587227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong JY, Qin LQ. Dietary glycemic index, glycemic load, and risk of breast cancer: meta-analysis of prospective cohort studies. Breast Cancer Res Treat. 2011;126:287–294. doi: 10.1007/s10549-011-1343-3. [DOI] [PubMed] [Google Scholar]
- 14.Yun SH, Kim K, Nam SJ, Kong G, Kim MK. The association of carbohydrate intake, glycemic load, glycemic index, and selected rice foods with breast cancer risk: a case-control study in South Korea. Asia Pac J Clin Nutr. 2010;19:383–392. [PubMed] [Google Scholar]
- 15.Amadou A, Degoul J, Hainaut P, Chajes V, et al. Dietary Carbohydrate, Glycemic Index, Glycemic Load, and Breast Cancer Risk Among Mexican Women. Epidemiology. 2015;26:917–924. doi: 10.1097/EDE.0000000000000374. [DOI] [PubMed] [Google Scholar]
- 16.Mullie P, Koechlin A, Boniol M, Autier P, Boyle P. Relation between Breast Cancer and High Glycemic Index or Glycemic Load: A Meta-analysis of Prospective Cohort Studies. Crit Rev Food Sci Nutr. 2015 doi: 10.1080/10408398.2012.718723. 0. [DOI] [PubMed] [Google Scholar]
- 17.Turati F, Galeone C, Gandini S, Augustin LS, et al. High glycemic index and glycemic load are associated with moderately increased cancer risk. Mol Nutr Food Res. 2015;59:1384–1394. doi: 10.1002/mnfr.201400594. [DOI] [PubMed] [Google Scholar]
- 18.Lampe JW. Nutrition and cancer prevention: small-scale human studies for the 21st century. Cancer Epidemiol Biomarkers Prev. 2004;13:1987–1988. [PubMed] [Google Scholar]
- 19.Cespedes MV, Casanova I, Parreno M, Mangues R. Mouse models in oncogenesis and cancer therapy. Clin Transl Oncol. 2006;8:318–329. doi: 10.1007/s12094-006-0177-7. [DOI] [PubMed] [Google Scholar]
- 20.Sills RC, French JE, Cunningham ML. New models for assessing carcinogenesis: an ongoing process. Toxicol Lett. 2001;120:187–198. doi: 10.1016/s0378-4274(01)00293-4. [DOI] [PubMed] [Google Scholar]
- 21.Blackshear PE. Genetically engineered rodent models of mammary gland carcinogenesis: an overview. Toxicol Pathol. 2001;29:105–116. doi: 10.1080/019262301301418919. [DOI] [PubMed] [Google Scholar]
- 22.Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: Pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Haseman J, Melnick R, Tomatis L, Huff J. Carcinogenesis bioassays: study duration and biological relevance. Food Chem Toxicol. 2001;39:739–744. doi: 10.1016/s0278-6915(01)00010-2. [DOI] [PubMed] [Google Scholar]
- 24.Pariza MW. Animal studies: summary, gaps, and future research. Am J Clin Nutr. 1997;66:1539S–1540S. doi: 10.1093/ajcn/66.6.1539S. [DOI] [PubMed] [Google Scholar]
- 25.Fenton JI, Hord NG. Stage matters: choosing relevant model systems to address hypotheses in diet and cancer chemoprevention research. Carcinogenesis. 2006;27:893–902. doi: 10.1093/carcin/bgi355. [DOI] [PubMed] [Google Scholar]
- 26.Maronpot RR, Flake G, Huff AJ. Relevance of Animal Carcinogenesis Findings to Human Cancer Predictions and Prevention. Toxicologic Pathology. 2004;32:40–48. doi: 10.1080/01926230490425003. [DOI] [PubMed] [Google Scholar]
- 27.Report of Group B of the American Cancer Society Research Workshop on Cancer and Nutrition: Panel on Animal Studies. Cancer Research. 1993;53:2452s–2454s. [PubMed] [Google Scholar]
- 28.Hoffmann I. Transcending reductionism in nutrition research. The American Journal of Clinical Nutrition. 2003;78:514S–516S. doi: 10.1093/ajcn/78.3.514S. [DOI] [PubMed] [Google Scholar]
- 29.Thompson HJ. Diet, Nutrition, and Cancer: Development of Hypotheses and Their Evaluation in Animal Studies. Cancer Research. 1993;53:2442s–2445s. [PubMed] [Google Scholar]
- 30.Jacobs DR, Tapsell LC. Food, Not Nutrients, Is the Fundamental Unit in Nutrition. Nutrition Reviews. 2007;65:439–450. doi: 10.1111/j.1753-4887.2007.tb00269.x. [DOI] [PubMed] [Google Scholar]
- 31.Runchey SS, Pollak MN, Valsta LM, Coronado GD, et al. Glycemic load effect on fasting and post-prandial serum glucose, insulin, IGF-1 and IGFBP-3 in a randomized, controlled feeding study. Eur J Clin Nutr. 2012;66:1146–1152. doi: 10.1038/ejcn.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang KT, Lampe JW, Schwarz Y, Breymeyer KL, et al. Low glycemic load experimental diet more satiating than high glycemic load diet. Nutr Cancer. 2012;64:666–673. doi: 10.1080/01635581.2012.676143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neuhouser ML, Schwarz Y, Wang C, Breymeyer K, et al. A low-glycemic load diet reduces serum C-reactive protein and modestly increases adiponectin in overweight and obese adults. J Nutr. 2012;142:369–374. doi: 10.3945/jn.111.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Runchey SS, Valsta LM, Schwarz Y, Wang C, et al. Effect of low- and high-glycemic load on circulating incretins in a randomized clinical trial. Metabolism. 2013;62:188–195. doi: 10.1016/j.metabol.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson HJ, McGinley JN, Rothhammer K, Singh M. Rapid induction of mammary intraductal proliferations, ductal carcinoma in situ and carcinomas by the injection of sexually immature female rats with 1-methyl-1-nitrosourea. Carcinogenesis. 1995;16:2407–2411. doi: 10.1093/carcin/16.10.2407. [DOI] [PubMed] [Google Scholar]
- 36.Tamashiro KLK, Terrillion CE, Hyun J, Koenig JI, Moran TH. Prenatal Stress or High-Fat Diet Increases Susceptibility to Diet-Induced Obesity in Rat Offspring. Diabetes. 2009;58:1116–1125. doi: 10.2337/db08-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh M, McGinley JN, Thompson HJ. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab Invest. 2000;80:221–231. doi: 10.1038/labinvest.3780025. [DOI] [PubMed] [Google Scholar]
- 38.Snedecor GW, Cochran WG, editors. Statistical Methods. 8. Iowa State University Press; Ames, IA: 1989. [Google Scholar]
- 39.Trygg J, Wold S. Orthogonal projections to latent structures (O-PLS) Journal of Chemometrics. 2002;16:119–128. [Google Scholar]
- 40.Wiklund S. Multivariate Analysis for Omics. Umetrics; Umea, Sweden: Sep 3, 2008. Multivariate Data Analysis and Modeling in “omics”. Ref Type: Conference Proceeding. [Google Scholar]
- 41.Wiklund S, Johansson E, Sjostrom L, Mellerowicz EJ, et al. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal Chem. 2008;80:115–122. doi: 10.1021/ac0713510. [DOI] [PubMed] [Google Scholar]
- 42.User’s Guide to Simca-P, Simca-P+ 11. Umetrics AB; Umea, Sweden: 2005. [Google Scholar]
- 43.Morrison DF, editor. Multivariate Statistical Methods. 3. McGraw-Hill Publishing Co; New York: 1990. [Google Scholar]
- 44.Gabrielsson J, Jonsson H, Airiau C, Schmidt B, et al. OPLS methodology for analysis of pre-processing effects on spectroscopic data. Chemometrics and Intelligent Laboratory Systems. 2006;84:153–158. [Google Scholar]
- 45.Zhang G, Hamaker BR. Slowly digestible starch: concept, mechanism, and proposed extended glycemic index. Crit Rev Food Sci Nutr. 2009;49:852–867. doi: 10.1080/10408390903372466. [DOI] [PubMed] [Google Scholar]
- 46.Englyst KN, Liu S, Englyst HN. Nutritional characterization and measurement of dietary carbohydrates. Eur J Clin Nutr. 2007;61(Suppl 1):S19–S39. doi: 10.1038/sj.ejcn.1602937. [DOI] [PubMed] [Google Scholar]
- 47.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Ozcan L, Tabas I. Role of Endoplasmic Reticulum Stress in Metabolic Disease and Other Disorders. Annu Rev Med. 2012;63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothwell NJ, Stock MJ. The cafeteria diet as a tool for studies of thermogenesis. J Nutr. 1988;118:925–928. doi: 10.1093/jn/118.8.925. [DOI] [PubMed] [Google Scholar]
- 50.McDaniel SM, O’Neill C, Metz RP, Tarbutton E, et al. Whole-food sources of vitamin A more effectively inhibit female rat sexual maturation, mammary gland development, and mammary carcinogenesis than retinyl palmitate. J Nutr. 2007;137:1415–1422. doi: 10.1093/jn/137.6.1415. [DOI] [PubMed] [Google Scholar]
- 51.Rozen P, Liberman V, Lubin F, Angel S, et al. A new dietary model to study colorectal carcinogenesis: experimental design, food preparation, and experimental findings. Nutr Cancer. 1996;25:79–100. doi: 10.1080/01635589609514430. [DOI] [PubMed] [Google Scholar]
- 52.Nardella C, Lunardi A, Patnaik A, Cantley LC, Pandolfi PP. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 2011;1:108–116. doi: 10.1158/2159-8290.CD-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clohessy JG, de SE. Infrastructure needs for translational integration of mouse and human trials. Cold Spring Harb Protoc. 2013;2013 doi: 10.1101/pdb.top078782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.