

Abstract
The apelinergic system includes a series of endogenous peptides apelin, ELABELA/TODDLER and their 7-transmembrane G-protein coupled apelin receptor (APJ, AGTRL-1, APLNR). The APJ receptor is an attractive therapeutic target because of its involvement in cardiovascular diseases and potentially other disorders including liver fibrosis, obesity, diabetes, and neuroprotection. To date, pharmacological characterization of the APJ receptor has been limited due to the lack of small molecule functional agonists or antagonists. Through focused screening we identified a drug-like small molecule agonist hit 1 with a functional EC50 value of 21.5 ± 5 µM and binding affinity (Ki) of 5.2 ± 0.5 µM. Initial structure-activity studies afforded compound 22 having a 27-fold enhancement in potency and the first sub-micromolar full agonist with an EC50 value of 800 ± 0.1 nM and Ki of 1.3± 0.3 µM. Preliminary SAR, synthetic methodology, and in vitro pharmacological characterization indicate this scaffold will serve as a favorable starting point for further refinement of APJ potency and selectivity.

Keywords: APJ small molecule agonist, pyrazole, apelin, AGTRL1, APLNR
Graphical Abstract

1. Introduction
The APJ (APLNR gene product) receptor is a 380 amino acid class A G protein-coupled receptor (GPCR).1 The amino acid sequence of the APJ receptor is highly conserved across a number of species including human, bovine, rat and mouse suggesting a critical role for this receptor in physiology.2 The APJ receptor shares moderate sequence homology with the angiotensin 2 receptor 1 (AT1). However, none of the known AT1 ligands or angiotensin II bind APJ.1
In 1998, the APJ receptor was de-orphanized when its first endogenous ligand, apelin, was identified by Tatemoto and coworkers in bovine stomach extracts.3 Several forms of apelin peptides are obtained by cleavage of pre-pro-apelin, a 77 amino acid precursor peptide.4–7 These peptides include apelin-36, apelin-17, apelin-13, apelin-12 and pyr-apelin-13. Pyr-apelin-13 is the most abundant form in plasma and binds with high affinity to APJ and functions as a full agonist.4 Recently, a second endogenous peptidergic agonist of APJ named TODDLER/ELABELA was identified.8,9
In CNS, APLNR mRNA is expressed in neurons of the paraventricular and supraoptic nucleus of the hypothalamus.10–12 Messenger RNA (mRNA) encoding both APLNR and apelin are highly expressed peripherally in tissues including gastrointestinal tract, adipose, kidney, liver, lung and heart.2,5,13 However, questions remain related to APJ expression levels in various cell types within these organs. Interestingly, in recent studies using a transgenic mouse model wherein nuclear lacZ was knocked into the endogenous APLNR locus indicated that lacZ expression was largely restricted to cardiomyocytes and small capillaries of most tissues.14 Thus, highly vascularized tissues and myocardium in rodents are expected to express APJ in agreement with past observations.15,16
However, studies in humans with apelin peptides have produced some interesting and contradictory data. Japp et al. conducted studies in healthy human volunteers to investigate direct vascular effects of apelin-36 and pyr-apelin-13 infusion into forearm vessels.17 Both apelin isoforms caused reproducible vasodilation in arterial vessels but no such effect was noted in dorsal hand veins. The vasodilatory effect of pyr-apelin-13 infusion within the arteries was attributable to nitric oxide production. The lack of effect within the venous bed was in sharp contrast to what could be predicted based upon rodent studies discussed above and possibly points to species differences and/or disease related activation of the apelinergic system and underscores the need to test more stable and drug-like pharmacological agents. Follow-up studies by the same group in heart disease patients indicated that acute apelin administration caused peripheral and coronary vasodilation and increased cardiac output.18 Additional studies in humans indicated that APJ agonism produced sustained cardiovascular effects that were preserved in the presence of renin-angiotensin system activation or heart failure.19 Taken together, APJ has emerged as an intriguing target for cardiovascular diseases.
APJ has also emerged as an attractive target because of its possible involvement in fluid homeostasis,20,21 liver fibrosis,22 obesity,23–28 diabetes29–31, neuroprotection,32–34 immune response,35,36 nociception37 and others. Pathophysiological roles played by APJ are currently being elucidated using various apelin peptides.21,29,38,39 However, in vivo pharmacological studies are difficult due to the short half-life of apelin-13 and limited availability of drug-like small molecule probes.
A non-apelin based APJ agonist E339-3D6 has been recently described. The molecular weight of this compound is >1000 kDa and contains peptidergic fragments. E339-3D6 also contains a fluorescent probe that has been shown to be critical for potency at the receptor.40 Further structure activity relationships on E339-3D6 afforded more potent agonists (Ki = 0.089 µM) biased toward cAMP production that retain the critical fluorophore.41 In addition to E339-3D6, a low molecular weight small molecule APJ full agonist ML233 (EC50 = 3.7 µM) was reported to be selective over AT1.42 Current attempts to improve agonist potency have proven challenging despite two rounds of optimization.42 Recent patent disclosures from Sanofi Aventis and Sanford-Burnham Medical Research Institute reported small molecule agonists based on the substituted benzimidazole and triazole core scaffold respectively.43,44 These are the only reports of drug like small molecule agonists to date. Therefore, novel small molecule agonists of APJ with improved pharmacological properties are needed.
Considering the limitations of available tool compounds to study the APJ receptor, our laboratory initiated a program to discover novel, drug-like, small molecule agonists using a primary calcium mobilization assay45 and a secondary radioligand binding assay.40,41 Based on protein sequence, the AT1 receptor shares the highest overall homology with the APJ receptor.1 Using this information, we hypothesized that some of the structural features common to AT1 receptor lig-ands might be a good starting point for identification of APJ small molecules. Further refinement of our compound selection was based on an evaluation of the putative binding site homology among related GPCR’s.
Chemogenomic analysis of 30 critical GPCR residues within the transmembrane cavity places APJ in the chemoattractant receptor cluster.46 Using binding site cavity alignment Surgand and colleagues depict a very hydrophobic APJ core with conserved aromatic residues on helix 6 (F6.44, W6.48, and Y6.51) in addition to a positively charged K6.55 that resides toward the extracellular face of helix 6. Other residues on helix 3 that surround the binding cavity include a series of hydrophobic and lipophilic residues (F3.33, M3.36, and V3.40). Coupling this data with common structural features of AT1 ligands suggested a more focused screen of ligands having a lipophilic core with anionic features would be appropriate.
Our initial evaluation focused on AT1 antagonists including losartan and valsartan. Since none of these analogs possessed agonist activity we proceeded to an in house library of targeted peptidergic GPCR compounds possessing lipophilic cores with a carboxylic acid moiety using a high-throughput calcium mobilization assay.45 Approximately 100 compounds from our focused library were screened and four hits belonging to one structural family were identified.
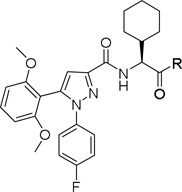
Compound 1 was identified as a functional APJ agonist with EC50 value of 21.5 ± 5 µM and Ki of 5.2 ± 0.5 µM using 125I radiolabeled pyr-apelin-13 (pyr-apelin-13) (Figure 1).40,41 Radioligand displacement studies indicated that 1 was a competitive orthosteric ligand of APJ that produced 100% displacement of 125I radiolabeled pyr-apelin-13 (pyr-apelin-13) as indicated in Figure 1A. However, compound 1 also had off-target NTR2 receptor activity (EC50 = 271 nM, Ki = 622 nM).47 After confirmation of 1 as a valid hit we began scaffold modifications at sites A-C in order to determine receptor tolerances at each site and enhance selectivity for APJ versus NTR2.
Figure 1.

A. Chemical structure of initial hit 1, identified through HTS screen and sites of chemical modification explored that resulted in new lead 22; B. Radioligand binding of compound 1 using using 125I radiolabeled pyr-apelin-13
2. Chemistry
A general scheme for the synthesis of analog 22 is shown in Scheme 1. 2,6-Dimethoxy acetophenone 32 was condensed with diethyl oxalate to afford the sodium salt of diketone 33 in quantitative yield. Reaction of 33 with 4-fluorophenyl hydrazine hydrochloride and concentrated hydrochloric acid in refluxing acetic acid provided 1, 5-pyrazoles 34a and 1,3-pyrazoles 34b in a ratio of 4:1. Transformation of 34a to acid 2 was achieved by alkaline hydrolysis using lithium hydroxide monohydrate. Carboxamides were obtained through parallel solution-phase synthesis by coupling acids with selected amines in presence of HBTU as coupling reagent. Intermediate 35 was obtained by coupling tert-butyl (S)-3-amino-5-cyclohexylpentanoate with 2. Tert-butyl ester in 35 was simply deprotected by trifluoroacetic acid to provide 22.
3. Results and Discussion
Modification of Site A
As shown in Scheme 1 the synthesis of 2 provides a mixture of isomers at N1 (34a) and N2 (34b) in a ratio of 4:1 that are readily separable using normal phase chromatography. Confirmation that 2 bears the 4-Fluoro-Phenyl ring substituted on the N1 nitrogen was obtained through X-Ray crystallography (Figure 2).
Scheme 1.

Synthesis of compound 22
Reagents and conditions: a) Diethyl oxalate, MeONa, MeOH, reflux, 6 h, 90%; b) 4-Fluorophenyl hydra-zine hydrochloride, glacial acetic acid, conc. HCl, reflux, 3.5 h, 61%; c) LiOH, MeOH/THF/H2O, rt, 18 h, 87%; d) Tert-butyl (S)-3-amino-5-cyclohexylpentanoate, HBTU, Et3N, CH3CN, rt, 3 h, 79%; e) TFA, DCM, rt, 4 h, 80%
Figure 2.

Crystal structure of compound 2
Evaluation of compound 1a and 3 for calcium mobilization demonstrated movement of the 4-Fluoro-Phenyl group to the N2 position and isomerization of the R-cyclohexyl glycine significantly reduce activity. Methyl ester 4 and compound 5 lacking a carboxylic acid group were also evaluated and did not possess activity confirming the significance of carboxylic acid group for APJ potency.
Many potential therapeutic indications have been postulated for apelin agonists. Of those, neuroprotection32–34 is one that may require therapeutic exposure to the central nervous system (CNS). In general, carboxylic acids do not penetrate the CNS well. Therefore, a small set of amides were prepared to determine if replacement of the terminal carboxylic acid would be detrimental to APJ potency. Primary amide 6 was devoid of activity as was dimethyl amide 7, and N-benzyl amide 8. The addition of more acidic protons from hydroxamic acid 9 was also not tolerated. Additional analogs having glycine methyl ester substitution 10 and glycine acid 11 were also inactive. These data demonstrated that the car-boxyl OH group is an important mediator of receptor activation in this series.
Modification of Site B
Based on the results from our initial screen of structurally related scaffolds, the 2,6-dimethoxyphenyl substituent appeared critical. Confirmation of this facet was obtained through the synthesis of analogs 12–15 (Table 2). Removal of both methyl groups from the dimethoxy group (12) caused a complete loss of agonist potency. Similarly, removal of just one methyl groups (13) also resulted in a loss of potency. In order to evaluate if the increase in polarity from the phenolic groups was responsible for the loss of activity, unsubstituted phenyl (14) was evaluated. This suggested that the 2,6-dimethoxy phenyl group was critical for activity. Moreover, replacement of the aryl group with methyl 15 was inactive. Overall, this suggests the 2, 6-dimethoxy phenyl moiety is an important contributor of agonist activity at the APJ receptor.
Table 2.
Structural modification at Site B
 | ||
|---|---|---|
| Compd. | Ar/R | EC50(µM ) |
| 12 | 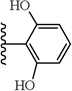 |
>30 |
| 13 | 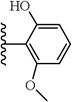 |
>30 |
| 14 | Ph | >30 |
| 15 | CH3 | >30 |
Modification of Site C
A chemically diverse set of amino acid scaffolds were substituted for cyclohexyl glycine (Table 3) to explore the effects of side chain hydrophobic and polar requirements for receptor activity. These results indicated that polar side-chains were not tolerated (16, 17, 18) and hydrophobic substituents such as isobutyl 19 did not retain APJ potency. We then substituted β-Leu (20) for Leu (19) to determine if elongation of the carboxylic acid group by one methylene would enhance binding and agonist potency. This modification resulted in a two-fold enhancement in potency (20, EC50 = 9.4 µM) with a roughly 9-fold increase in affinity (20, Ki = 0.60 µM) compared to 1. Similarly, substitution of β-cyclohexyl glycine 21 for cyclohexyl glycine (1) resulted in a similar potency enhancement (EC50 = 9.9 µM) and an improved Ki of 1.4 µM. Interestingly, extending the hydrophobic cyclohexyl ring by two methylene units improved the potency by 12 fold (22, EC50 = 0.80 µM) compared to 21. Since the original hit came from an NTR2 library, we examined the activity of 21 and 22 at both NTR1 and NTR2. Neither compound had any significant NTR1 activity either as an agonist or antagonist. These compounds behaved as partial agonists at NTR2 with 21 having an EC50 value of ~345 nM with 57% Emax and 22 having an EC50 value of ~3406 nM with Emax of ~23%. Therefore, these compounds demonstrated significantly reduced activity at NTR2 compared with 1 and these data indicate that selectivity over NTR1 and 2 can be further increased with additional structural modifications. An additional series of analogs that altered placement of the carboxyl and saturation of the hydrophobic group possessed limited activity at the APJ receptor (23–31). Overall the SAR modifications on the core scaffold indicate that 22 can serve as a new lead for further refinement as a selective APJ agonist.
Table 3.
Structural modification at Site C
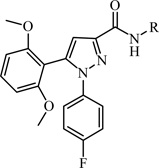 | |||
|---|---|---|---|
| Compd. | R | EC50
* (µM) ±SEM |
Ki * (µM)± SEM |
| 16 | 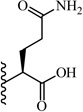 |
>30 | |
| 17 | 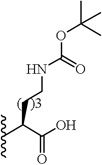 |
>30 | |
| 18 | 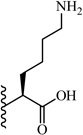 |
>30 | |
| 19 | 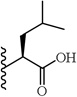 |
>30 | |
| 20 | 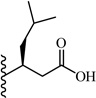 |
9.4 ± 3.4 | 0.6 ± 0.10 |
| 21 | 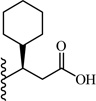 |
9.9 ± 2.6 | 1.4 ± 0.2 |
| 22 | 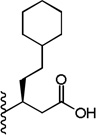 |
0.8 ± 0.1 | 1.3 ± 0.3 |
| 23 | 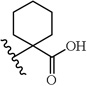 |
>30 | |
| 24 | 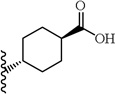 |
>30 | |
| 25 | 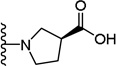 |
>30 | |
| 26 | 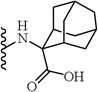 |
>30 | |
| 27 | 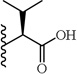 |
>30 | |
| 28 |  |
>30 | |
| 29 | 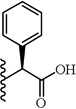 |
>30 | |
| 30 |  |
>30 | |
| 31 | 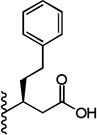 |
3 ± 0.2 | 3 ± 0.6 |
EC50 and Ki values are averages of multiple experiments (where n = 3, unless otherwise stated) ± standard error of the mean.
4. Conclusion
In summary, compound 1 was found through focused screening to be a specific APJ receptor ligand that served as novel scaffold to explore the importance of various structural features (sites A-C). Conversion of the carboxylic acid in 1 to a variety of amides, (6–11) resulted in analogs with no activity at the APJ receptor. This suggests the carboxylic acid plays an important role in receptor interactions and potentially activation. Modification of site B (12–15) demonstrated the significance of a 2,6-dimethoxyphenyl substitution on APJ agonist potency. Modification of site C demonstrated that polar side-chains were not tolerated (16, 17, 18) and hydrophobic sub-stituents such as isobutyl (19) did not retain APJ potency. Elongation of the carboxylic acid by one methylene retained agonist potency for compounds 20 (EC50 = 9.4 µM) and 21 (EC50 = 9.9 µM). Interestingly, extending the hydrophobic cyclohexyl ring by two methylene (22) improved the potency by 12 fold (EC50 = 0.80 µM). More importantly, radioligand binding studies on compounds 1 (Ki = 5.2 µM), 21 (Ki = 1.4 µM), 22 (Ki = 1.3 µM), and 20 (Ki = 0.60 µM) indicate these ligands specifically act at the APJ receptor and the affinity can be enhanced through structural modification. In conclusion, we have identified and characterized one of the first drug-like small molecule (mw <500) APJ agonists (1, 20, 22) that serves as an ideal starting scaffold for the development of more potent and selective APJ agonists having improved drug-like properties.
5. Experimental Procedures
5.1 Chemistry
5.1.1 General
Reagents and starting materials were obtained from commercial suppliers and were used without purification. Reactions were conducted under N2 atmosphere using ovendried glassware. All solvents and chemicals used were reagent grade. Anhydrous tetrahydrofuran (THF), dichloromethane (DCM), and N, N-dimethylformamide (DMF) were purchased from Fisher Scientific and used as such. Flash column chromatography was carried out using a Teledyne ISCO Combiflash Rf system and Redisep Rf pre-packed HP silica columns. Purity and characterization of compounds were established by combination of HPLC, TLC, LC-MS, and NMR analytical technique described below. 1H NMR spectra were recorded on a Bruker Avance DPX300 (300 MHz) spectrometer using CHCl3-d, MeOH-d, DMSO-d6 with tetramethylsilane (TMS) (0.00 ppm) as the internal reference. Chemical shifts are reported in ppm relative to the solvent signal, and coupling constants (J) values are reported in Hertz (Hz). Thin-layer chromatography (TLC) was performed on precoated silica gel GF Uniplates from Analtech and spots were visualized with UV light or I2 detection or phosphomolybdic acid stain. Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). Optical rotations were measured on an Auto Pol III polarimeter at the sodium D line. Analytical and preparative HPLC was performed on an automated Varian ProStar HPLC system equipped with a Xterra® C18 RP18 (4.6 × 100 mm i. d., 3.5 µm) column, with a flow rate of 1 mL/min.; λmax = 254 nm; mobile phase A: H2O containing 0.1% TFA and mobile phase B: CH3CN containing 0.1% TFA. Purity of the target compounds was determined to be ≥ 95% by HPLC. Chemical names were generated using ChemDraw Ultra (CambridgeSoft, version 10.0).
S)-2-Cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (400 mg, 0.168 mmol) was dissolved in THF (15 mL). To the solution was added HATU (444 mg, 0.168 mmol) and triethylamine (0.80 mL, 5.482 mmol). The resulting mixture was stirred at room temperature for 15 minutes. L-Cyclohexylglycine-tert-butyl ester hydrochloride (320 mg, 1.285 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4. The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-tert-butyl 2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetate as white solid (75 mg, 79 %). Following the tert-butyl ester deprotection procedure 22, the title compound 1 was obtained from (S)-tert-butyl 2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetate. 94% yield; white solid; [α]D25 = + 9.03 (c 0.15, MeOH); 1H NMR (MeOH-d4, 300 MHz) δ 1.11-1.41 (m, 5 H), 1.60-1.86 (m, 5 H), 1.89-2.04 (m, 1 H), 3.60 (br. s., 6 H), 4.57 (d, J=6.03 Hz, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.80 (s, 1 H), 7.02-7.10 (m, 2 H), 7.25 - 7.38 (m, 4 H). MS (ESI) m/z: Calcd. for C26H28FN3O5 481.20 [M]+, found 482.5 [M+H]+.
(S)-2-Cyclohexyl-2-(3-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-5-carboxamido)acetic acid (1a)
Using the procedure described in 1, the title compound 1a was obtained from 3-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-5-carboxylic acid (34b). white solid. 1H NMR (CDCl3 , 300 MHz) δ 0.78 - 1.20 (m, 5 H), 1.53 - 1.79 (m, 5 H), 1.81 - 1.94 (m, 1 H), 3.79 (s, 6 H), 4.61 (dd, J=8.29, 4.90 Hz, 1 H), 6.40 (d, J=8.67 Hz, 1 H), 6.64 (d, J=8.29 Hz, 2 H), 6.88 (s, 1 H), 7.11 (t, J=8.48 Hz, 2 H), 7.26 -7.34 (m, 1 H), 7.54 (dd, J=8.85, 4.71 Hz, 2 H). MS (ESI) m/z: Calcd. for C26H28FN3O5 481.20 [M]+, found 482.5 [M+H]+.
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2)
Lithium hydroxide monohydrate (324 mg, 7.717 mmol) in 2 mL of water was added to a solution of methyl 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylate (34a) (1.1 g, 3.086 mmol) in MeOH (11 mL) and THF (3 mL). The mixture was stirred at room temperature for 3 h. The reaction mixture was then concentrated to about half the volume and then extracted with ether (2 × 25 mL). The aqueous layer was acidified with 1 N HCl and extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4. The solvent was evaporated in vacuo to give the title compound 2 as white solid (872 mg, 87 %). 1H NMR (CDCl3 , 300 MHz) δ 3.60 (s, 6 H), 6.51 (d, J=9.0 Hz, 2 H), 7.01 (s, 1H). 7.00-6.97 (m, 2 H), 7.27-7.31 (m, 3 H). MS (ESI) m/z: Calcd. for C18H15FN2O4 342.10 [M]+, found 343.3 [M+H]+, 341.6 [M-H]−.
(R)-2-Cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (3)
Following the procedure described in 1, the title compound 3 was obtained using 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) and D-cyclohexylglycine methyl ester hydrochloride . For hydrolysis of methyl ester, (R)-methyl 2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetate (50 mg, 0.100 mmol) was dissolved in 1,2-dichloroethane (2 mL) and after addition of trimethyltin hydroxide (55 mg, 0.300 mmol), the mixture was heated at 80 °C until TLC analysis indicated a complete reaction. After completion of the reaction, the mixture was concentrated in vacuo, and the residue was taken up in ethyl acetate (15 mL). The organic layer was washed with HCl (5%) (3 × 5-15 mL). The organic layer was then washed with brine (5-15 mL) and dried over sodium sulfate. Removal of the solvent in vacuo afforded the crude residue. The residue was purified by silica gel flash chromatography (MeOH:DCM) to give (R)-2-Cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (3) as white solid. 37% yield; [α]D25 = −9.52 (c 0.10, MeOH); 1H NMR (CDCl3 , 300 MHz) δ 1.06 - 1.37 (m, 5 H), 1.58 - 1.90 (m, 5 H), 1.92 - 2.06 (m, 1 H), 3.54 (s, 3 H), 3.61 (s, 3 H), 4.73 (dd, J=8.67, 5.65 Hz, 1 H), 6.50 (t, J=8.67 Hz, 2 H), 6.93 - 7.04 (m, 3 H), 7.22 - 7.36 (m, 3 H), 7.48 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C26H28FN3O5 481.20 [M]+, found 482.4 [M+H]+.
(S)-Methyl 2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetate (4)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (700 mg, 2.045 mmol) was dissolved in THF (17 mL). To the solution was added HATU (778 mg, 2.045 mmol) and triethylamine (0.850 mL, 6.13 mmol). The resulting mixture was stirred at room temperature for 15 minutes. L-Cyclohexylglycine methyl ester hydrochloride (425 mg, 2.045 mmol) was added and stirred at room temperature for 16 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4. The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetate (4) as white solid. 94% yield; 1H NMR (CDCl3 , 300 MHz) δ 1.06 -1.37 (m, 5 H), 1.61 - 1.82 (m, 5 H), 1.84 - 1.99 (m, 1 H), 3.53 (s, 3 H), 3.62 (s, 3 H), 3.76 (s, 3 H), 4.76 (dd, J=9.04, 6.03 Hz, 1 H), 6.47 - 6.54 (m, 2 H), 6.94 (s, 1 H), 6.97 - 7.01 (m, 2 H), 7.25-7.33 (m, 3 H), 7.43 (d, J=9.04 Hz, 1 H). MS (ESI) m/z: Calcd. for C27H30FN3O5 495.22 [M]+, found 496.5 [M+H]+.
N-(Cyclohexylmethyl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (5)
Following the procedure described in 4, compound 5 was obtained using 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) and cyclohexyl methylamine. 82% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 0.93 - 1.07 (m, 2 H), 1.14 - 1.32 (m, 3 H), 1.52 - 1.87 (m, 6 H), 3.30 (t, J=6.40 Hz, 2 H), 3.58 (s, 6 H), 6.50 (d, J=8.67 Hz, 2 H), 6.92 - 7.00 (m, 3 H), 7.05 (t, J=6.03 Hz, 1 H), 7.22 - 7.33 (m, 3 H). MS (ESI) m/z: Calcd. for C25H28FN3O3 437.21 [M]+, found 438.5 [M+H]+.
(S)-N-(2-Amino-1-cyclohexyl-2-oxoethyl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (6)
Following the procedure described in 4, compound 6 was obtained using (S)-2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1) and 2 M ammonia in methanol. 50 % yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.03 -1.40 (m, 5 H), 1.63 - 1.92 (m, 5 H), 1.93-2.09 (m, 1 H), 3.54 (s, 3 H), 3.62 (s, 3 H), 4.48 (dd, J=8.67, 7.16 Hz, 1 H), 5.44 (br. s., 1 H), 6.16 (br. s., 1 H), 6.51 (t, J=9.23 Hz, 2 H), 6.90 -7.03 (m, 2 H), 7.24 - 7.34 (m, 4 H), 7.42 (d, J=9.04 Hz, 1 H). MS (ESI) m/z: Calcd. for C26H29FN4O4 480.22 [M]+, found 481.4 [M+H]+.
(S)-N-(1-Cyclohexyl-2-(dimethylamino)-2-oxoethyl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (7)
Following the procedure described in 4, compound 7 was obtained from (S)-2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1) and N, N’-dimethyl hydrochloride. 69% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.02 - 1.35 (m, 5 H), 1.61 - 1.82 (m, 5 H), 1.83-1.94 (m, 1 H), 2.99 (s, 3 H), 3.20 (s, 3 H), 3.50 (s, 3 H), 3.64 (s, 3 H), 4.98 -5.07 (m, 1 H), 6.47 (d, J=8.29 Hz, 1 H), 6.53 (d, J=8.29 Hz, 1 H), 6.89 - 6.99 (m, 2 H), 7.22 - 7.34 (m, 4 H), 7.59 (d, J=9.42 Hz, 1 H). MS (ESI) m/z: Calcd. for C28H33FN4O4 508.25 [M]+, found 509.5 [M+H]+.
(S)-N-(2-(Benzylamino)-1-cyclohexyl-2-oxoethyl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (8)
Following the procedure described in 4, compound 8 was obtained from (S)-2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1a) and benzylamine. 79 % yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.00 - 1.40 (m, 5 H), 1.62 - 1.91 (m, 5 H), 1.93 - 2.16 (m, 1 H), 3.53 (s, 3 H), 3.62 (s, 3 H), 4.39 - 4.56 (m, 3 H), 6.43 - 6.58 (m, 3 H), 6.92 - 7.02 (m, 3 H), 6.90 (s, 1 H), 7.20 - 7.37 (m, 7 H), 7.45 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C33H35FN4O4 570.26 [M]+, found 571.9 [M+H]+.
(S)-N-(1-Cyclohexyl-2-(hydroxyamino)-2-oxoethyl)-5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (9)
Following the procedure described in 4 compound 9 was obtained from (S)-2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1a) and hydroxylamine hydrochloride. 27 % yield; white solid; 1H NMR (MeOH-d4 , 300 MHz) δ 1.02 - 1.38 (m, 5 H), 1.62-1.72 (m, 1 H), 1.73 - 1.95 (m, 5 H), 3.61 (s, 6 H), 5.45 - 5.54 (m, 1 H), 6.62 (d, J=8.29 Hz, 2 H), 6.78 (s, 1 H), 7.00 - 7.09 (m, 2 H), 7.23 - 7.39 (m, 4 H). MS (ESI) m/z: Calcd. for C26H29FN4O5 496.21 [M]+, found 495.4 [M-H]−.
(S)-Methyl 2-(2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetamido)acetate (10)
Following the procedure described in 4, compound 10 was obtained using (S)-2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (1) and glycine methyl ester hydrochloride. 85% yield; white solid; 1H NMR (CDCl3 , 300 MHz) 1.04 - 1.36 (m, 5 H), 1.62 - 1.92 (m, 5 H), 1.94 - 2.08 (m, 1 H), 3.54 (s, 3 H), 3.62 (s, 3 H), 3.75 (s, 3 H), 4.07 (t, J=5.09 Hz, 2 H), 4.49 (dd, J=8.85, 7.35 Hz, 1 H), 6.51 (t, J=9.80 Hz, 2 H), 6.58 (t, J=5.09 Hz, 1 H), 6.92 - 6.95 (m, 1 H), 6.97 (d, J=8.67 Hz, 1 H), 7.21 - 7.33 (m, 4 H), 7.43 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C29H33 FN4O6 552.24 [M]+, found 553.4 [M+H]+.
(S)-2-(2-Cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetamido)acetic acid (11)
Following the lithium hydroxide hydrolysis procedure 2, compound 11 was obtained from (S)-methyl 2-(2-cyclohexyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetamido)acetate (10). 95% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.07-1.40 (m, 5 H), 1.62 -1.72(m, 1 H), 1.73 - 1.97 (m, 5 H), 3.59 (s, 3 H), 3.62 (s, 3H), 3.86-4.04 (s, 2H), 4.49-4.58 (m, 1 H), 6.62 (d, J=9.0 Hz, 2 H), 6.80 (s, 1H), 7.00-7.10 (m, 2H), 7.25-7.39 (m, 3H), 7.97 (d, J=9.0 Hz, 1 H ), 8.55 (br. s., 1 H). MS (ESI) m/z: Calcd. for C28H31FN4O6 538.22 [M]+, found 537.5 [M-H]-.
(S)-2-Cyclohexyl-2-(5-(2,6-dihydroxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)acetic acid (12)
Following the procedure described in 1, the title compound 12 was obtained from 5-(2,6-dihydroxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid and L-Cyclohexylglycine-tert-butyl ester hydrochloride. 1H NMR (MeOH-d4 , 300 MHz) δ 1.08 - 1.41 (m, 5 H), 1.61-1.86 (m, 5 H), 1.89-2.05 (m, 1 H), 4.48-4.64 (m, 1 H), 6.24 - 6.32 (m, 2 H), 6.85 (s, 1 H), 6.96 - 7.09 (m, 3 H), 7.37-7.45 (m, 2 H). MS (ESI) m/z: Calcd. for C24H24FN3O5 453.17 [M]+, found 454.4 [M+H]+.
2-Cyclohexyl-2-(1-(4-fluorophenyl)-5-(2-hydroxy-6-methoxyphenyl)-1H-pyrazole-3-carboxamido)acetic acid (13)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0438 mmol) was dissolved in DCM (6 mL). To the solution was added 1M BBr3 solution in DCM dropwise at 0 C. Reaction mixture was then stirred at room temperature for 18 hrs. Water was added to the reaction mixture and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. Crude residue had a mixture of 1-(4-fluorophenyl)-5-(2-hydroxy-6-methoxyphenyl)-1H-pyrazole-3-carboxylic acid and 5-(2,6-dihydroxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid. Following the procedure described in 1, the title compound 13 was obtained after purification with preparative TLC (Ether/Hexane (8:2). 1H NMR (MeOH-d4 , 300 MHz) δ 1.10 - 1.43 (m, 5 H), 1.63 - 1.88 (m, 5 H), 1.90-2.06 (m, 1 H), 3.53 (s, 3 H), 4.53 - 4.60 (m, 1 H), 6.44 (t, J=8.67 Hz, 2 H), 6.83 (s, 1 H), 7.01 - 7.10 (m, 2 H), 7.18 (t, J=8.29 Hz, 1 H), 7.30 - 7.41 (m, 2 H), 7.94 (d, J=9.0 Hz, 1 H). MS (ESI) m/z: Calcd. for C25H26FN3O5 467.19 [M]+, found 468.5 [M+H]+.
(S)-2-Cyclohexyl-2-(1-(4-fluorophenyl)-5-phenyl-1H-pyrazole-3-carboxamido)acetic acid (14)
Following the procedure described in 1, compound 14 was obtained using 1-(4-fluorophenyl)-5-phenyl-1H-pyrazole-3-carboxylic acid. 85% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.06 -1.40 (m, 5 H), 1.59-1.90 (m, 5 H), 1.93-2.07 (m, 1H), 4.73 (dd, J=8.67, 5.65 Hz, 1 H), 7.04 (s, 1 H), 7.06 - 7.12 (m, 2H), 7.16 - 7.23 (m, 2 H) 7.28 - 7.37 (m, 5 H), 7.42 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C24H24FN3O3 421.18 [M]+, found 422.5 [M+H]+.
(S)-2-Cyclohexyl-2-(1-(4-fluorophenyl)-5-methyl-1H-pyrazole-3-carboxamido)acetic acid (15)
Following the procedure described in 1, compound 15 was obtained using 1-(4-fluorophenyl)-5-methyl-1H-pyrazole-3-carboxylic acid. 74% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.05 - 1.35 (m, 5 H), 1.59 - 1.87 (m, 5 H), 1.90-2.05 (m, 1 H), 2.31 (s, 3 H), 4.66 (dd, J=8.67, 5.65 Hz, 1 H), 6.73 (s, 1 H), 7.16 - 7.25 (m, 2 H), 7.35 (d, J=8.67 Hz, 1 H), 7.40 - 7.48 (m, 2 H). MS (ESI) m/z: Calcd. for C19H22FN3O3 359.16 [M]+, found 360.2 [M+H]+.
(S)-5-Amino-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-oxopentanoic acid (16)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (100 mg, 0.292 mmol) was dissolved in THF (7 mL). To the solution was added HATU (111 mg, 0.292 mmol) and triethylamine (0.20 mL, 1.461 mmol). The resulting mixture was stirred at room temperature for 15 minutes. L-glutamine α-4-nitrobenzyl ester hydrobromide (116 mg, 0.32 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-4-nitrobenzyl 5-amino-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-oxopentanoate as white solid (113 mg, 65 %). Following the trimethyltin hydroxide hydrolysis procedure 3, compound 16 was obtained from (S)-4-nitrobenzyl 5-amino-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-oxopentanoate. 75 % yield; white solid. 1H NMR (CDCl3 , 300 MHz) δ 2.18 - 2.46 (m, 2 H), 2.47 - 2.65 (m, 2 H), 3.55 (s, 3 H), 3.60 (s, 3 H), 4.73 - 4.87 (m, 1 H), 6.42-6.61 (m, 3 H), 6.90 - 7.03 (m, 2 H), 6.93 (s, 1 H), 7.20-7.37 (m, 4 H), 7.88 (d, J=7.16 Hz, 1 H). MS (ESI) m/z: Calcd. for C23H23FN4O6 470.16 [M]+, found 471.4 [M+H]+.
(S)-6-((Tert-butoxycarbonyl)amino)-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoic acid (17)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added HATU (167 mg, 0.438 mmol) and triethylamine (0.30 mL, 2.19 mmol). The resulting mixture was stirred at room temperature for 20 minutes. H-Lys(Boc)-OMe hydrochloride (143 mg, 0.482 mmol) was added and stirred at room temperature for 3 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 6-((tert-butoxycarbonyl)amino)-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoate as white solid (145 mg, 57%). Using the trimethyltin hydroxide hydrolysis procedure 3, compound 17 was obtained from (S)-methyl 6-((tert-butoxycarbonyl)amino)-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoate. 93 % yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.12 - 1.39 (m, 2 H), 1.42 (s, 9 H), 1.45-1.62 (m., 2 H), 1.74-195 (m, 1 H), 1.96-2.16 (m, 1 H), 3.00-3.21 (m, 2 H), 3.60 (s, 3 H), 3.56 (s, 3 H), 4.56-4.84 (m, 1 H), 6.50 (d, J=5.27 Hz, 2 H), 6.91 - 7.00 (m, 3 H), 7.20 - 7.33 (m, 4 H), 7.49 (br. s., 1 H). MS (ESI) m/z: Calcd. for C29H35FN4O7 570.25 [M]+, found 571.8 [M+H]+.
(S)-6-Amino-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoic acid trifluoroacetate (18)
Following the tert-butyl ester deprotection procedure 22, compound 18 was obtained from (S)-6-((tert-butoxycarbonyl)amino)-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoic acid (17). 78% yield; white solid; 1H NMR (MeOH-d4 , 300 MHz) δ 1.49-1.62 (m, 2 H), 1.65-1.79 (m, 2 H), 1.85-1.97 (m, 1 H), 2.01-2.13 (m, 1 H), 2.81 (bs, 2H), 2.93 (t, J=9.0 Hz, 3 H), 3.62 (s, 6 H), 4.64-4.71 (m, 1 H), 6.63 (d, J=9.0 Hz, 2 H), 6.81 (s, 1 H), 7.06 (t, J=9.0 Hz, 2 H), 7.27-7.32 (m, 2 H), 7.36 (t, J=9.0 Hz, 1H). MS (ESI) m/z: Calcd. for C24H27FN4O5 470.20 [M]+, found 471.5 [M+H]+.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-methylpentanoic acid (19)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added HATU (167 mg, 0.438 mmol) and triethylamine (0.18 mL, 1.314 mmol). The resulting mixture was stirred at room temperature for 20 minutes. L-leucine methyl ester hydrochloride (80 mg, 0.438 mmol) was added and stirred at room temperature for 4 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-methylpentanoate as white solid (135 mg, 66 %). Using the trimethyltin hydroxide hydrolysis procedure 3, compound 19 was obtained from (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-methylpentanoate. 82 % yield; white solid. 1H NMR (CDCl3 , 300 MHz) δ 0.99 (dd, J=6.03, 2.64 Hz, 6 H), 1.69 - 1.92 (m, 3 H), 3.61 (s, 3 H), 3.55 (s, 3 H), 4.74 - 4.86 (m, 1 H), 6.50 (t, J=6.78 Hz, 2 H), 6.93 - 7.01 (m, 3 H), 7.23 - 7.36 (m, 4 H). MS (ESI) m/z: Calcd. for C24H26FN3O5 455.19 [M]+, found 456.4 [M+H]+.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-methylpentanoic acid (20)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (6 mL). To the solution was added benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (213 mg, 0.482 mmol) and triethylamine (0.18 mL, 1.314 mmol). The resulting mixture was stirred at room temperature for 15 minutes. (S)-Tert-butyl 3-amino-5-methylhexanoate (97 mg, 0.482 mmol) in 0.5 mL of THF was added dropwise, and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4, followed by filtration. The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give the title compound as white solid (145 mg, 63 %). Using the tert-butyl ester deprotection procedure 22, the title compound 20 was obtained from (S)-tert-butyl 3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-methylhexanoate. 81% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 0.96 (t, J=6.78 Hz, 6 H), 1.40 -1.51 (m, 1 H), 1.62 - 1.82 (m, 2 H), 2.70 (d, J=5.27 Hz, 2 H), 3.52 (s, 3 H), 3.59 (m, 3 H), 4.47-4.60 (m, 1 H), 6.50 (d, J=8.67 Hz, 2 H), 6.93 - 7.02 (m, 2 H), 7.22 - 7.35 (m, 5 H). MS m/z: Calcd. for C25H28FN3O5 469.20 [M]+, found 470.6 [M+H]+.
(S)-3-Cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)propanoic acid (21)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (123 mg, 0.359 mmol) was dissolved in THF (6 mL). To the solution was added BOP (146 mg, 0.329 mmol) and triethylamine (0.150 mL, 1.077 mmol). The resulting mixture was stirred at room temperature for 15 minutes. (S)-Tert-butyl 3-amino-3-cyclohexylpropanoate (75 mg, 0.329 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-tert-butyl 3-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)propanoate as colorless foam (115 mg, 58 %). Using the tert-butyl ester deprotection procedure 22, the title compound 21 was obtained from (S)-tert-butyl 3-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)propanoate. 31% yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 1.03 - 1.29 (m, 5 H), 1.60-1.94 (m, 6 H), 2.62-2.79 (m, 2 H), 3.56 (s, 3 H), 3.59 (s, 3 H), 4.22-4.31 (m, 1 H), 6.50 (d, J=9.0 Hz, 2 H), 6.93-6.99 (m, 2 H), 6.95 (s, 1 H), 7.23-7.41 (m, 4 H). MS (ESI) m/z: Calcd. for C27H30FN3O5 495.22 [M]+, found 494.8 [M-H]−.
(S)-5-Cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)pentanoic acid (22)
Following the procedure described in 17, (S)-Tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)pentanoate (35) was obtained using 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) and tert-butyl (S)-3-amino-5-cyclohexylpentanoate. Trifluoroacetic acid (0.7 mL) was added dropwise to a solution of (S)-tert-butyl5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)pentanoate (35) (70 mg, 0.120 mmol) in CH2Cl2 (1.5 mL). The reaction mixture was stirred at room temperature for 1.5 h and the solvent was evaporated in vacuo. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give the title compound 22 as white solid. 80% yield; 1H NMR (CDCl3, 300 MHz) δ 0.77-0.98 (m, 2 H), 1.08-1.40 (m, 6 H), 1.57-1.78 (m, 7 H), 2.70 (d, J=5.46 Hz, 2 H), 3.57 (s, 3 H), 3.58 (s, 3 H), 4.32-4.44 (m, 1 H), 6.50 (d, J=8.10 Hz, 2 H), 6.90 - 7.05 (m, 3 H), 7.24 (d, J=5.09 Hz, 2 H), 7.27 - 7.36 (m, 2 H). MS (ESI) m/z: Calcd. for C29H34FN3O5 523.60 [M]+, found 522.7 [M-H]−.
1-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)cyclohexanecarboxylic acid (23)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added HATU (167 mg, 0.438 mmol) and triethylamine (0.30 mL, 2.19 mmol). The resulting mixture was stirred at room temperature for 20 minutes. Ethyl 1-aminocyclohexane-1-carboxylate hydrochloride (100 mg, 0.482 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 6-((tert-butoxycarbonyl)amino)-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)hexanoate as white solid (145 mg, 67%). Using the lithium hydroxide hydrolysis procedure 2, compound 23 was obtained from ethyl 1-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)cyclohexanecarboxylate. 83% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.29 - 1.46 (m, 2 H), 1.47 - 1.81 (m, 4 H), 1.95 - 2.13 (m, 2 H), 2.21-2.34 (m, 2 H), 3.59 (s, 6 H), 6.52 (d, J=8.29 Hz, 2 H), 6.95 - 7.02 (m, 2 H), 7.17 - 7.36 (m, 5 H). MS (ESI) m/z: Calcd. for C25H26FN3O5 467.19 [M]+, found 468.5 [M+H]+.
Trans-4-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)cyclohexanecarboxylic acid (24)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added BOP (193 mg, 0.438 mmol) and triethylamine (0.182 mL, 1.314 mmol). The resulting mixture was stirred at room temperature for 20 minutes. Trans-methyl-4-aminocyclohexanecarboxylate hydrochloride (93 mg, 0.482 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give trans-methyl-4-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)cyclohexane-1-carboxylate. Following the lithium hydroxide hydrolysis procedure 2, compound 24 was obtained from trans-methyl-2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)benzoate. 80% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 1.21 - 1.42 (m, 2 H), 1.52 - 1.75 (m, 2 H), 2.01 - 2.24 (m, 4 H), 2.26 -2.39 (m, 1 H), 3.58 (s, 6 H), 3.92 - 4.08 (m, 1 H), 6.50 (d, J=8.67 Hz, 2 H), 6.87 (d, J=8.67 Hz, 1 H), 6.93 - 7.01 (m, 3 H), 7.22 - 7.33 (m, 3 H). MS m/z: MS (ESI) m/z: Calcd. for C25H26FN3O5 467.19 [M]+, found 468.5 [M+H]+.
(S)-1-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carbonyl)pyrrolidine-3-carboxylic acid (25)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.305 mmol) was dissolved in THF (7 mL). To the solution was added HATU (174 mg, 0.305 mmol) and triethylamine (0.32 mL, 1.525 mmol). The resulting mixture was stirred at room temperature for 15 minutes. L-Proline methyl ester hydrochloride (83 mg, 0.335 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 1-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carbonyl)pyrrolidine-3-carboxylate as white solid (115 mg, 58 %). Using the trimethyltin hydroxide hydrolysis procedure 3, compound 25 was obtained from (S)-methyl 1-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carbonyl)pyrrolidine-3-carboxylate. 45 % yield; white solid. 1H NMR (MeOH-d4, 300 MHz) δ 1.86 - 2.14 (m, 1 H), 2.16 - 2.48 (m, 1 H), 3.54 (s, 3 H), 3.60 (s, 3 H), 3.69 - 3.90 (m, 2 H), 4.15 - 4.24 (m, 1 H), 4.56-4.67 (m, 1 H), 5.35 (dd, J=8.67, 2.64 Hz, 1 H), 6.54 - 6.68 (m, 2 H), 6.81 (s, 1 H), 6.94 - 7.10 (m, 2 H), 7.21 -7.40 (m, 3 H). MS (ESI) m/z: Calcd. for C23H22FN3O5 439.15 [M]+, found 440.4 [M+H]+.
2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)adamantane-2-carboxylic acid (26)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) was heated under reflux in SOCl2 for 2 hr followed by cooling and concentration in vac-uo. The residue was then dissolved in toluene and evaporated (3×) to remove HCl. The residue was then taken up in dry THF and added to a well agitated mixture of 5:1 THF:water (15 mL/g of acid chloride) containing 1.1 eq 2-aminoadamantane-carboxylic acid and 2.2 eq of NaOH at 5 °C. Following the addition, the stirring was continued for 18 hr at ambient temp. After this time, the mixture is made acidic with 1 N HCl and extracted (3×) with CH2Cl2, dried over sodium sulfate and evaporated. The residue was chromato-graphed using a 0-15% CH2Cl2:MeOH gradient. Evaporation provided the title compound 26. 71% yield; off-white solid; 1H NMR (CDCl3) δ 7.20-7.36 (m, 4 H), 6.90-7.04 (m, 3 H), 6.51 (d, J = 8.5 Hz, 2 H), 3.54-3.63 (m, 6 H), 2.71-2.81 (m, 2 H), 2.23 (d, J = 12.8 Hz, 2 H), 2.07 (d, J = 12.2 Hz, 2 H), 1.68-1.95 (m, 8 H). MS (ESI) m/z: Calcd. for C29H30FN3O5 519.22 [M]+, found 518.9 (M-H)−.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-methylbutanoic acid (27)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added HATU (167 mg, 0.438 mmol) and triethylamine (0.30 mL, 0.2.19 mmol). The resulting mixture was stirred at room temperature for 20 minutes. L-Valine methyl ester hydrochloride (80 mg, 0.482 mmol) was added and stirred at room temperature for 3 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-methylbutanoate as white solid (135 mg, 68 %). Following the trimethyltin hydroxide hydrolysis procedure 3, compound 27 was obtained from (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-methylbutanoate. 62 % yield; white solid. 1H NMR (MeOH-d4, 300 MHz) δ 1.04 (t, J=6.59 Hz, 6 H), 2.24 - 2.38 (m, 1 H), 3.61 (s, 6 H), 4.55 (d, J=5.27 Hz, 1 H), 6.62 (d, J=8.67 Hz, 2 H), 6.80 (s, 1 H), 6.99 - 7.10 (m, 2 H), 7.25 - 7.38 (m, 3 H). MS (ESI) m/z: Calcd. for C23H24FN3O5 441.17 [M]+, found 442.3 [M+H]+.
2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)benzoic acid (28)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (100 mg, 0.292 mmol) was dissolved in THF (7 mL). To the solution was added HATU (111 mg, 0.292 mmol) and triethylamine (0.122 mL, 0.876 mmol). The resulting mixture was stirred at room temperature for 20 minutes. Methyl 2-aminobenzoate trifluoroacetate (85 mg, 0.321 mmol) was added and stirred at room temperature for 18 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4.The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)benzoate as white solid (18 mg, 13%). Using the lithium hydroxide hydrolysis procedure 2, compound 28 was obtained from methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)benzoate. 70% yield; white solid; 1H NMR (CDCl3 , 300 MHz) δ 3.59 (s, 6 H), 6.54 (d, J=6.10, 2 H), 7.00 (t, J=6.10, 2 H), 7.05 (s, 1 H), 7.14 - 7.21 (m, 1 H), 7.31 - 7.41 (m, 3 H), 7.62 - 7.71 (m, 1 H), 8.10 (dd, J=8.10, 1.70 Hz, 1 H), 9.00 (dd, J=8.67, 0.75 Hz, 1 H), 12.38 (s, 1 H). MS (ESI) m/z: Calcd. for C25H20FN3O5 461.14 [M]+, found 462.6 [M+H]+.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-2-phenylacetic acid (29)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (150 mg, 0.438 mmol) was dissolved in THF (7 mL). To the solution was added HATU (167 mg, 0.438 mmol) and triethylamine (0.30 mL, 2.191 mmol). The resulting mixture was stirred at room temperature for 20 minutes. (S)-(+)-2-phenylglycine methyl ester hydrochloride (90 mg, 0.482 mmol) was added and stirred at room temperature for 2 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-2-phenylacetate as white solid (145 mg, 68 %). Using the trimethyltin hydroxide hydrolysis procedure 3, compound 29 was obtained from (S)-methyl 2-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-2-phenylacetate. 60 % yield; white solid. 1H NMR (MeOH-d4 , 300 MHz) δ 3.60 (s, 6 H), 5.51-5.59 (m, 1 H), 6.61 (d, J=8.67 Hz, 2 H), 6.78 (s, 1 H), 7.00 - 7.09 (m, 2 H), 7.22 -7.40 (m, 6 H), 7.51 (d, J=7.54 Hz, 2 H). MS (ESI) m/z: Calcd. for C26H22FN3O5 475.15 [M]+, found 476.5 [M+H]+.
(S)-2-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanoic acid (30)
The title compound 30 was synthesized on a wang resin by standard protocol. Wang resin (100 mg, 1.11 mmol/ g, 0.11 mmol) was swollen in CH2Cl2/DMF (8:2) for 30 minutes, and the amino acid was coupled to the resin by use of Fmoc-L-Phe (5.0 equiv), HBTU (2.0 equiv), and HOBt (2.0 equiv) in DMF with a reaction time of 1 h. The Fmoc group was cleaved with piperidine in DMF (20 %). After deprotection of the Fmoc group, 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) was coupled using HBTU (2.0 equiv), and HOBt (2.0 equiv) in DMF with a reaction time of 1 h. Cleavage of the product from the resin was achieved by treatment with TFA/H2O/triisopropylsilane (95:2.5:2.5) for 1 h and precipitation by adding cold ether to the solution. The solid title compound was filtered and washed with cold ether and dried (21 mg). white solid. 1H NMR (MeOH-d4, 300 MHz) δ 3.13 - 3.20 (m, 1 H), 3.59 (s, 6 H), 4.76 - 4.92 (m, 2 H), 6.61 (d, J=8.29 Hz, 2 H), 6.75 (s, 1 H), 6.95 - 7.09 (m, 2 H), 7.16 -7.29 (m, 7 H), 7.35 (t, J=8.48 Hz, 2 H). MS (ESI) m/z: Calcd. for C27H24FN3O5 489.17 [M]+, found 490.6 [M+H]+.
(S)-3-(5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-phenylpentanoic acid (31)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (45 mg, 0.131 mmol) was dissolved in THF (6 mL). To the solution was added BOP (69 mg, 0.157 mmol) and triethylamine (0.05 mL, 0.327 mmol). The resulting mixture was stirred at room temperature for 15 minutes. (S)-Tert-butyl 3-amino-5-phenylpentanoate (34 mg, 0.144 mmol) was added and stirred at room temperature for 3 h. THF was evaporated in vacuo, water was added to the residue and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with water, brine and then dried with Na2SO4 .The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give (S)-tert-butyl 3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-phenylpentanoate. Using the tert-butyl ester deprotection procedure 22, the title compound 31 was obtained from (S)-tert-butyl 3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)-5-phenylpentanoate. 77% yield; white solid; 1H NMR (CDCl3, 300 MHz) δ 1.92-2.16 (m, 2 H), 2.67-2.84 (m, 2 H), 2.74 (d, J=5.46 Hz, 2 H), 3.57 (s, 3 H), 3.59 (s, 3 H), 4.46-4.56 (m, 1 H), 6.51 (d, J=8.10 Hz, 2 H), 6.86 - 7.05 (m, 3 H), 7.13 - 7.25 (m, 5 H), 7.27 -7.35 (m, 3 H), 7.44 (d, J=8.67 Hz, 1 H). MS (ESI) m/z: Calcd. for C29H28FN3O5 517.55 [M]+, found 516.8 [M-H]−.
Sodium salt of methyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (33)
Following the procedure as reported in the thesis titled - Quéré, L.
Etude de l`interaction ligand-récepteur neurotensinergique. University of Namur. 1995
Methyl 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylate (34a)
Sodium salt of methyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate (33) (1.5 g, 5.204 mmol) and commercially available 4-fluorophenylhydrazine hydrochloride (0.85 g, 5.204 mmol) was mixed with glacial acetic acid (25 mL) and conc. HCl (0.6 mL). The reaction mixture was heated to reflux for 3.5 h. After cooling, reaction mixture was poured into water (25 mL). The aqueous layer was extracted with CH2Cl2 (3 × 30 mL) and the combined CH2Cl2 layer was washed with saturated aqueous NaHCO3. The organic layer was then washed with saturated brine, dried over Na2SO4, followed by filteration. The solvent was evaporated in vacuo to give crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give desired isomer methyl 5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylate (34a) as light yellow solid (1.1 g, 61 %). Methyl 3-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-5-carboxylate (34b) was obtained as as light yellow foam (120 mg). 1H NMR (CDCl3 , 300 MHz) δ 3.59 (s, 6 H), 3.96 (s, 3 H), 6.50 (d, J=9.0 Hz, 2 H), 6.90-6.97 (m, 2 H), 6.96 (s, 1H), 7.34-7.24 (m, 3 H). MS (ESI) m/z: Calcd. for C19H17FN2O4 356.12 [M]+, found 357.4 [M+H]+.
(S)-Tert-butyl 5-cyclohexyl-3-(5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxamido)pentanoate (35)
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic acid (2) (1g, 3.286 mmol) was dissolved in acetonitrile (30 mL). To the solution was added HBTU (1.75 g, 4.63 mmol) and tert-butyl (S)-3-amino-5-cyclohexylpentanoate (0.88 g, 3.450 mmol) followed by dropwise addition of triethylamine (0.911 mL, 6.572 mmol). The resulting mixture was stirred at room temperature for 3 h. Reaction mixture was quenched with saturated NaHCO3 solution (10 mL) and acetonitrile was removed in vacuo. Ethyl acetate was added to the residue and washed with saturated NaHCO3 solution. The aqueous layer was re-extracted with EtOAc (2 × 30 mL). The combined organic layers were washed with brine and then dried with Na2SO4, followed by filtration. The solvent was evaporated in vacuo to give the crude residue. The residue was purified by silica gel flash chromatography (EtOAc:Hex) to give the title compound as white solid (1.5 g, 79 %). 1H NMR (CDCl3 , 300 MHz) δ 0.80-0.96 (m, 2 H), 1.11-1.88 (m, 6 H), 1.46 (s, 9 H), 1.58 -1.76 (m, 7 H), 2.54 (dd, J=5.46, 1.41 Hz, 2 H), 3.56 (s, 3 H), 3.59 (s, 3 H), 4.35-4.47 (m, 1 H), 6.50 (d, J=7.72 Hz, 2 H), 6.89 - 7.03 (m, 3 H), 7.22 -7.26 (m, 2 H), 7.27-7.37 (m, 2 H). MS (ESI) m/z: Calcd. for C33H42FN3O5 579.70 [M]+, found 580.7 [M+H]+.
5.2 X-ray Crystallographic Analysis
X-ray Crystallographic Analysis. Single-crystal x-ray diffraction data on compound (2) was collected using MoKα radiation and a Bruker APEX 2 CCD area detector. The crystal was prepared for data collection by coating with high viscosity microscope oil. The oil coated crystal was mounted on a micromesh mount (Mitergen, Inc.) and transferred to the diffractometer, and a data set was collected at 150 K. The 0.529 × 0.422 × 0.368 mm3 crystal was triclinic in space group P-1, with unit cell dimensions a = 8.550(5), b = 12.619(7), c = 16.694(10) Å, a = 91.803(9)°, β = 95.702(8)°, and g = 109.750(8)°. Data was 96.3% complete to 25.53°θ (~0.82 Å) with an average redundancy of 2.12. The data crystal was twinned with three components in a 14.6:3.9:0.9 ratio (based on mean I/s), and only non-overlapped reflections from the major twin component were used in structure solution and refinement. The structure was solved by direct methods and refined by full-matrix least-squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Corrections were applied for Lorentz, polarization, and absorption effects. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C–H distance set at 0.96 Å. The final anisotropic full matrix least-squares refinement on F2 with 463 variables and two restraints converged at R1 = 6.81% for the observed data and wR2 = 19.00% for all data. Atomic coordinates for (2) have been deposited with the Cambridge Crystallographic Data Centre (deposition number 1048741). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax:+44(0)-1223–336033 or depos-it@ccdc.cam.ac.uk
5.3 Calcium mobilization assay for APJ, NTR1 and NTR2
Cells stably expressing human APJ-Gαq16 were removed from the flasks using a non-enzymatic solution (Cell-stripper, Mediatech Inc., Orlando, FL) and quenched with DMEM/F12, 10 % FBS, centrifuged, and re-suspended in the serum-containing media. Cells were counted with a hemocytometer and 30,000 cells were transferred to each well of a black Costar 96-well optical bottom plate (Corning Corporation, Corning, NY). Each plate was incubated at 37 C for 24 h to confluence. The culture media was removed from the plates and cells were subsequently loaded with a fluorescent calcium probe (Calcium 5 dye; Molecular Devices, Sunnyvale, CA) in an HBSS-based buffer containing 20 mM HEPES, 1% BSA and 10 µM probenecid (Sigma) in a total volume of 225 µL. Cells were incubated at 37 C for 1 h and then stimulated with various compounds or pyr-apelin-13 (Anaspec, Freemont, CA) at various concentrations using a FLIPR Tetra plate-reader, which automatically added the agonist at 10x concentration to each well after reading baseline values for ~17 sec. Agonist-mediated change in fluorescence (488 nm excitation, 525 nm emission) was monitored in each well at 1 sec intervals for 90 sec. Data were collected using Softmax version 4.8 (MDS Analytical Technologies) and analyzed using Prism software (GraphPad, La Jolla, CA). Nonlinear regression analysis was performed to fit data and obtain maximum response (Emax), EC50, correlation coefficient (r2), and other parameters. All experiments were performed in duplicate at least three times to ensure reproducibility and data reported as mean ±standard error, unless noted otherwise.
Assays for rat neurotensin receptors 1 and 2 (NTR1 and NTR2) followed a very similar format and have been described.47,48
5.4 Radioligand displacement
Competitive inhibition binding was performed using [125I]-apelin-13 (Perkin Elmer, Akron, OH). Eleven concentrations (ranging from 10−11 M to 3.16×10−5 M) of compound were incubated in assay buffer (0.025 M HEPES, (pH=7.4), 0.01 M MgCl2, 0.001 M CaCl2, 0.5% fatty acid-free BSA) with 7.5×10−11 M [125I]-apelin-13 (Perkin-Elmer) in the presence of 0.125 µg of CHO-K1 cell membrane homogenates expressing the human apelin receptor (Perkin Elmer). Nonspecific binding (NSB) was determined in the presence of 10−6 M unlabeled apelin-13 (Anaspec). The assays were run in 1.4 mL polypropylene tubes (Thermo Scientific, Waltham, MA) in a 96-well format. All assays were incubated at room temperature on a shaking platform for two hours. Incubation was terminated and bound radioligand was separated from free radioligand by rapid vacuum filtration over 0.5% PEI treated GF/C filter plates using ice-cold 50 mM Tris-HCl (pH=7.4) in a Brandel Scientific (Gaithersburg, MD) 96-well harvester. Filter plates were allowed to dry and Microscint 20 scintillation cocktail (Perkin-Elmer) was added to each well. Bound radioactivity was determined with a TopCount 12-detector instrument (Packard Instruments) using standard scintillation counting techniques. The binding data from each assay plate were normalized to NSB and total counts per minute (CPM) bound values prior to analysis. The Ki values were calculated from a three-parameter logistic curve fit to the data with Prism (version 6.0, GraphPad Software, Inc., San Diego, CA) using 2.1×10−10 M as the Kd for [125I]-apelin-13.
Figure 3.

Structures of compound 1a, 3, 4 and 5
Table 1.
Structural modification at site A
 | ||
|---|---|---|
| Compd. | R | EC50 (µM) |
| 6 | >30 | |
| 7 | >30 | |
| 8 | 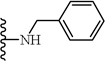 |
>30 |
| 9 | >30 | |
| 10 | 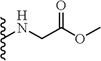 |
>30 |
| 11 | 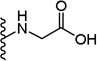 |
>30 |
Acknowledgments
The authors thank Dr. Angela Giddings, for help with various assays and to Dr. Danni Harris for detailed discussion of data. The X-ray crystallographic work was supported by NIDA through Interagency Agreement #Y1-DA1101 with the Naval Research Laboratory (NRL).
Funding Sources
This work was supported NICHD grant 1R01HD079547-01A1 to Rangan Maitra and Scott Runyon.
ABBREVIATIONS
- AT1
Angiotensin 1
- CB1
Cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- NPS
Neuropeptide S receptor
- NTR-1
Neurotensin receptor 1
- BOP
Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- LiOH.H2O
Lithium hydroxide monohydrate
- TFA
Trifluroacetic acid
- HCl
Hydrochloric acid
- Et3N
Triethylamine
- DCM
Dichloromethane
- THF
Tetrahydrofuran
- MeOH
Methanol
- SAR
Structure Activity Relationship
- TLC
thin layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. Gene. 1993;136:355. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- 2.Habata Y, Fujii R, Hosoya M, Fukusumi S, Kawamata Y, Hinuma S, Kitada C, Nishizawa N, Murosaki S, Kurokawa T, Onda H, Tatemoto K, Fujino M. Biochim Biophys Acta. 1999;1452:25. doi: 10.1016/s0167-4889(99)00114-7. [DOI] [PubMed] [Google Scholar]
- 3.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Biochem Biophys Res Commun. 1998;251:471. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 4.Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, Kitada C, Honda S, Kurokawa T, Onda H, Nishimura O, Fujino M. J Biol Chem. 2000;275:21061. doi: 10.1074/jbc.M908417199. [DOI] [PubMed] [Google Scholar]
- 5.Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S, Nishizawa N, Kitada C, Onda H, Nishimura O, Fujino M. Biochim Biophys Acta. 2001;1538:162. doi: 10.1016/s0167-4889(00)00143-9. [DOI] [PubMed] [Google Scholar]
- 6.Langelaan DN, Bebbington EM, Reddy T, Rainey JK. Biochemistry. 2009;48:537. doi: 10.1021/bi801864b. [DOI] [PubMed] [Google Scholar]
- 7.Lee DK, Saldivia VR, Nguyen T, Cheng R, George SR, O’Dowd BF. Endocrinology. 2005;146:231. doi: 10.1210/en.2004-0359. [DOI] [PubMed] [Google Scholar]
- 8.Chng SC, Ho L, Tian J, Reversade B. Dev Cell. 2013;27:672. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Science. 2014;343:746. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Carroll AM, Don AL, Lolait SJ. J Neuroendocrinol. 2003;15:1095. doi: 10.1046/j.1365-2826.2003.01102.x. [DOI] [PubMed] [Google Scholar]
- 11.O’Carroll AM, Lolait SJ. J Neuroendocrinol. 2003;15:661. doi: 10.1046/j.1365-2826.2003.01044.x. [DOI] [PubMed] [Google Scholar]
- 12.De Mota N, Lenkei Z, Llorens-Cortes C. Neuroendocrinology. 2000;72:400. doi: 10.1159/000054609. [DOI] [PubMed] [Google Scholar]
- 13.Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O’Dowd BF. J Neurochem. 2000;74:34. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 14.Lathen C, Zhang Y, Chow J, Singh M, Lin G, Nigam V, Ashraf YA, Yuan JX, Robbins IM, Thistlethwaite PA. Circulation. 2014;130:1179. doi: 10.1161/CIRCULATIONAHA.113.007822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleinz MJ, Davenport AP. Regul Pept. 2004;118:119. doi: 10.1016/j.regpep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Kleinz MJ, Skepper JN, Davenport AP. Regul Pept. 2005;126:233. doi: 10.1016/j.regpep.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 17.Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, Sharma S, Neilson I, Webb DJ, Megson IL, Flapan AD, Newby DE. J Am Coll Cardiol. 2008;52:908. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, Johnston NR, Denvir MA, Megson IL, Flapan AD, Newby DE. Circulation. 2010;121:1818. doi: 10.1161/CIRCULATIONAHA.109.911339. [DOI] [PubMed] [Google Scholar]
- 19.Barnes GD, Alam S, Carter G, Pedersen CM, Lee KM, Hubbard TJ, Veitch S, Jeong H, White A, Cruden NL, Huson L, Japp AG, Newby DE. Circ Heart Fail. 2013;6:482. doi: 10.1161/CIRCHEARTFAILURE.111.000077. [DOI] [PubMed] [Google Scholar]
- 20.De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C. Proc Natl Acad Sci U S A. 2004;101:10464. doi: 10.1073/pnas.0403518101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitra A, Katovich MJ, Mecca A, Rowland NE. Physiol Behav. 2006;89:221. doi: 10.1016/j.physbeh.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Reichenbach V, Ros J, Fernandez-Varo G, Casals G, Melgar-Lesmes P, Campos T, Makriyannis A, Morales-Ruiz M, Jimenez W. J Pharmacol Exp Ther. 2012;340:629. doi: 10.1124/jpet.111.188078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke KJ, Whitaker KW, Reyes TM. J Neuroendocrinol. 2009;21:83. doi: 10.1111/j.1365-2826.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- 24.O’Shea M, Hansen MJ, Tatemoto K, Morris MJ. Nutr Neurosci. 2003;6:163. doi: 10.1080/1028415031000111273. [DOI] [PubMed] [Google Scholar]
- 25.Sunter D, Hewson AK, Dickson SL. Neurosci Lett. 2003;353:1. doi: 10.1016/s0304-3940(03)00351-3. [DOI] [PubMed] [Google Scholar]
- 26.Taheri S, Murphy K, Cohen M, Sujkovic E, Kennedy A, Dhillo W, Dakin C, Sajedi A, Ghatei M, Bloom S. Biochem Biophys Res Commun. 2002;291:1208. doi: 10.1006/bbrc.2002.6575. [DOI] [PubMed] [Google Scholar]
- 27.Valle A, Hoggard N, Adams AC, Roca P, Speakman JR. J Neuroendocrinol. 2008;20:79. doi: 10.1111/j.1365-2826.2007.01617.x. [DOI] [PubMed] [Google Scholar]
- 28.Volkoff H, Wyatt JL. Peptides. 2009;30:1434. doi: 10.1016/j.peptides.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Dray C, Knauf C, Daviaud D, Waget A, Boucher J, Buleon M, Cani PD, Attane C, Guigne C, Carpene C, Burcelin R, Castan-Laurell I, Valet P. Cell Metab. 2008;8:437. doi: 10.1016/j.cmet.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Yue P, Jin H, Aillaud M, Deng AC, Azuma J, Asagami T, Kundu RK, Reaven GM, Quertermous T, Tsao PS. Am J Physiol-Endoc M. 2010;298:E59. doi: 10.1152/ajpendo.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue P, Asagami T, Kundu RK, Yee YG, Glassford AJ, Azuma J, Quertermous T, Tsao PS. Circulation. 2007;116:247. [Google Scholar]
- 32.O’Donnell LA, Agrawal A, Sabnekar P, Dichter MA, Lynch DR, Kolson DL. J Neurochem. 2007;102:1905. doi: 10.1111/j.1471-4159.2007.04645.x. [DOI] [PubMed] [Google Scholar]
- 33.Cheng B, Chen J, Bai B, Xin Q. Peptides. 2012;37:171. doi: 10.1016/j.peptides.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Zeng XJ, Yu SP, Zhang L, Wei L. Exp Cell Res. 2010;316:1773. doi: 10.1016/j.yexcr.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horiuchi Y, Fujii T, Kamimura Y, Kawashima K. J Neuroimmunol. 2003;144:46. doi: 10.1016/j.jneuroim.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 36.Cayabyab M, Hinuma S, Farzan M, Choe H, Fukusumi S, Kitada C, Nishizawa N, Hosoya M, Nishimura O, Messele T, Pollakis G, Goudsmit J, Fujino M, Sodroski J. J Virol. 2000;74:11972. doi: 10.1128/jvi.74.24.11972-11976.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu N, Wang H, Fan L, Chen Q. Peptides. 2009;30:1153. doi: 10.1016/j.peptides.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. Hypertension. 2009;54:598. doi: 10.1161/HYPERTENSIONAHA.109.134619. [DOI] [PubMed] [Google Scholar]
- 39.Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, Fujimiya M. Regul Pept. 2001;99:87. doi: 10.1016/s0167-0115(01)00236-1. [DOI] [PubMed] [Google Scholar]
- 40.Iturrioz X, Alvear-Perez R, De Mota N, Franchet C, Guillier F, Leroux V, Dabire H, Le Jouan M, Chabane H, Gerbier R, Bonnet D, Berdeaux A, Maigret B, Galzi JL, Hibert M, Llorens-Cortes C. Faseb J. 2010;24:1506. doi: 10.1096/fj.09-140715. [DOI] [PubMed] [Google Scholar]
- 41.Margathe JF, Iturrioz X, Alvear-Perez R, Marsol C, Riche S, Chabane H, Tounsi N, Kuhry M, Heissler D, Hibert M, Llorens-Cortes C, Bonnet D. J Med Chem. 2014;57:2908. doi: 10.1021/jm401789v. [DOI] [PubMed] [Google Scholar]
- 42.Khan P, Maloney PR, Hedrick M, Gosalia P, Milewski M, Li L, Roth GP, Sergienko E, Suyama E, Sugarman E, Nguyen K, Mehta A, Vasile S, Su Y, Shi S, Stonich D, Nguyen H, Zeng FY, Novo AM, Vicchiarelli M, Diwan J, Chung TDY, Pinkerton AB, Smith LH. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD: 2010. [Google Scholar]
- 43.Hachtel SW, Wolfart P, Weston J, Mueller M, Defossa E, Mertsch K, Weng JH, Binnie RA, Abdul-Latif F, Bock WJ, Walser A. Sanofi[Fr]: WO2014044738 A1. 2014 [Google Scholar]
- 44.Pinkerton ABSLH. Sanford-Burnham Medical Research Institute: WO2015184011 A2. 2015 [Google Scholar]
- 45.Giddings ARSP, Thomas J, Tajuba J, Bortoff K, Maitra R. International Journal of High Throughput Screening. 2010;1:39. [Google Scholar]
- 46.Surgand JS, Rodrigo J, Kellenberger E, Rognan D. Proteins. 2006;62:509. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- 47.Thomas JB, Giddings AM, Wiethe RW, Olepu S, Warner KR, Sarret P, Gendron L, Longpre JM, Zhang YN, Runyon SP, Gilmour BP. J Med Chem. 2014;57:5318. doi: 10.1021/jm5003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas JB, Navarro H, Warner KR, Gilmour B. Bioorg Med Chem Lett. 2009;19:1438. doi: 10.1016/j.bmcl.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]