

Abstract
In this manuscript we highlight the potential of stereospecific nickel-catalyzed cross-coupling reactions for applications in the pharmaceutical industry. Using an inexpensive and sustainable nickel catalyst, we report a gram-scale Kumada cross-coupling reaction. Reactions are highly stereospecific and proceed with inversion at the benzylic position. We also expand the scope of our reaction to incorporate isotopically labeled substituents.
The emergence of reliable cross-coupling protocols has had a substantial effect on the pharmaceutical industry.1 Transition-metal catalyzed reactions, particularly those involving palladium and copper catalysts, are widely used to synthesize libraries of compounds for SAR studies.1,2 The prominence of cross-coupling reactions is due to in part to the generality of the methods, the broad functional group tolerance, and the commercial availability of the starting materials. However, the ubiquity of Csp2–Csp2 cross-couplings can cause a bias toward planar compounds that lack complex stereochemical information.3 One strategy to enter underutilized areas of chemical space is incorporation of increased sp3 character and stereogenic centers into the backbones of molecules.3 For example, combretastatin A-4 (1, Figure 1) is a potent tubulin-binding anticancer agent. Analogues such as 2 replace the alkene linkage with a stereocenter.4 Incorporation stereogenic centers is a strategy to identify molecules that have greater selectivity for their target proteins and present fewer off-target interactions.3 A cross-coupling protocol that could reliably form Csp3–Csp3 bonds with control of absolute and relative stereochemistry could provide synthetic access to new lead compounds for drug discovery efforts.
Figure 1.

Tubulin-binding compounds.
Recently, our laboratory has developed a series of stereospecific nickel-catalyzed alkyl cross coupling reactions that form new Csp3–Csp3 bonds.5 In these transformations, an enantioenriched benzylic ether or ester undergoes a cross-coupling reaction in the presence of an achiral nickel catalyst (Figure 2). Reactions proceed with a high degree of stereochemical fidelity and inversion at the benzylic center. Our lab has successfully developed Kumada,6 Suzuki,7 and Negishi8 cross-coupling reactions that incorporate methyl, n-alkyl and aryl substituents. These reactions can be employed, for example, to introduce a benzylic methyl substituent, a moiety that can provide a compound with increased potency and protection against metabolism.9 Additionally, as a nonprecious metal, nickel catalysts provide a sustainable alternative to palladium catalysts and are particularly well-suited to cross-coupling reactions that connect sp3-centers.10 In this manuscript, we describe the expansion of our nickel-catalyzed Kumada cross-coupling reaction to certain pharmaceutical applications. We began by increasing the scale of our methodology to the gram-scale. We also describe incorporation a new class of nucleophiles, Grignard reagents that carry isotopically labeled alkyl and aryl groups, for synthesis of compounds bearing stable isotope tracers.
Figure 2.

Stereospecific cross-coupling reactions of benzylic ethers and esters.
To challenge our methodology with a preparative-scale Kumada cross-coupling reaction, we first selected a representative substrate. We have recently developed ring-opening reactions of aryl-substituted tetrahydropyrans (THPs) and tetrahydrofurans (THFs) that occur in conjunction with stereospecific Csp3–Csp3 bond formation.6d This Kumada cross-coupling reaction provides the distinct diversity elements necessary for a diversity oriented synthesis (DOS) approach,11 as a novel appendage (hydroxyl functional handle) is formed in conjunction with a skeletal rearrangement of the molecule (ring-opening). Requisite starting materials are prepared in one step with high diastereoselectivity from the corresponding aldehydes via clay-mediated Prins reaction.12 We were happy to see that the Kumada reaction works well on a multigram scale. Utilizing an air-stable preformed catalyst, 2-aryltetrahydropyran 3 undergoes the cross-coupling reaction to afford the ring-opened product 4 with 2.5 mol % catalyst loading (Scheme 1).13 Consistent with our previous studies, the reaction was highly stereospecific, with clean inversion at the benzylic center.
Scheme 1. Cross-Coupling of Aryl-Substituted Tetrahydropyran 3 with Methylmagnesium Iodide on a 5 g Scale.

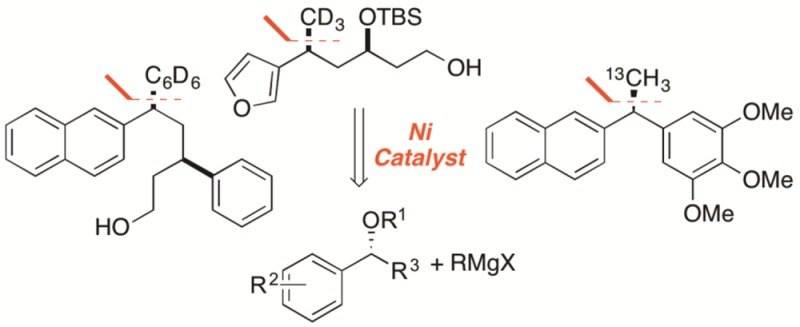
One previously unexplored application of our cross-coupling methodology involves the incorporation of isotopic tracers into biologically relevant molecules. Tracers that utilize stable isotopes (e.g., 2H, 13C) have a range of applications, including stable isotope dilution assays, metabolite identification, and chronic administration studies.14 Importantly, stable isotopes do not have the health risks associated with radioactive isotopes and therefore can be used in studies of pregnant women and young children.15 Stable isotope dilution assays (SIDA), for example, rely on a mixture of isotopomers that are traced through living systems and can be used to elucidate complex biological pathways.16 These studies typically require coadministration of both enriched and nonenriched substrates. Therefore, a synthetic method that could easily incorporate labeled or nonlabeled substituents would suit these applications.
We hypothesized that our Kumada-type cross-coupling reaction could be employed in synthesis of biologically relevant compounds and their isotopologues. The desired isotopomer can be synthesized by proper choice of the alkyl halide precursor. Isotopically labeled alkyl and aryl halides are commercially available and are readily transformed into the corresponding Grignard reagents (Scheme 2). To test this hypothesis, we turned to enantioenriched diarylethane 2, which displays activity against colon cancer lines.4 Our laboratory has previously synthesized 2 by Kumada coupling of ether 5 with methylmagnesium iodide.6a Cross-coupling of 5 with 13C-methylmagnesium iodide afforded 13C-2 in 60% yield with >99% enantiospecificity. As anticipated, the yield was similar to that which we had previously reported with unlabeled reagent.5
Scheme 2. Cross-Couplings of Isotopically-Labeled Grignard Reagents.

Reactions carried out on a 0.2 mmol scale.
Yield after two steps. See SI for more details.
With 13C-2 in hand, we turned to incorporating stable isotope tracers with a mass increase ≥3 amu, as is typically required for stable isotope dilution assay applications.14a,17 Another common stable isotope is deuterium (2H), and a variety of deuterated alkyl and aryl halides are commercially available. Kumada cross-coupling of 3-furyl tetrahydropyran 6 with d3-methylmagnesium iodide provided CD3-labeled 7 in 84% yield. This acyclic product had no loss of stereochemical fidelity, with a dr of >20:1 for both substrate and product, and possesses numerous reactive functional group handles. Aryl Grignard reagents can also be incorporating using our methodology;6b,6c thus we sought to incorporate a d5-phenyl tracer. Compound 3 underwent Kumada cross-coupling with d5-phenylmagnesium bromide, which yielded 8 in good yield and high dr. The yields of both 7 and 8 were comparable to those of their respective nonlabeled analogues.6d
In conclusion, the stereospecific nickel-catalyzed Kumada cross-coupling reaction of benzylic ethers has potential for applications in pharmaceutical chemistry settings. The transformation can be carried out on a synthetically useful scale with low loadings of an inexpensive base metal catalyst. Cross-coupling reactions of benzylic ethers proceed in high yields with excellent stereospecificity on gram-scale. Our protocol can be used to prepare biologically active molecules and their isotopologues using a single set of reaction conditions and commercially available reagents.
Experimental Section
General Notes
a. Catalyst Choice
For each class of Grignard reagent employed, a specific nickel precatalyst will provide optimal yield in the cross-coupling reaction.
i. Reactions of Aryl and n-Alkyl Grignard Reagents
All aryl and n-alkyl Grignard reagents are successfully incorporated by utilizing a Ni(dppe)Cl2 precatalyst; this precatalyst is bench stable and provides high ease-of-use since reactions setup does not require use of a glovebox. Ni(dppe)Cl2 is a fine, red powder that produces a black, opaque reaction mixture after addition of Grignard reagent.
ii. Reactions of Methylmagnesium Iodide
For cross-coupling reactions involving methyl Grignard reagent, catalysts prepared in situ from Ni(cod)2 and a bidentate phosphine ligand afford high yields of the desired product. Ni(cod)2 is a yellow, granular solid which forms reaction mixtures of various hues, depending on choice of ligand. Generally, rac-BINAP is our ligand of choice; however, in cases of heteroaromatic-containing substrates, DPEPhos affords higher yields.18 Ni(cod)2 is air sensitive and best stored in glovebox freezer. If a less sensitive catalyst precursor is desired, a preformed (rac-BINAP)NiCl2 complex or combination of Ni(acac)2, cyclooctadiene, and the requisite phosphine ligand can be employed.19,20 Across a range of conditions (e.g., employing 0.2–25 mmol of ether starting material and 1–10 mol % catalyst loading), the air stable Ni(II) salt, (rac-BINAP)NiCl2, afforded the desired product 4 in similar yields to the Ni(cod)2/rac-BINAP precatalyst system.
b. Grignard Reagent Preparation
Grignard reagents are prepared as solutions in Et2O. Grignard reagents prepared in THF do not provide product, likely due to inhibition of the nickel catalyst by THF. In some cases, particularly in reactions employing aryl Grignard reagents, we have noted that higher molarity Grignard reagents provide a higher yield of the desired product. In reactions of methylmagnesium iodide, Grignard molarity is typically between 2.0 and 3.5 M in Et2O. For sluggish substrates, addition of an equivalent of MgI2 frequently accelerates the cross-coupling reaction.5
c. Workup
After quenching with methanol, the reaction can be worked up in one of two ways to remove residual nickel catalyst. The first method is by aqueous extraction. For this method, the reaction mixture is diluted in diethyl ether, washed with water (×3), dried over Na2SO4, and concentrated in vacuo. Alternatively, workup can be accomplished by quenching the reaction with methanol (to consume excess Grignard reagent) and elution of the mixture through a plug of silica gel eluting with neat Et2O.
d. Purification
After workup, products are typically purified by silica gel flash column chromatography. In certain instances the reaction mixture contains a small amount (typically <5%) of a styrene byproduct that results from β-hydride elimination. When necessary, such styrenes can be removed by two methods. The first method is by flash column chromatography employing silver-impregnated silica gel.21 Alternatively, Upjohn dihydroxylation of the unpurified reaction mixture (employing 1 mol % Os relative to styrene) and facile separation by standard silica gel chromatography may be employed.22
A representative procedure for the cross-coupling reaction is detailed below. For complete experimental details see the Supporting Information.
Cross-Coupling Reaction to Afford Alcohol 4 (Scheme 1)
A flame-dried 500 mL round-bottom flask equipped with a stir bar was charged with tetrahydropyran 3 (5.00 g, 17.3 mmol, 1.00 equiv) and (rac-BINAP)NiCl2 (323 mg, 0.433 mmol, 0.0250 equiv). The flask was evacuated and backfilled with N2 three times, at which point anhydrous toluene (200 mL) was added, followed by slow addition of methylmagnesium iodide (14.4 mL, 43.3 mmol, 3.00 M in Et2O, 2.50 equiv). After 36 h the reaction was quenched with methanol (90 mL), filtered through a plug of silica gel (eluting with neat Et2O, 500 mL), and concentrated in vacuo. Phenyltrimethylsilane (PhTMS) was added as internal standard, and a 1H NMR yield was calculated to be 73% with a small amount of styrene byproduct (2.6 mmol, 15%).
An oxidative workup was employed for the facile removal of the styrene byproduct. The unpurified reaction mixture was added to a 50 mL round-bottom flask, which was then charged with N-methylmorpholine N-oxide (NMO, 323 mg, 2.83 mmol, 1.10 equiv), osmium tetroxide (161 μL, 0.0259 mmol, 4% solution in H2O, 1 mol % relative to styrene), 6 mL of acetone, and 2 mL of water. The reaction was allowed to stir open to air for 24 h, at which point saturated NaHCO3 (15 mL) was added, and the mixture was filtered over Celite. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organics were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The product was purified by flash column chromatography (20% EtOAc/hexanes) to afford the title compound as a yellow oil (3.67 g, 12.1 mmol, dr >20:1, 70%).
Acknowledgments
This work was supported by NIH NIGMS (R01GM100212).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.oprd.5b00148.
Experimental procedures and characterization data for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Johansson-Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- a Zapf A.; Beller M. Top. Catal. 2002, 19, 101–109. 10.1023/A:1013889401432. [DOI] [Google Scholar]; b Yin J.Selected Applications of Pd- and Cu-Catalyzed Carbon-Heteroatom Cross-Coupling Reactions in the Pharmaceutical Industry. In Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, 1st ed.; Crawley M. L., Trost B. M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2005; pp 97–163. [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- a Messaoudi S.; Hamze A.; Provot O.; Tréguier B.; Rodrigo De Losada J.; Bignon J.; Liu J.-M.; Wdzieczak-Bakala J.; Thoret S.; Dubois J.; Brion J.-D.; Alami M. ChemMedChem 2011, 6, 488–497. 10.1002/cmdc.201000456. [DOI] [PubMed] [Google Scholar]; b Alami M.; Messaoudi S.; Hamze A.; Provot O.; Brion J.-D.; Liu J.-M.; Bignon J.; Bakala J.. Dihydro-iso-ca-4 and analogues: potent cytotoxics, inhibitors of tubulin polymerization. Patent US/2011/0160228 A1, Dec 10, 2009.
- Tollefson E. J.; Hanna L. E.; Jarvo E. R. Acc. Chem. Res. 2015, 48, 2344–2353. 10.1021/acs.accounts.5b00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Taylor B. L. H.; Swift E. C.; Waetzig J. D.; Jarvo E. R. J. Am. Chem. Soc. 2011, 133, 389–391. 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; b Taylor B. L. H.; Harris M. R.; Jarvo E. R. Angew. Chem., Int. Ed. 2012, 51, 7790–7793. 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]; c Yonova I. M.; Johnson A. G.; Osborne C. A.; Moore C. E.; Morrissette N. S.; Jarvo E. R. Angew. Chem., Int. Ed. 2014, 53, 2422–2427. 10.1002/anie.201308666. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tollefson E. J.; Dawson D. D.; Osborne C. A.; Jarvo E. R. J. Am. Chem. Soc. 2014, 136, 14951–14958. 10.1021/ja5076426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Harris M. R.; Hanna L. E.; Greene M. A.; Moore C. E.; Jarvo E. R. J. Am. Chem. Soc. 2013, 135, 3303–3306. 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b For a related reaction, seeZhou Q.; Srinivas H. D.; Dasgupta S.; Watson M. P. J. Am. Chem. Soc. 2013, 135, 3307–3310. 10.1021/ja312087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewska H. M.; Swift E. C.; Jarvo E. R. J. Am. Chem. Soc. 2013, 135, 9083–9090. 10.1021/ja4034999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E. J.; Kümmerle A. E.; Fraga C. A. M. Chem. Rev. 2011, 111, 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- a Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mesganaw T.; Garg N. K. Org. Process Res. Dev. 2013, 17, 29–39. 10.1021/op300236f. [DOI] [Google Scholar]; c For lead reference of stereoconvergent cross-coupling reactions using a chiral nickel catalyst, see:Liang Y.; Fu G. C. J. Am. Chem. Soc. 2014, 136, 5520–5524. 10.1021/ja501815p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Geist E.; Kirschning A.; Schmidt T. Nat. Prod. Rep. 2014, 31, 441–448. 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- Burke M. D.; Schreiber S. L. Angew. Chem., Int. Ed. 2004, 43, 46–58. 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- Dintzner M. R.; Maresh J. J.; Kinzie C. R.; Arena A. F.; Speltz T. J. Chem. Educ. 2012, 89, 265–267. 10.1021/ed200256w. [DOI] [Google Scholar]
- A 1.00 g reaction of THP 3 employing 1 mol% (rac-BINAP)NiCl2 afforded 65% yield of desired product 4 after a 3 day reaction period.
- a Haskins N. J. Biomed. Mass Spectrom. 1982, 9, 269–277. 10.1002/bms.1200090702. [DOI] [PubMed] [Google Scholar]; b Wolfe R. R.; Chinkes D. L.. Basic Characteristics of Isotopic Tracers. In Isotopic Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2005; pp 1–9. [Google Scholar]
- Pacy P. J.; Cheng K. N.; Thompson G. N.; Halliday D. Ann. Nutr. Metab. 1989, 33, 65–78. 10.1159/000177523. [DOI] [PubMed] [Google Scholar]
- Rychlik M.; Asam S. Anal. Bioanal. Chem. 2008, 390, 617–628. 10.1007/s00216-007-1717-x. [DOI] [PubMed] [Google Scholar]
- Wiltshire H. R.; Prior K. J.; Trach F.; Schlageter M.; Schönenberger H. J. Labelled Compd. Radiopharm. 1998, XLI, 1103–1126. . [DOI] [Google Scholar]
- For examples and a discussion, see reference (5).
- For preparation of air-stable Ni(II) salts, see:Standley E. A.; Smith S. J.; Müller P.; Jamison T. F. Organometallics 2014, 33, 2012–2018. 10.1021/om500156q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For example employing a combination of Ni(acac)2 and cyclooctadiene as ligand, see:Tollefson E. J.; Erickson L. W.; Jarvo E. R. J. Am. Chem. Soc. 2015, 137, 9760–9763. 10.1021/jacs.5b03870. [DOI] [PubMed] [Google Scholar]
- Lawrence B. M. J. Chromatogr. 1968, 38, 535. 10.1016/0021-9673(68)85084-8. [DOI] [PubMed] [Google Scholar]
- Li J. J.; Limberakis C. Pflum D. A.. Modern Organic Synthesis in the Laboratory; Oxford University Press: New York, NY, 2007; p 73. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.