

Abstract
Molecular modeling of receptors for adenosine and nucleotide (P2) receptors with docked ligand, based on mutagenesis, was carried out. Adenosine 3′,5′-bisphosphate derivatives act as selective P2Y1 antagonists/partial agonists. The ribose moiety was replaced with carbocyclics, smaller and larger rings, conformationally constrained rings, and acyclics, producing compounds that retained receptor affinity. Conformational constraints were built into the ribose rings of nucleoside and nucleotide ligands using the methanocarba approach, i.e. fused cyclopropane and cyclopentane rings in place of ribose, suggesting a preference for the Northern (N) conformation among ligands for P2Y1 and A1 and A3ARs.
Modulation of adenosine receptors (P1) and nucleotide (P2) receptors by selective agonists and antagonists (1,2) has the potential for the treatment of wide range of diseases, including those of the cardiovascular, inflammatory, and central nervous systems. There are four subtypes of adenosine receptors (A1, A2A, A2B, and A3), all of which are G protein-coupled receptors (GPCRs) generally coupled to adenylate cyclase. Extracellular nucleotides, principally ATP, ADP, UTP, and UDP, act through two families of membrane-bound P2 receptors: P2Y subtypes, GPCRs which are activated by both adenine and uracil nucleotides and generally coupled to phospholipase C; and P2X subtypes, ligand-gated ion channels which are activated principally by adenine nucleotides (2). As many as seven subtypes have been cloned within each family. Agonists of adenosine and P2 receptors are almost exclusively nucleosides and nucleotides, respectively, while antagonists of these receptors are structurally more diverse (1). In comparison to the adenosine receptors, much less is known about the specific effects of P2 receptors, largely due to the lack of selective ligands.
We are currently designing and synthesizing novel ligands for both adenosine and P2 receptors. Recent methods utilized in these investigations include: conformationally constraining the ribose, or ribose-like, moiety of nucleosides and nucleotides to freeze a conformation that may provide favorable affinity and/or selectivity at P1 and P2 receptors (3,4); modifying known receptor antagonists (5–7); use of a template approach based on the pyridine family for the design of novel adenosine antagonists (8); and the screening of chemical libraries in conjunction with molecular modeling (9).
RECEPTOR MODELING AND MUTAGENESIS AS TOOLS IN LIGAND DEVELOPMENT FOR GPCRs
GPCRs represent a large family of many hundreds of gene products that share common structural motif, i.e. they contain seven membrane-spanning helical domains (TMs) (10,11). A large fraction of the pharmaceutical agents currently in use consist of synthetic agents that modulate of the action of GPCRs, either as agonists or antagonists. Thus, it is of great interest to computationally explore aspects of ligand binding common to these protein targets, as an aid in the design of ligands. We have used mutagenesis and molecular modeling in conjunction with ligand modification to define putative binding sites (i.e. for small molecules, within the TM domains). Conformational considerations of both receptors and their ligands are important in structure-based drug design.
We carried out such efforts for both adenosine and P2 receptors. We have used alanine scanning mutagenesis of both human P2Y1 and A2A receptors to show the importance of specific residues in molecular recognition (12,13). Lacking, until recently (14), a high resolution crystallographic structure for any GPCR, molecular modeling by homology of the receptors was carried out using a low resolution rhodopsin template. Ligands were docked in orientations based on mutagenesis results both with purine receptors and with other GPCRs, assuming that identified were consistent with both mutagenesis results and known ligand specificities. To obtain an energetically refined 3D structure of the complex, we introduced a “cross docking” procedure (13), which was designed to simulate the reorganization of the native receptor induced by the ligand. In order to ascertain which residues of the human P2Y1 receptor were involved in ligand recognition, we have mutated both the TMs (3, 5, 6, and 7) and the ELs. A cluster of positively charged amino acids, Lys and Arg, near the exofacial side of TMs 3 and 7 and to a lesser extent TM6, putatively coordinated the phosphate moieties of nucleotide agonists and antagonists (15). Two essential disulfide bridges in the extracellular domains were identified, and several charged residues in ELs 2 (E209) and 3 (R287) have been shown to be critical for receptor activation. This suggested that the role of the ELs in ligand recognition was as important as that of the TMs. The presence of “meta-binding sites” in the P2Y1 receptor, in which the nucleotide binds to distal site(s) on its way to the principal TM binding site, were predicted using both mutagenesis and molecular modeling. These secondary binding sites may serve to guide the ligand in its approach to the TM binding site and reduce the energy barrier to complex formation.
With simplified pharmacophores, some of which are rigid and apparently at the pharmacological boundary between agonists and antagonists (see below), we are exploring the steric and electronic constraints of the receptor binding site, and the structural basis of receptor activation.
RIBOSE-MODIFIED NUCLEOSIDE AND NUCLEOTIDE ANALOGUES: P2Y1 RECEPTOR LIGANDS HAVING NON-RIGID RIBOSE-LIKE RINGS
The P2Y1 receptor is a GPCR that stimulates PLC in response to adenine nucleotides and is present in the heart, smooth muscles, prostate, ovary, brain, and platelets. For example, a selective antagonist of P2 receptors in platelets, such as the P2Y1 receptor, could be useful as an antithrombotic agent (16). A selective agonist could potentially be useful as an insulin secretagogue or hypotensive agent (17).
Initial findings by Boyer et al. (18) indicated that adenosine bisphosphates (at either 3′,5′ or 2′,5′ positions) acted as selective antagonists or partial agonists at the P2Y1 receptor. We have modified these lead compounds, resulting in a series of derivatives of 2′- or 3′-deoxyadenosine 3′,5′-bisphosphate that acted as potent and selective P2Y1 antagonists, or in some cases, depending on subtle structural changes, agonists or partial agonists (3,6,7). Figure 1 shows various types of modifications of nucleotide-based ligands in the development of P2Y1 receptor antagonists. The analogue MRS 2179 (2′-deoxy-N6-methyladenosine-3′,5′-bisphosphate), shown to be a more potent competitive antagonist, contains an intact ribose ring, but has N6-methyl and 2′-deoxy modifications, leading to higher affinity and lower agonist efficacy. MRS 2179 was also found to block rat P2X1 receptors (19). A 2′-substitution (2′-OH or ether instead of 2′-H) increased agonist efficacy over 2′-H. The 2-position of the adenine ring accommodated substitution, such as amino, Cl, or methylthio. Even longer chain thioether were shown to be of moderate affinity at the P2Y1 receptor, in parallel to findings with ATP-related agonists, in which long chain 2-thioethers enhanced potency. The N6-position appeared to occupy a small hydrophobic pocket on the receptor binding site, since substitution was limited to Me or Et (6). There was a precipitous drop in affinity upon substitution of the N6-position with Pr.
Figure 1.

Structural modifcations of the nucleoside moiety of nucleotide ligands of P2 receptors, highlighting the SAR for P2Y1 receptor antagonists. IC50 values for antagonism of the effects of 30 nM 2-MeSADP on phospholipase C at the turkey erythrocyte P2Y1 receptor are indicated (3,6,7).
Molecular modeling of ligands docked to a putative receptor binding site predicted that the ribose endocyclic oxygen of adenine nucleotides that acted as P2Y1 agonists or antagonists antagonists was not involved in specific and essential H-bonding (13). As a result, we substituted the ribose moiety in the antagonists series with a variety of cyclic (3) (carbocylics, smaller and larger rings, conformationally constrained rings) and acyclic moieties (20), without loss of affinity for the receptor.
A simple carbocyclic substitution of the ribose of MRS 2179 resulted in a high affinity agonist, displaying an IC50 value at the turkey erythrocyte P2Y1 receptor of 0.148 μM (Fig. 1) (7). Ring expansion of the ribose in the 2-Cl-N6-Me series (in the form of an anhydrohexitol derivative, MRS 2283, IC50 0.566 μM) and ring contraction (in the form of a cyclobutyl derivative, MRS 2264, IC50 0.805 μM) has further emphasized the flexibility of substitution for the ribose moiety of P2Y receptor ligands (3). An acyclic modification of the ribose ring was found to preserve affinity at the P2Y1 receptor (20). MRS 2286 (2-[2-(2-chloro-6-methylaminopurin-9-yl)-ethyl]-propane-1,3-bisoxy(diammonium-phosphate)) was an antagonist at the turkey erythrocyte P2Y1 receptor with an IC50 value of 0.84 μM, and no agonist affinity was observed. Furthermore, MRS 2286 was inactive at P2X1 receptors (19).
NUCLEOSIDES AND NUCLEOTIDES HAVING METHANOCARBA RINGS AS LIGANDS FOR ADENOSINE AND P2 RECEPTORS
Conformational constraints were incorporated into the ribose-like rings of purine nucleosides and nucleotides to probe the effects on receptor affinity and/or selectivity. In general, the ribose rings of nucleosides and nucleotides may adopt a range of conformations as described by a “pseudorotational cycle” (21). The Northern ((N), 2′-exo, 3′-endo) and Southern ((S), 2′-endo, 3′-exo) conformations are the most relevant to the biological activities observed for nucleosides and nucleotides in association with DNA, RNA, and various enzymes. Do the adenosine and P2 receptors prefer either one of these specific conformations of the ring?
In order to approach this question experimentally, we designed a series of adenosine and P2 receptor ligands containing conformationally rigid ribose-like rings, based on carbocyclic rings. The modification selected was the methanocarba approach, in which a fused cyclopropane moiety constrains the pseudosugar (cyclopentane) ring of the nucleoside to either a (N)- or (S)- conformation (Fig. 2) (21), as defined in the pseudorotational cycle. Such analogues helped to define the role of sugar puckering in stabilizing the active receptor-bound conformation, and thereby allowed identification of a favored isomer.
Figure 2.

Effects on turkey erythrocyte P2Y1 receptor-mediated phosphoinositide metabolism by methanocarba nucleotide analogues (3). The two structural variations restrict the ring pucker, i.e. hold the ribose-like ring (pseudosugar) in either Northern (N) or Southern (S) envelope conformations. PLC response is indicated on the y-axis, either in the presence (for antagonist MRS 2279, ▲, or partial agonist MRS 2266, ●) or absence (for full agonist MRS 2268, ▽, or partial agonist MRS 2266, ○) of the agonist 2-MeSADP, 30 nM.
A preference for the (N) conformation of ribose at both the P2Y1 receptor (3) and at adenosine receptors was defined using methanocarba analogues (4). MRS 2268, the (N)-methanocarba analogue of 2′-deoxyadenosine-3′,5′-bisphosphate, was a potent P2Y1 agonist (EC50 = 155 nM), 120-fold more potent than the corresponding Southern (S) isomer, MRS 2266. MRS 2268 alone elicited a full activation of PLC (Fig. 2), while MRS 2266 alone produced a partial agonist response and partially antagonized full activation of the enzyme by 2-methylthioadenosine 5′-diphosphate (2-MeSADP). However, the corresponding 2-Cl-N6-methyl-(N)-methanocarba analogue, MRS 2279 ((1R,2S,4S,5S)-1-[(phosphato)methyl]-4-(2-chloro-6-aminopurin-9-yl) bicyclo [3.1.0]-hexane-2-phosphate tetraammonium salt), fully antagonized the effects of 2-MeSADP with IC50 of 52 nM.
Adenine modifications of the (N)methanocarba analogues were consistent with the findings in the ribose series. Thus the presence of an N6-Me group in these bisphosphate analogues tended to transform a partial or full agonist into an antagonist displaying no agonist activity. This effect was hinted in the ribose series, but was more pronounced in the methanocarba series. As in the ribose series, the 2-chloro modification enhanced affinity.
The same methanocarba approach was extended to ARs (Fig. 3) (4). Methanocarba modification of the ribose moiety of adenosine analogues was a general approach for the design of A1 and A3AR agonists having favorable pharmaco-dynamic properties. While simple carbocyclic substitution of adenosine agonists greatly diminished potency, methanocarba-adenosine analogues defined the role of sugar puckering in stabilizing the active AR-bound conformation, and thereby allowed identification of a favored isomer. In binding assays at A1, A2A, and A3ARs, (N)-methanocarba-adenosine had a higher affinity than the (S)-analogue, particularly at the human A3AR ((N)-/(S)- affinity ratio of 150). (N)-Methanocarba analogues containing various N6-substituents, in which the parent compounds were potent agonists at either A1 (e.g. cyclopentyl) or A3ARs (e.g. 3-iodobenzyl), were synthesized. The N6-cyclopentyl derivatives were A1AR-selective in radioligand binding assays. For example, the 2-chloro analogue MRS 1761 (Fig. 3), although slightly less potent than the ribose analogue, CCPA, was 390-fold selective for rat A1 versus A2AARs. The N6- cyclopentyl derivatives maintained high efficacy at recombinant human but not rat A1ARs, as indicated by stimulation of binding of [35S]GTP-γ-S. The (N)-methanocarba N6-(3-iodobenzyl)adenosine (MRS 1743) and its 2-chloro derivative (MRS 1760) had Ki values of 4.1 and 2.2 nM at A3ARs, respectively, and were more selective than the ribose analogues. They proved to be partial agonists at A3ARs (Fig. 4). Partial agonism combined with high functional potency at A3ARs (EC50 <1 nM) may produce tissue selectivity. In conclusion, at least three ARs favor the ribose (N)-conformation.
Figure 3.

Structures of N6-substituted N-methanocarba adenosine derivatives optimized for interaction with A1 (N6-cyclopentyl) or A3 (N6-(3-iodobenzyl)) receptors (4). Ki values (nM) in radioligand binding assays at four subtypes of adenosine receptors are shown.
Figure 4.
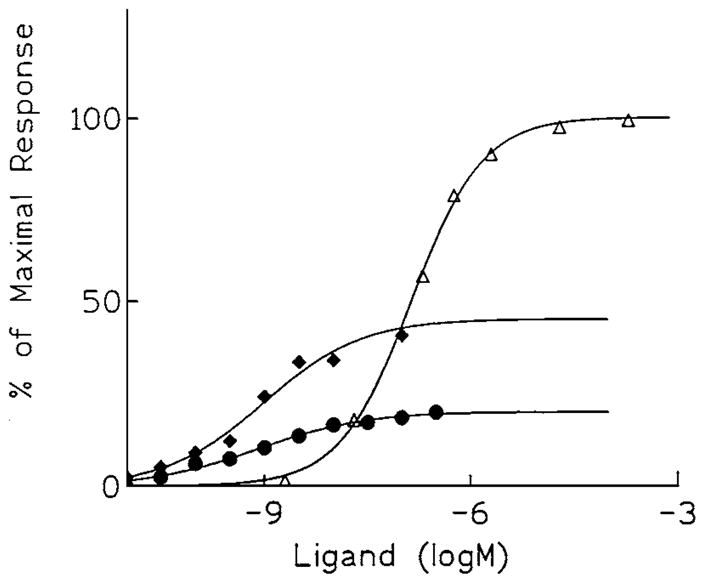
Concentration-response curves for stimulation of binding of [35S]GTP-γ-S by the full agonist 5′-N-ethylcarboxamidoadenosine (△), or two A3-selective (N)mc derivatives (4), which proved to be partial agonists (MRS 1743, ◆; MRS 1760, ●), in membranes prepared from CHO cells stably expressing human brain A3 receptors. EC50 values (nM): NECA, 155; MRS 1743, 0.70; MRS 1760, 0.67.
References
- 1.Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Trends Pharmacol Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA, Knutsen LJS. P1 and P2 purine and pyrimidine receptors. In: Abbracchio MP, Williams M, editors. Handbook of Experimental Pharmacology. 2000. in press. [Google Scholar]
- 3.Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez VE, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. J Med Chem. 2000;43:829–842. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson KA, Ji X-d, Li AH, Melman N, Siddiqi MA, Shin KJ, Marquez VE, Ravi RG. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim Y-C, Ji X-d, Melman N, Linden J, Jacobson KA. J Med Chem. 2000;43:1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. J Med Chem. 1998;41:183–190. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nandanan E, Camaioni E, Jang SY, Kim YC, Cristalli G, Herdewijn P, Secrist JA, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. J Med Chem. 1999;42:1625–1638. doi: 10.1021/jm980657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li A-H, Moro S, Melman N, Ji X-d, Jacobson KA. J Med Chem. 1998;41:3186–3201. doi: 10.1021/jm980093j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webb TR, Melman N, Lvovskiy D, Ji X-d, Jacobson KA. Bioorg Med Chem Lett. 2000;10:31–34. doi: 10.1016/s0960-894x(99)00583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Rhee AM, Jacobson KA. Drug Devel Res. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osman R, Colson A, Perlman JH, Laakonen LJ, Gershengorn MC. In: Structure-function analysis of G protein-coupled receptors. Wess J, editor. 1999. pp. 59–84. [Google Scholar]
- 12.Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA. J Med Chem. 1997;40:2588–2595. doi: 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 15.Moro S, Hoffmann C, Jacobson KA. Biochem. 1999;38:3498–3507. doi: 10.1021/bi982369v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon C, Hechler B, Freund M, Eckly A, Vial C, Ohlmann P, Dierich A, LeMeur M, Cazenave JP, Gachet C. J Clin Invest. 1999;104:1731–1737. doi: 10.1172/JCI8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer B, Chulkin A, Boyer JL, Harden TK, Gendron FP, Beaudoin AR, Chapal J, Hillaire-Buys D, Petit PJ. Med Chem. 1999;42:3636–3646. doi: 10.1021/jm990158y. [DOI] [PubMed] [Google Scholar]
- 18.Boyer JL, Romero-Avila T, Schachter JB, Harden TK. Mol Pharmacol. 1996;50:1323–1329. [PubMed] [Google Scholar]
- 19.Brown SG, King BF, Kim YC, Burnstock G, Jacobson KA. Drug Devel Res. 2000;49:253–259. doi: 10.1002/1098-2299(200004)49:4<253::AID-DDR4>3.0.CO;2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YC, Gallo-Rodriguez C, Jang SY, Nandanan E, Adams M, Harden TK, Boyer JL, Jacobson KA. J Med Chem. 2000;43:746–755. doi: 10.1021/jm9905211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]