

Abstract
A central challenge in the field of metabolic engineering is the efficient identification of a metabolic pathway genotype that maximizes specific productivity over a robust range of process conditions. Here we review current methods for optimizing specific productivity of metabolic pathways in living cells. New tools for library generation, computational analysis of pathway sequence-flux space, and high-throughput screening and selection techniques are discussed.
Keywords: Metabolic Engineering, Protein Engineering, Synthetic Biology, Deep Mutational Scanning, Computational Optimization
Introduction
Microorganisms have the potential to produce many chemicals of use to society1,2. In some cases, production from heterologous microorganisms is more sustainable than purifying the chemical from natural sources. Examples include harvesting Pacific yew trees or Chinese wormwood for taxol3 or the anti-malarial artemisinin4, respectively. Additionally, the ability to create renewable and sustainable biofuels and biochemicals is increasingly attractive given concerns about climate change and peak oil5.
An organism producing a desired product may not exist, or a given strain may not be suitable for required economical processing conditions. Because of this, reconstructed pathways are often implanted into chassis microorganisms5. Some of these pathways include those specific for biofuels (ethanol5, isobutanol6, 1-butanol7, 1,4-butanediol8), polymer monomers (polylactic acid9, isoprene10, 3-hydroxypropionic acid11) and pharmaceutically active ingredients (precursors for taxol3 or opioids12). However, in many cases product toxicity or transport limits end titers, product recovery from aqueous fermentation broths is inefficient, or the volumetric productivity is below that required for a cost-effective process. Combined, these limitations temper the promise of sustainable replacement of the palette of petrochemicals and naturally extracted specialty chemicals currently in use by society.
In particular, the specific productivities of most engineered metabolic pathways are far below that which is needed for industrial production. Some implanted pathways have limited flux because of substantial thermodynamic reversibility at key steps13. Additionally, pathway enzymes transplanted into heterologous hosts often have poor expression because of weak catalytic efficiency14, poor protein solubility, or membrane targeting issues12,15. Host-specific problems include cofactor accessibility16, siphoning of pathway intermediates, intermediate toxicity and post-translational flux regulation of key precursors12,17,18. Furthermore, the performance of a specific engineered metabolic pathway may differ between host strains19, media formulations12, temperatures and oxygen conditions20.
A grand challenge in the field of metabolic engineering is the accurate and efficient identification of a pathway genotype that maximizes specific productivity over a robust range of process conditions. Attempts to improve specific productivity have largely focused on screening individual pathway enzymes for activity or balancing gene expression by testing libraries of elements like promoters and ribosome binding sites (RBS)21–24. Error-prone polymerase chain reaction (PCR) mutagenesis of pathway enzymes has also been used to find activity-improving mutations17,18. However, pathway optimization by total enumeration becomes unwieldy, as balancing activity at multiple nodes leads to a combinatorial explosion. Consider a plasmid-encoded pathway composed of a series of expression elements (e.g. promoters, RBS, terminators) and pathway gene variants (Fig. 1a). A pathway library comprised of a single enzyme of average length25 driven by 10 alternative promoter and 10 alternative RBS sequences can be covered by testing 102 variants. A library containing the above gene expression genotypes with all possible single non-synonymous mutations to the enzyme now contains 6 × 105 variants. Testing the same number of variants using a two-enzyme pathway requires a theoretical coverage of 3.6 × 1011 variants, which is too large a sequence space to cover under most conditions. The combinatorial problem only becomes more acute with more pathway enzymes. To partially circumvent this combinatorial intractability, modular pathway design has been used to partition individual enzymatic steps into reaction groups. Then, expression of the resulting modules are balanced, not individual enzymes12,26–28 (Fig. 1b). Alternative ways to explore sequence-flux space include computational predictions from small training sets29–31 (Fig. 1c) or high-throughput screening or selection techniques32–36 (Fig. 1d).
Figure 1.
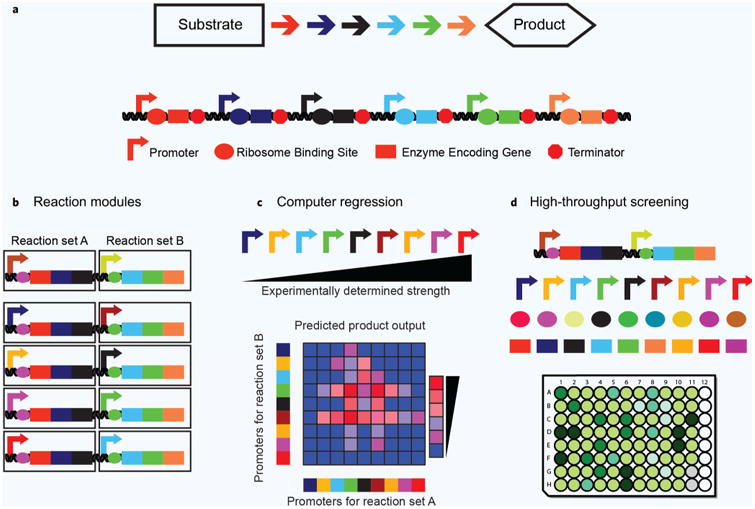
There are three main strategies to sample sequence-flux space in metabolic pathways. (a) A model pathway. Each arrow represents a different reaction chemistry used to convert an initial substrate into a value-added product. Expression elements like promoters and ribosome binding sites (RBS) facilitate expression of the enzyme-encoding gene sequences. (b) Individual reaction chemistries are grouped into modules. Expression of these modules is varied, reducing the combinatorial search space. (c) Given a training set of different sequences with a given output, models can be harnessed to predict optimal pathway expression levels. (d) Advanced DNA assembly methods can be used to create unique pathway variants that are then assessed using high-throughput screens or selections.
The focus of this review is on new technologies that identify highly productive and robust synthetic metabolic pathways. This review will not cover continuous evolution37, whole-genome engineering38 or computational pathway design39 — the interested reader can find excellent reviews on some of these topics elsewhere40–43. We begin by describing pathway evaluation of isogenic cultures. We next describe computational approaches to predict high-performing genotypes with a limited training set of pathway variants. Next, we consider high-throughput methods to assess metabolic pathways, including population-based screens or selections. New enabling techniques for DNA library construction, sequencing and evaluation will be described throughout.
Evaluation of Isogenic Cultures
One way to evaluate pathway variants is through the use of isogenic cultures. In a typical set-up, a library containing a combinatorial library of expression elements or enzyme variants is created, and clonal variants are tested individually. Lu et al. optimized a xylose fermentation pathway in Saccharomyces cerevisiae by shuffling promoters of various strength in front of each pathway enzyme44. Different promoter combinations were made and tested individually for ethanol productivity and enzymatic activity. Solomon et al. tested different expression levels of glucokinase (glk) and galactose permease (galp) to enable glycolytic uptake in Escherichia coli independent of the phosphotransferase system45. Carbon flux was modulated by controlling expression of glk and galp under control of synthetic constitutive promoters. Juminaga et al. constructed a pathway for L-tyrosine production in E. coli MG1655 by modifying plasmid copy numbers, promoter strength, gene codon usage, and placement of genes in operons46. The best pathway variant had a volumetric productivity of 55 mg L-tyrosine/L/hr, representing 80% of the theoretical yield. Ajikumar et al. optimized a pathway for overproduction of taxadiene, a key taxol precursor3. The authors used a modular approach by separating the pathway into two operons, with one containing the methylerythritolphosphate pathway and the other containing genes encoding the downstream terpenoid-producing enzymes. The promoter strength in front of each operon was systematically varied and taxadiene product measured. Notably, the taxadiene production landscape was highly non-linear in response to operon expression.
Similar isogenic approaches can be used to engineer key rate-determining enzymes or transporters in implanted metabolic pathways. Zhang et al. used site-directed mutagenesis of active site residues of the enzymes KivD and LeuA47. Fermentations of E. coli harboring pathways with different combinations of KivD/LeuA variants were tested for quantification of desired alcohol products. Leonard et al. generated combinatorial mutations in the downstream enzymes geranylgeranyl diphosphate synthase and levopimaradiene synthase to tune the selectivity and increase the productivity of levopimaradiene production in E. coli48. The best strain had a maximum volumetric productivity of 7.3 mg levopimaradiene per L per h. Lee et al. improved xylose utilization in S. cerevisiae by directed evolution of xylose isomerase49. After three rounds of error-prone PCR and screening they isolated a mutant with a 61-fold improvement in aerobic growth rate and an eight-fold improvement in ethanol production and xylose consumption. Screening pathway variants is not only limited to enzymes. Young et al. demonstrated the tunability of yeast sugar transporters through a combination of motif-based design and saturation mutagenesis50. This approach was used to identify xylose-specific fungal molecular transporters, which when expressed improved xylose utilization by S. cerevisiae.
Computational Predictions Using Empirical Training Sets
Adjusting the right balance of enzyme specific activities within a pathway is crucial as the fitness cost of protein expression51, catabolism of pathway intermediates, and off-product reactions can all lower specific productivities. While there have been many admirable attempts to forward engineering biological systems and parts22,24,52–54 and analytical equations describing pathway flux have been formulated55, tuning metabolic pathways is largely still an empirical exercise. Because of this, predictive computational models have been used to predict high productivity portions of sequence-flux space given sparse flux datasets resulting from testing isogenic cultures. Lee et al. used a linear regression model trained on empirical data to relate enzyme expression levels to product titers in a violacein biosynthetic pathway29. This simple model could accurately predict promoter combinations resulting in the production of violacein or one of the three alternative products. Another approach to computationally model and improve pathway performance is to correlate targeted proteomics and metabolite data. George et al. generated isopentenol pathway variants with differing promoters, operon organization, and codon-usage30. They then used HPLC and LC-MS to quantify glucose, organic acids, and pathway intermediates and used selected reaction monitoring mass spectrometry to quantify all proteins in their pathway. Spearman rank correlations were calculated from values of protein area and metabolite concentrations. Based on these relationships, individual variants were reconstructed and tested in time-course experiments to test model predictions. While this method may not capture complex regulatory interactions like feedback inhibition, other methods like 13C metabolic flux analysis studies are more than capable of doing so56,57. In one example, Feng et al. tested different xylose reductase, xylitol dehydrogenase and xylulose kinase variants in a yeast xylose pathway and used 13C metabolic flux analysis to determine if the different cofactor requirements of the enzyme variants had any effect on growth and/or ethanol production58. They found that production of ethanol was unaffected by the cofactor requirements of the xylose pathway. However, the cofactor balanced xylose pathway allowed growth under a wider variety of conditions. Farasat et al. developed a sequence-expression-activity mapping method to find optimal expression conditions with desired activity for a carotenoid biosynthetic pathway31. In a first step, an RBS calculator is used to make a library that spans a large range of protein expression space. Next, a subset of the library is tested for activity and used as a training set for a computational model. A new library is then constructed with targeted expression within a narrow window specified by the model. Zelcbuch et al. performed an iterative assembly of three fluorescent reporters, each with an associated RBS, into an operon59. This initial search reduced the expression search space for a balanced astaxanthin pathway. In a clever approach, they were able to haplotype the individual non-local RBS sequences included within the operon by sequencing a downstream barcode built using iterative restriction and ligation steps.
Enabling DNA Construction Methods
New genetic modification methods like DNA Assembler60, Golden Gate assembly61, Gibson cloning62, SLIC63, site-specific recombination or VEGAS assembly64 enable efficient construction of pathway variants with an array of different enzymes, promoters and RBS sequences. Smanski et al. utilized Gibson cloning62 and Golden Gate assembly61 to refactor the Klebsiella oxytoca nitrogen fixation gene cluster32 by systematically varying the expression levels of individual genes in the complete 16-gene pathway. Performance of their clusters was assessed by RNA-seq for expression levels and nitrogenase activity assays. The best of the 122 full-length pathways tested resulted in recovery of 57% of the wild-type activity. Layton and Trinh used Gibson cloning to make ester fermentative pathways in E. coli33. Their modular design of their pathway allowed quick replacement of RBS and promoter sequences. Oliver et al. improved 2,3-butanediol production in cyanobacteria by using SLIC to swap different RBS sequences in front of each pathway enzyme34. Colloms et al. used serine integrase site-specific recombination to rank gene order and RBS sites for a more efficient production of violacein and lycopene35. Du et al. used S. cerevisiae native homologous recombination to swap promoters of various expression strength in front of relevant genes65. This was used to improve xylose and cellobiose utilization pathways. Kim et al. used a similar approach to balance the flux of a xylose utilizing pathway for biofuel production36. Importantly, the optimal pathway was strongly dependent on both the host genotype but also the sugar composition of the growth medium. Latimer et al. combinatorially tested promoters of the eight-gene pathway for xylose utilization in Saccharomyces cerevisiae20. Library plasmids were made with Golden Gate assembly. Similar to results above, they found that the enrichment of specific yeast promoters in their library after selection was dependent on the number of genes expressed, the culture media conditions, and the cofactor dependence of the enzymes.
Alternative High-Throughput Screening Methods
Many of the above examples utilized medium-throughput plate-based screening or a growth based selection in order to sort variants. There have been recent developments to employ fluorescence-activated cell sorting (FACS) or microfluidic sorting technology in cases without an observable growth phenotype. For example, Wang et al. cultured xylose-consuming strains in droplets and microfluidic sorting based on the fluorescence of oxidized extracellular metabolites66. Michener et al. utilized FACS to screen improved variants of caffeine demethylase using a designed RNA biosensor67. The RNA biosensor is a combination of ribozyme and aptamer located in the 3′ UTR of a fluorescent reporter gene. When the aptamer is bound to a desired ligand, the ribozyme misfolds leading to lower RNA cleavage rates and increasing the fluorescent output. Tang et al. utilized FACS to screen for E. coli clones with enhanced triacetic acid lactone (TAL) production using a an engineered TAL fluorescent reporter68. Jha et al. used a FACS screen to identify E. coli clones with increased enzymatic production of 3,4 dihydroxy benzoate69. In these above examples, the limitation is developing a fluorescent reporter that is coupled to intracellular concentrations of a target metabolite.
Population-Based Measurements
One limitation of high-throughput screening is the inability to haplotype a unique pathway sequence to an output phenotype. Typically, only a few “winners” of the selection are sequenced. This is sub-optimal for two reasons. First, the winner variants depend strongly on the exact selection or screening conditions used, and so a selection must be repeated for each change of fermentation condition or host genotype. This limitation precludes use of multi-objective optimization (Pareto optimization) techniques into pathway redesign. Recent contributions from the Salis lab show the utility of Pareto optimization to improve translation elongation rates and mRNA stability of a single construct across multiple bacterial species70,71. Secondly, high-throughput methods do not allow coverage of a complete sequence-flux space for even moderate-length pathways, and losing crucial genotypic information of the pathway makes it impossible to use the powerful computational analyses and prediction tools that have been demonstrated for low-throughput pathways. We envision population-based measurements that can more thoroughly search sequence-flux space and also identify Pareto optimal genotypes that are robust to different processing conditions (Fig. 2).
Figure 2.
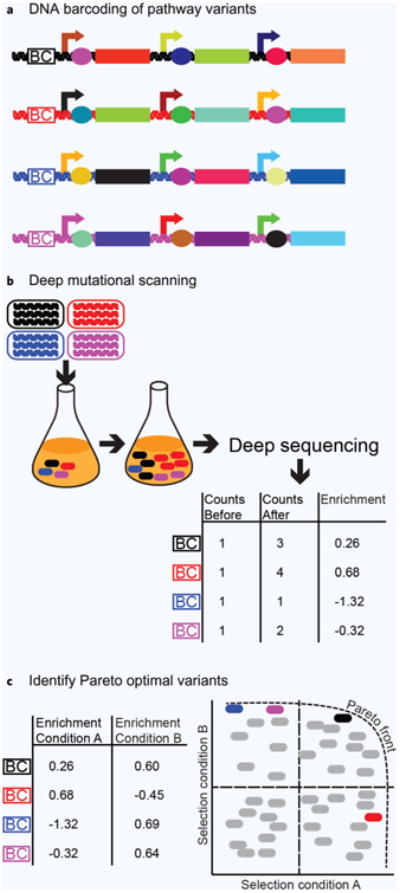
Population-based measurements of pathways enable thorough search of flux space. (a) DNA barcoding methods allow long DNA constructs to be uniquely identified by short sequences. (b) Deep mutational scanning quantifies the enrichment of individual DNA variants after a selection. The enrichment relating the change in frequency of an individual variant can be related to specific productivity. (c) Comparing the fitness of individual variants between different selection conditions allows one to find Pareto optimal pathway sequences. This enables the identification of pathways supporting high specific productivity under robust processing conditions.
Recent advances in deep sequencing technology allow the ability to track tens of thousands of pathway variants in a high throughput screen. Most of such methods rely upon “barcoding” individual cells with a short unique identifier DNA sequence (Fig. 2a). A growth selection is performed, and these populations are deep sequenced at the barcode locus. The change in frequency of an individual barcode can be related to the fitness of that unique variant72,73. While in principle such techniques could be used to track individual metabolic pathway variants, most demonstrations have been for studies on evolution. Smith et al. developed a barcode sequencing method (Bar-seq), which they validated by performing growth selections of a mixed culture containing yeast deletion strains74. The barcode abundance after selection was determined for each deletion strain by deep sequencing the entire population. More recently, Levy et al. barcoded 500,000 lineages of Saccharomyces cerevisiae and used a growth selection to track time-dependent changes in fitness among the population75. Chubiz et al. introduced FREQ-Seq, a method to barcode and determine allele frequencies from a mixed population72. FREQ-Seq was used to map seven variants of the enzyme Tet(X2), conferring tetracycline resistance, in ten different evolving populations76.
Frequency analysis of variants within a population can be used to assess if a single variant improves, reduces, or has no effect on function. This approach has been used for evaluation of yeast translation initiation sites77 and bacterial promoter strengths21 by coupling these upstream elements to fluorescent reporter proteins. Subsequently, populations are sorted by FACS. In fact, massively parallel sequence-function mapping is now commonplace in determining the sequence effects on function for proteins78. For example, this methodology has been used to improve the affinity of engineered protein binders to Influenza79. The question remains how to leverage impressive deep sequencing technology to improve implanted metabolic pathways.
In principle, high-throughput sequence-function mapping can be used to determine metabolic pathways supporting higher or lower flux, provided that it is coupled to a selectable phenotype like growth. A new approach called FluxScan was recently reported80 that maps the sequence determinants of flux in living cells (Fig. 2b). First, a selection is designed to allow growth if and only if flux is routed through the implanted pathway. A mutational library is then created and transformed into the strain of interest. After a growth selection is performed for 4–10 generations, the entire population is deep sequenced and compared with the population before selection. The frequency change of each variant can be calculated and converted to a flux value. To demonstrate this method, Klesmith et al. determined the effect of flux for over 8,000 single-point mutants in a pyrolysis oil catabolic pathway80. One designed pathway incorporating 15 beneficial mutations identified from FluxScan supported a 15-fold improvement in growth rate on levoglucosan, a chief pyrolysis oil constituent.
One significant technical challenge with FluxScan and related deep sequencing approaches is the inability to cover the complete length of metabolic pathways: current long read lengths of the Illumina platform are approximately 300 bp, whereas full operons can exceed 10 kb. One method to escape this limitation is to sequence small contiguous regions of sequence (a gene tile) able to fit on a single read. This “tiling” is then repeated along the length of the entire gene encoding sequence81. Other potential solutions to extend deep sequencing include coupling a pre-defined barcode sequence to a given pathway variant using clever DNA construction approaches32,59 or to utilize the next generation of long-read, highly accurate haplotype sequencing technologies82,83.
The ideal outcome of any experiment should be to find Pareto optimal pathway sequences that are robust and transferrable to any process condition. Single isogenic culturing conditions are not suited for this task as each pathway variant would individually have to be tested under each process condition to determine the resulting phenotype. Therefore, high-throughput population-based measurements are more capable to resolve the fitness of each sequence variant under each process condition (Fig. 2c) provided that they are performed under diverse conditions. Sequence variants from current high-throughput genomic methods that originate from different process conditions highlight this open problem. Gall et al. used the SCALEs method to map the gene expression in E. coli that conferred an advantage in the presence of 1-naphthol84. They show that genes with enhanced expression depend on the type of culturing method used. Only 25% of clones that were reproducibly enriched in serial transfer cultures were similarly enriched in single batch cultures. Similarly, Warner et al. used TRMR and found differential gene expression that was dependent on the four different growth conditions under evaluation85. While these examples highlight gene expression on the genomic scale, the problem also applies to expression and gene variants on the individual pathway level. An example of this is from the aforementioned FluxScan study where the enrichment of individual mutations from the enzyme levoglucosan kinase was strongly dependent on the biophysical properties of the starting enzyme variant from each selection80. Being able to measure the pathway phenotype from different expression elements and individual gene variants under different growth conditions should allow the elucidation of pathway sequences that are optimal over a range of diverse conditions and determine why other sequences fail when these conditions change. If implemented, these high-throughput population-based measurements can help train computational models by providing empirical data and similarly computational models can help reduce the sequence search space for a more targeted population-based screen.
Outlook
In the near future we believe that robust, high performing pathways can be efficiently identified. New DNA assembly technologies allow for construction of large libraries of pathway variants covering a large range of protein expressions and activities. The number of unique pathway variants that can be made far exceeds that which can be accurately validated using existing technology. In this review, we have covered current methods to reduce the search space.
There is no general method that can assess any metabolic pathway, as there are limitations to each of the main approaches. There are two practical limitations that must be surmounted. First, the relationship between gene expression and pathway flux is highly non-linear. Second, a specific genotype may only support high productivity in a narrow range of process conditions. We suggest that marrying computational modeling with empirical datasets resulting from population-based measurements will allow a more efficient discovery of Pareto optimal gene encoding, expression, or regulatory sequences.
Acknowledgments
The authors acknowledge support from the National Science Foundation (Awards CBET-1254238, CBET-1236120).
Footnotes
Author Contributions: J.R.K. and T.A.W. wrote the review article.
References
- 1.Keasling JD. Synthetic biology for synthetic chemistry. ACS Chem Biol. 2008;3:64–76. doi: 10.1021/cb7002434. [DOI] [PubMed] [Google Scholar]
- 2.Markham KA, Alper HS. Synthetic biology for specialty chemicals. Annu Rev Chem Biomol Eng. 2014;6:35–52. doi: 10.1146/annurev-chembioeng-061114-123303. [DOI] [PubMed] [Google Scholar]
- 3.Ajikumar PK, et al. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paddon CJ, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 5.Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Microbial engineering for the production of advanced biofuels. Nature. 2012;488:320–328. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- 6.Trinh CT, Li J, Blanch HW, Clark DS. Redesigning Escherichia coli metabolism for anaerobic production of isobutanol. Appl Environ Microbiol. 2011;77:4894–4904. doi: 10.1128/AEM.00382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atsumi S, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng. 2008;10:305–311. doi: 10.1016/j.ymben.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Yim H, et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol. 2011;7:445–452. doi: 10.1038/nchembio.580. [DOI] [PubMed] [Google Scholar]
- 9.Jung YK, Kim TY, Park SJ, Lee SY. Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng. 2010;105:161–171. doi: 10.1002/bit.22548. [DOI] [PubMed] [Google Scholar]
- 10.Lindberg P, Park S, Melis A. Engineering a platform for photosynthetic isoprene production in cyanobacteria, using Synechocystis as the model organism. Metab Eng. 2010;12:70–79. doi: 10.1016/j.ymben.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Borodina I, et al. Establishing a synthetic pathway for high-level production of 3-hydroxypropionic acid in Saccharomyces cerevisiae via β-alanine. Metab Eng. 2015;27:57–64. doi: 10.1016/j.ymben.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Thodey K, Galanie S, Smolke CD. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat Chem Biol. 2014;10:837–844. doi: 10.1038/nchembio.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bond-Watts BB, Bellerose RJ, Chang MCY. Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol. 2011;7:222–227. doi: 10.1038/nchembio.537. [DOI] [PubMed] [Google Scholar]
- 14.Milo R, Last RL. Achieving diversity in the face of constraints: Lessons from metabolism. Science. 2012;336:1663–1667. doi: 10.1126/science.1217665. [DOI] [PubMed] [Google Scholar]
- 15.Trenchard IJ, Smolke CD. Engineering strategies for the fermentative production of plant alkaloids in yeast. Metab Eng. 2015;30:96–104. doi: 10.1016/j.ymben.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avalos JL, Fink GR, Stephanopoulos G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat Biotechnol. 2013;31:335–341. doi: 10.1038/nbt.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastian S, et al. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab Eng. 2011;13:345–352. doi: 10.1016/j.ymben.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 18.DeLoache WC, et al. An enzyme-coupled biosensor enables (S)-reticuline production in yeast from glucose. Nat Chem Biol. 2015;11:465–471. doi: 10.1038/nchembio.1816. [DOI] [PubMed] [Google Scholar]
- 19.Paddon CJ, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 20.Latimer LN, et al. Employing a combinatorial expression approach to characterize xylose utilization in Saccharomyces cerevisiae. Metab Eng. 2014;25:20–29. doi: 10.1016/j.ymben.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Kosuri S, et al. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci USA. 2013;110:14024–14029. doi: 10.1073/pnas.1301301110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mutalik VK, et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- 23.Lee ME, DeLoache WC, Cervantes B, Dueber JE. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol. 2015;4:975–986. doi: 10.1021/sb500366v. [DOI] [PubMed] [Google Scholar]
- 24.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skovgaard M, Jensen LJ, Brunak S, Ussery D, Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 2001;17:425–428. doi: 10.1016/s0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- 26.Sheppard MJ, Kunjapur AM, Wenck SJ, Prather KLJ. Retro-biosynthetic screening of a modular pathway design achieves selective route for microbial synthesis of 4-methyl-pentanol. Nat Commun. 2014;5:5031. doi: 10.1038/ncomms6031. [DOI] [PubMed] [Google Scholar]
- 27.Tseng HC, Prather KLJ. Controlled biosynthesis of odd-chain fuels and chemicals via engineered modular metabolic pathways. Proc Natl Acad Sci USA. 2012;109:17925–17930. doi: 10.1073/pnas.1209002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou K, Qiao K, Edgar S, Stephanopoulos G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat Biotechnol. 2015;33:377–383. doi: 10.1038/nbt.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee ME, Aswani A, Han AS, Tomlin CJ, Dueber JE. Expression-level optimization of a multi-enzyme pathway in the absence of a high-throughput assay. Nucleic Acids Res. 2013;41:10668–10678. doi: 10.1093/nar/gkt809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.George KW, et al. Correlation analysis of targeted proteins and metabolites to assess and engineer microbial isopentenol production. Biotechnol Bioeng. 2014;111:1648–1658. doi: 10.1002/bit.25226. [DOI] [PubMed] [Google Scholar]
- 31.Farasat I, et al. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol Syst Biol. 2014;10:731. doi: 10.15252/msb.20134955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smanski MJ, et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat Biotechnol. 2014;32:1241–1249. doi: 10.1038/nbt.3063. [DOI] [PubMed] [Google Scholar]
- 33.Layton DS, Trinh CT. Engineering modular ester fermentative pathways in Escherichia coli. Metab Eng. 2014;26:77–88. doi: 10.1016/j.ymben.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Oliver JWK, Machado IMP, Yoneda H, Atsumi S. Combinatorial optimization of cyanobacterial 2,3-butanediol production. Metab Eng. 2014;22:76–82. doi: 10.1016/j.ymben.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Colloms SD, et al. Rapid metabolic pathway assembly and modification using serine integrase site-specific recombination. Nucleic Acids Res. 2014;42:e23. doi: 10.1093/nar/gkt1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim B, Du J, Eriksen DT, Zhao H. Combinatorial design of a highly efficient xylose-utilizing pathway in Saccharomyces cerevisiae for the production of cellulosic biofuels. Appl Environ Microbiol. 2013;79:931–941. doi: 10.1128/AEM.02736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeitoun RI, et al. Multiplexed tracking of combinatorial genomic mutations in engineered cell populations. Nat Biotechnol. 2015;33:631–637. doi: 10.1038/nbt.3177. [DOI] [PubMed] [Google Scholar]
- 39.Henry CS, Broadbelt LJ, Hatzimanikatis V. Discovery and analysis of novel metabolic pathways for the biosynthesis of industrial chemicals: 3-hydroxypropanoate. Biotechnol Bioeng. 2010;106:462–473. doi: 10.1002/bit.22673. [DOI] [PubMed] [Google Scholar]
- 40.Medema MH, van Raaphorst R, Takano E, Breitling R. Computational tools for the synthetic design of biochemical pathways. Nat Rev Microbiol. 2012;10:191–202. doi: 10.1038/nrmicro2717. [DOI] [PubMed] [Google Scholar]
- 41.Oberhardt MA, Palsson BØ, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5320 doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woolston BM, Edgar S, Stephanopoulos G. Metabolic engineering: Past and future. Annu Rev Chem Biomol Eng. 2013;4:259–288. doi: 10.1146/annurev-chembioeng-061312-103312. [DOI] [PubMed] [Google Scholar]
- 43.Goldsmith M, Tawfik DS. Directed enzyme evolution: Beyond the low-hanging fruit. Curr Opin Struct Biol. 2012;22:406–412. doi: 10.1016/j.sbi.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Lu C, Jeffries T. Shuffling of promoters for multiple genes to optimize xylose fermentation in an engineered Saccharomyces cerevisiae strain. Appl Environ Microbiol. 2007;73:6072–6077. doi: 10.1128/AEM.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solomon KV, Moon TS, Ma B, Sanders TM, Prather KLJ. Tuning primary metabolism for heterologous pathway productivity. ACS Synth Biol. 2013;2:126–135. doi: 10.1021/sb300055e. [DOI] [PubMed] [Google Scholar]
- 46.Juminaga D, et al. Modular engineering of L-tyrosine production in Escherichia coli. Appl Environ Microbiol. 2012;78:89–98. doi: 10.1128/AEM.06017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K, Sawaya MR, Eisenberg DS, Liao JC. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc Natl Acad Sci USA. 2008;105:20653–20658. doi: 10.1073/pnas.0807157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leonard E, et al. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc Natl Acad Sci USA. 2010;107:13654–13659. doi: 10.1073/pnas.1006138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SM, Jellison T, Alper HS. Directed evolution of xylose isomerase for improved xylose catabolism and fermentation in the yeast Saccharomyces cerevisiae. Appl Environ Microbiol. 2012;78:5708–5716. doi: 10.1128/AEM.01419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young EM, Tong A, Bui H, Spofford C, Alper HS. Rewiring yeast sugar transporter preference through modifying a conserved protein motif. Proc Natl Acad Sci USA. 2014;111:131–136. doi: 10.1073/pnas.1311970111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bienick MS, et al. The interrelationship between promoter strength, gene expression, and growth rate. PLoS ONE. 2014;9:e109105. doi: 10.1371/journal.pone.0109105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brewster RC, Jones DL, Phillips R. Tuning promoter strength through RNA polymerase binding site design in Escherichia coli. PLoS Comput Biol. 2012;8:e1002811. doi: 10.1371/journal.pcbi.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mutalik VK, et al. Quantitative estimation of activity and quality for collections of functional genetic elements. Nat Methods. 2013;10:347–353. doi: 10.1038/nmeth.2403. [DOI] [PubMed] [Google Scholar]
- 54.Guimaraes JC, Rocha M, Arkin AP, Cambray G. D-Tailor: Automated analysis and design of DNA sequences. Bioinformatics. 2014;30:1087–1094. doi: 10.1093/bioinformatics/btt742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flamholz A, Noor E, Bar-Even A, Liebermeister W, Milo R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc Natl Acad Sci USA. 2013;110:10039–10044. doi: 10.1073/pnas.1215283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McAtee AG, Jazmin LJ, Young JD. Application of isotope labeling experiments and 13C flux analysis to enable rational pathway engineering. Curr Opin Biotechnol. 2015;36:50–56. doi: 10.1016/j.copbio.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 57.Young JD. 13C metabolic flux analysis of recombinant expression hosts. Curr Opin Biotechnol. 2014;30:238–245. doi: 10.1016/j.copbio.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Feng X, Zhao H. Investigating xylose metabolism in recombinant Saccharomyces cerevisiae via 13C metabolic flux analysis. Microb Cell Fact. 2013;12:114. doi: 10.1186/1475-2859-12-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zelcbuch L, et al. Spanning high-dimensional expression space using ribosome-binding site combinatorics. Nucleic Acids Res. 2013;41:e98. doi: 10.1093/nar/gkt151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden Gate shuffling: A one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 63.Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell LA, et al. Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du J, Yuan Y, Si T, Lian J, Zhao H. Customized optimization of metabolic pathways by combinatorial transcriptional engineering. Nucleic Acids Res. 2012;40:e142. doi: 10.1093/nar/gks549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang BL, et al. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat Biotechnol. 2014;32:473–478. doi: 10.1038/nbt.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michener JK, Smolke CD. High-throughput enzyme evolution in Saccharomyces cerevisiae using a synthetic RNA switch. Metab Eng. 2012;14:306–316. doi: 10.1016/j.ymben.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 68.Tang SY, et al. Screening for enhanced triacetic acid lactone production by recombinant Escherichia coli expressing a designed triacetic acid lactone reporter. J Am Chem Soc. 2013;135:10099–10103. doi: 10.1021/ja402654z. [DOI] [PubMed] [Google Scholar]
- 69.Jha RK, Kern TL, Fox DT, Strauss CEM. Engineering an Acinetobacter regulon for biosensing and high-throughput enzyme screening in E. coli via flow cytometry. Nucleic Acids Res. 2014;42:8150–8160. doi: 10.1093/nar/gku444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kushwaha M, Salis HM. A portable expression resource for engineering cross-species genetic circuits and pathways. Nat Commun. 2015;6 doi: 10.1038/ncomms8832. Article No.: 7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng CY, Farasat I, Maranas CD, Salis HM. Rational design of a synthetic Entner– Doudoroff pathway for improved and controllable NADPH regeneration. Metab Eng. 2015;29:86–96. doi: 10.1016/j.ymben.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 72.Chubiz LM, Lee MC, Delaney NF, Marx CJ. FREQ-Seq: A rapid, cost-effective, sequencing-based method to determine allele frequencies directly from mixed populations. PLoS ONE. 2012;7:e47959. doi: 10.1371/journal.pone.0047959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J. Massively parallel single-amino-acid mutagenesis. Nat Methods. 2015;12:203–206. doi: 10.1038/nmeth.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith AM, et al. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009;19:1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levy SF, et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 2015;519:181–186. doi: 10.1038/nature14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walkiewicz K, et al. Small changes in enzyme function can lead to surprisingly large fitness effects during adaptive evolution of antibiotic resistance. Proc Natl Acad Sci USA. 2012;109:21408–21413. doi: 10.1073/pnas.1209335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noderer WL, et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Mol Syst Biol. 2014;10:748. doi: 10.15252/msb.20145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fowler DM, Fields S. Deep mutational scanning: A new style of protein science. Nat Meth. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whitehead TA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klesmith JR, Bacik JP, Michalczyk R, Whitehead TA. Comprehensivesequence-flux mapping of a levoglucosan utilization pathway in E. coli. ACS Synth Biol. 2015;4:1235–1243. doi: 10.1021/acssynbio.5b00131. [DOI] [PubMed] [Google Scholar]
- 81.Kowalsky CA, et al. High-resolution sequence-function mapping of full-length proteins. PLoS ONE. 2015;10:e0118193. doi: 10.1371/journal.pone.0118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stapleton JA, et al. Haplotype-phased synthetic long reads from short-read sequencing. bioRxiv. 2015 doi: 10.1371/journal.pone.0147229. doi: http://dx.doi.org/10.1101/022897. [DOI] [PMC free article] [PubMed]
- 83.Hong L, et al. BAsE-Seq: A method for obtaining long viral haplotypes from short sequence reads. Genome Biol. 2014;15:517. doi: 10.1186/s13059-014-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gall S, Lynch MD, Sandoval NR, Gill RT. Parallel mapping of genotypes to phenotypes contributing to overall biological fitness. Metab Eng. 2008;10:382–393. doi: 10.1016/j.ymben.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 85.Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LBA, Gill RT. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol. 2010;28:856–862. doi: 10.1038/nbt.1653. [DOI] [PubMed] [Google Scholar]