

Abstract
Cdc27 is a core component of the anaphase-promoting complex/cyclosome (APC/C), a multi-subunit E3 ubiquitin ligase whose oscillatory activity is responsible for the metaphase-to-anaphase transition and mitotic exit. Here, in normal murine mammary gland epithelial cells (NMuMG), Cdc27 expression is controlled post-transcriptionally through the RNA binding protein heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1/PCBP1). shRNA-mediated knockdown of hnRNP E1 abrogates translational silencing of the Cdc27 transcript resulting in constitutive expression of Cdc27. Dysregulated expression of Cdc27 leads to premature activation of the G2/M-APC/C-Cdc20 complex resulting in the aberrant degradation of Cdh1/Fzr1, a co-factor of the G1 and late G2/M-APC/C and a substrate normally reserved for the SCF-βTRCP ligase. Loss of Cdh1 expression and of APC/C-Cdh1 activity, upon constitutive expression of Cdc27, results in mitotic aberrations and aneuploidy in NMuMG cells. Further, tissue microarray (TMA) of breast cancer patient tumor samples reveals high Cdc27 levels compared to non-neoplastic breast tissue and a significant correlation between disease recurrence and Cdc27 expression. These results suggest that dysregulation of hnRNP E1-mediated translational regulation of Cdc27 leads to chromosomal instability and aneuploidy, and that Cdc27 expression represents a significant predictor of breast cancer recurrence.
Implication Statement
The RNA binding protein hnRNP E1 mediates translational regulation of the cell cycle regulator Cdc27 and that dysregulation of Cdc27 leads to aneuploidy. In addition, high Cdc27 expression in breast cancer patient tumor specimens significantly predicts disease recurrence, suggesting a novel role for Cdc27 as a predictor of relapse.
Keywords: hnRNP E1 (heterogeneous ribonucleoprotein E1), PAK1 (p21-activated kinase 1), CDC27 (cell division cycle 27), APC/C (anaphase-promoting complex/cyclosome), Fzr1 (Fizzy-related homolog 1)
Introduction
Chromosomal instability has long been recognized as a hallmark of cancer. Cancer progresses as cells override the intrinsic system of checks and balances that normally prevents them from dividing in the presence of a damaged or aneuploid genome. Chromosomal instability is described as increased chromosome missegregation and often results in aneuploidy, or the condition of having too many or too few chromosomes. Under normal physiological conditions, cell cycle traverse is carefully controlled by sequential post-translational modifications, especially E3 ligase-mediated ubiquitination (1,2). The anaphase-promoting complex/cyclosome (APC/C) is a major E3 ligase complex that promotes the metaphase-to-anaphase transition, and its activation is inhibited until surveillance mechanisms within the cell sense proper metaphase alignment and bipolar spindle attachment of chromosomes (2,3). Activation of the APC/C occurs via interaction with a co-factor that confers specificity to the complex, either Cdh1/Fzr1 during G1 phase or Cdc20 during mitosis (4-6). Premature activation of the mitotic APC/C-Cdc20 leads to missegregation of chromosomes and significantly contributes to chromosomal instability (7-9). During the process of metastasis, transformed cells exploit chromosomal instability to increase their adaptability in changing microenvironments (10,11). The increased chromosomal instability also results in phenotypically heterogeneous tumors that are difficult to stratify and thus difficult to completely eradicate, as some subpopulations of cells develop drug resistance (10,11). It is therefore important to understand the mechanisms that drive chromosomal instability.
Intensive transcriptional array analyses have attempted to delineate disease-specific gene signatures in an effort to more accurately stratify and treat patients. However, these transcriptional profiles are limited because the transcriptome does not mirror the proteome and a large number of post-transcriptional regulatory processes escape these interrogations. We have previously described a transcript-selective translational regulatory pathway in which a ribonucleoprotein (mRNP) complex, consisting of heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1) and eukaryotic elongation factor 1A1 (eEF1A1), binds to 3′UTRs and silences translation of mRNA transcripts that mediate epithelial-to-mesenchymal transition (12-14). Activation of Akt2 leads to the phosphorylation of hnRNP E1, inducing the release of the mRNP complex from 3′-UTR element, resulting in the reversal of translational silencing and increased protein expression (12-14). In addition, others have shown that p21-activated kinase 1 (Pak1) phosphorylates hnRNP E1 in response to mitogenic stimulation (15) and that hypoxia-induced Akt phosphorylation of hnRNP E1 results in destabilization of eNOS mRNA through microRNA-mediated degradation (16). Previously, we have used polysomal profiling coupled with Affymetrix array analysis to identify mRNAs that interact with hnRNP E1 in the context of translationally regulated TGFβ-mediated EMT (12); however, we also observed that a large number of mRNAs interact with hnRNP E1 in a TGFβ-independent manner, and that most of these mRNA-regulated transcripts have been implicated in many cellular processes shown to be dysregulated during tumorigenesis. Amongst the transcripts identified are cell cycle, metabolic, cytoskeletal, adhesion, cytokines and receptors, and EMT-type mRNAs (12). Here, we investigate hnRNP E1 in relation to its role in cell cycle progression and protection of chromosomal stability by regulating translation of cell division cycle 27 (Cdc27) mRNA. Cdc27, a core component of the APC/C E3 ubiquitin ligase, interacts directly with co-activators Cdh1 or Cdc20 during G1 phase and mitosis, respectively (17). The APC/C is a multi-subunit cullin-RING E3 ubiquitin ligase that regulates metaphase-to-anaphase progression and controls S phase entry by catalyzing ubiquitination of cell cycle regulatory proteins, especially the cyclins. APC/C substrate specificity is controlled through recognition of short destruction motifs, the D-box and KEN-box, by the APC/C cofactors Cdh1 and Cdc20 (17). Cdc27, along with Cdc16 and Cdc23, contain tetratricopeptide repeat motifs and function to provide scaffolding support and facilitate to APC/C-substrate interactions (18). We show that loss of cell cycle dependent hnRNP E1-mediated translational regulation of Cdc27 leads to premature activation of the G2/M-APC/C-Cdc20 and its aberrant targeting and degradation of Cdh1, a substrate normally targeted by the SCF-βTRCP ligase. Loss of cell cycle dependent Cdh1 expression and of APC/C-Cdh1 activity, results in mitotic aberrations and aneuploidy in cells, suggesting that hnRNP E1-mediated translational regulation of Cdc27 is involved in genomic and chromosomal stability.
Materials and Methods
Antibodies and reagents
Rabbit α-APC3 (12530), α-phospho-Pak1 T423 (2601), α-phospho-RXXS*/T* (6950), α-Cdc20 (14866), α-cyclin B1 (4135) α-Myc tag (2278), and α-alpha-Tubulin Alexa Fluor 488 conjugate (5063) were obtained from Cell Signaling Technology. Mouse α-Hsp90 (sc-101494), α-GAPDH (sc-166574), and goat α-NCAM-L1 (sc-1508) were obtained from Santa Cruz Biotechnology. Mouse α-hnRNP E1 (H00005093-M01) was obtained from Abnova. Mouse α-Plk1 (ab17056), α-securin (ab3305), α-Fzr1 (ab3242), and α-Cdc27 (ab10538) were obtained from Abcam. Mouse α-Flag M2 antibody (F1804) was obtained from Sigma Aldrich. HRP-conjugated goat α-mouse (31430), rabbit α-goat (31402), goat α-rabbit (31460), and Alexa Fluor 647-conjugated chicken α-goat secondary antibodies were obtained from Thermo Scientific.
Plasmids
pSilencer-U6-puro was obtained from Life Technologies and pSilencer-shhnRNP E1 was generated in the laboratory. pLX304-V5-Cdc27 was obtained from DNASU (Biodesign Institute at Arizona State University; Tempe, AZ). pGEX-6P-Cdh1wt (570) was a gift from Jonathon Pines (Addgene plasmid # 39877).
Lentivirus production and cell infection
pLX304-V5-Cdc27 lentiviral particles were produced by transfecting HEK293T cells with lentiviral packaging vectors psPAX2 and pMD2.G (obtained from Medical University of South Carolina shRNA Shared Technology Resource) along with pLX304-V5-Cdc27 or pLX304-V5-empty vector. Lentivirus-containing culture medium was collected 24 and 48 h post-transfection and added to NMuMG cells with 8 μg/ml hexadimethrene bromide. Cells were selected in 1μg/ml blasticidin and stable clones were selected. Expression of V5-tagged Cdc27 was confirmed by α-V5 tag immunoblot.
Cell culture and synchronization
NMuMG cells were obtained from ATCC (Manassas, VA, USA) and used within ten passages in culture. E1KD cells were generated in the laboratory by shRNA-mediated silencing of hnRNP E1 in NMuMG cells (NMuMG cells obtained from Manassas, VA, USA; previously called SH14) and have been described (14). NMuMG-Cdc27 cells are a stable clone generated in the laboratory by lentiviral transduction of pLX304-V5-Cdc27 plasmid into NMuMG cells. Cells were maintained at 37°C, 5% CO2, and 95% humidity in DMEM supplemented with 10% FBS, 10 mg/ml insulin, antibiotics/antimycotics (100 units/ml penicillin G, 100 mg/ml streptomycin, and 0.25 mg/ml amphotericin B). Cells were synchronized at G0 by overnight culture in low serum-containing medium (0.5% FBS), the re-stimulated to enter the cell cycle by addition of 10% FBS. Cells were synchronized at G1/S using double-thymidine block: cells were cultured overnight in complete medium containing 2 mM thymidine, washed three times in sterile 1× PBS, released for 13 hours in complete medium, re-blocked with 2 mM thymidine for 16 hours, washed three times in sterile 1× PBS, then isolated immediately or released into S phase by culturing in complete medium. Cells were synchronized in mitosis by thymidine-nocodazole block: cells were cultured overnight in complete medium containing 2 mM thymidine, washed three times in sterile 1× PBS, then released directly into complete medium containing 40 ng/ml nocodazole.
Preparation of cell extracts
Whole cell extracts were prepared by lysing cells in RIPA lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) as previously described (14). Protein quantitation was performed by Bradford assay using known concentrations of BSA as a standard.
qRT-PCR
Total RNA was isolated from cells using Trizol reagent (Invitrogen) as per manufacturer's instructions. RT was performed using M-MLV RT kit (Sigma Aldrich) according to manufacturer's instructions using 2 μg total RNA. PCR was performed using Maxima HotStart DNA polymerase master mix (Thermo Scientific), gene specific primers (Supplementary Table S2), and 2 μl cDNA reaction. Data presented as qRT-PCR were quantified using Bio-Rad Image Lab software.
Immunoprecipitation assays
HnRNP E1 immunoprecipitations for both downstream RNA isolation/RT-PCR and immunoblotting were performed as described previously (12), using α-PCBP1 antibody (Abnova; H00005093M01) and Protein A Sepharose (GE Healthcare Life Sciences). Immunoprecipitated RNA was isolated from beads by addition of Trizol, followed by RT-PCR as described above. Immunoprecipitated proteins were isolated from beads by addition of Laemmli sample buffer, followed by SDS-PAGE resolution and immunoblotting.
In vitro ubiquitination assay
In vitro ubiquitination assay was performed as described previously (7), using APC/C immunopurified from mitotic- or G1-synchronized NMuMG, E1KD, or NMuMG-Cdc27 cells using α-Cdc16 antibody (sc-365636; Santa Cruz Biotechnology) and Protein G Sepharose (GE Healthcare Life Sciences). Immunopurified APC/C complexes were incubated with 200 nM recombinant substrate (myc-Cyclin B1 residues 1-102 or full-length GST-Cdh1), 1X ubiquitination reaction buffer (10 mM Tris-Cl pH 7.6, 5 mM MgCl2, 10 mM DTT, 1 mg/ml human recombinant ubiquitin, 10 mM phosphocreatine, 50 μg/ml creatine phosphokinase, 15 mM dATP, 0.15 mg/ml BSA), 250 nM UBE1 (ubiquitin activating enzyme) (Boston Biochem; Cambridge, MA, USA), 1 μM His-UbcH10 (ubiquitin conjugating enzyme), 1 μM His-Ube2S (ubiquitin conjugating enzyme), 1 mg/ml creatine phosphokinase (Sigma-Aldrich; St. Louis, MO, USA), to a volume of 10 μl with water. Reactions were incubated at 37°C for 1 hour with shaking at 1000 rotations per minute. To stop reactions, 5 μl Laemmli sample buffer was added and reactions were vortexed. Myc-Cyclin B1, His-UbcH10, and His-Ube2S were generous gifts of Dr. A. Tipton (Oklahoma Medical Research Foundation; Oklahoma City, OK, USA).
Tissue microarray
Assembly and immunohistochemistry staining of human breast tissue microarrays (TMAs) were performed by the Biorepository and Tissue Analysis core at MUSC. TMAs consisted of matched normal and tumor tissue as well as normal and metastatic lymph node biopsies from patients for whom survival data were available (Supplementary Table S3). For immunohistochemistry staining of formalin-fixed, paraffin-embedded TMAs, sections were deparaffinized in xylene, rehydrated in alcohol and processed as follows: sections were incubated with target retrieval solution (S2367; Dako) in a steamer for 45 min followed by 3% hydrogen peroxide solution for 10 min and protein block (X0909; Dako) for 20 min at room temperature. Sections were incubated overnight in a humidity chamber at 4°C with α-Cdc27 antibody [AF3.1] (ab10538; Abcam) or α-Fzr1 (ab3242; Abcam) followed by biotinylated secondary antibody (PK6106; Vector laboratories) for 30 min and ABC reagent for 30 min. Immunocomplexes of horseradish peroxidase were visualized by DAB reaction (K3468; Dako), and sections were counterstained with hematoxylin before mounting. Images of stained sections were taken using an Olympus DP72 8-bit RGB camera with Cellsens acquisition software.
Statistical analyses
Statistical comparisons between groups were performed using the Student's t test. Data were presented as mean ± s.e.m. All experiments were independently repeated in triplicate. Statistical analysis was performed by two-tailed Student's t-test of pair-wise comparisons. Kaplan–Meier survival curves were analyzed using MedCalc Statistical Software (MedCalc Software, Ostend, Belgium) with significance determined by Logrank test. P < 0.05 were considered statistically significant. Quantitation of RT-PCR and immunoblot data was performed using Bio-Rad ChemiDoc MP Imaging System and Bio-Rad Image Lab Software.
Results and Discussion
HnRNP E1 regulates translation of mRNAs involved in several biological processes
We previously employed polyribosomal profiling coupled to Affymetrix gene array to identify mRNAs translationally regulated by TGFβ and hnRNP E1 (12). In order to determine whether hnRNP E1-regulated mRNAs cluster to functionally related groups independent of TGFβ, DAVID functional annotation analysis was performed. DAVID analysis (19,20) allows for the categorization of genes within a group based on enrichment of biological pathways, and we found that nucleic acid metabolism and cell cycle pathways were significantly enriched (Fig. 1A). Because loss of cell cycle regulation is recognized as a critical step in cancer progression, we focused our attention on this group of genes. A heatmap of the Affymetrix array data showing relative expression of genes within the cell cycle pathways is shown in Figure 1B. Within the group of hnRNP E1-regulate cell cycle genes (Fig. 1B), Cdc27 was of particular interest because of its identity as a core component of the APC/C, a E3 ubiquitin ligase that plays an integral role in promoting proper division of genetic material in dividing cells, thus in maintaining chromosomal integrity. To validate that Cdc27 expression is regulated translationally by hnRNP E1, immunoblot and RT-PCR analyses were performed using whole cell extracts and total RNA, respectively, isolated form NMuMG and NMuMG cells in which hnRNP E1 is stably silenced by shRNA-mediated knockdown (E1KD cells). As shown (Fig. 1C), where there is little change in the mRNA levels of Cdc27 between the two cell types, Cdc27 protein is significantly increased in E1KD cells, suggesting that the observed increase in expression occurs post-transcriptionally. To validate the findings of the Affymetrix array, polysome profiling of NMuMG and E1KD cells was performed, and we found that Cdc27 mRNA sediments primarily to the monosomal fractions in NMuMG cells whereas in E1KD cells Cdc27 mRNA is associated primarily with the actively translating polysomal fractions (Fig. 1D). To account for changes in global translation, we compared polysome profile sedimentation of housekeeping gene GAPDH, and results indicate that global translation did not increase in E1KDs (Fig. 1D). These results were consistent in the human mammary luminal epithelial (HMLE) cell line, in that Cdc27 mRNA expression remained unchanged between HMLE and HMLE-E1KD cells, while an increase in Cdc27 protein was observed (Fig. 1E). Cdc27 mRNA sediments exclusively to the polysome fractions in HMLE-E1KD cells compared to HMLE cells, further suggesting that translational regulation of Cdc27 occurs in normal human mammary epithelial cells (Fig. 1F).
Figure 1. HnRNP E1 regulates translation of mRNAs involved in many biological processes.
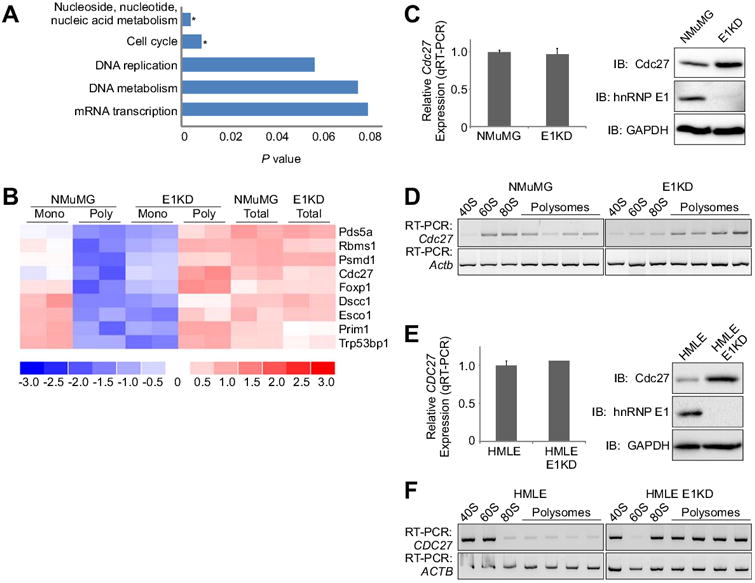
(A) DAVID analysis of translationally regulated genes identified in Affymetrix array of monosomal NMuMG genes and polysomal E1KD genes, with results given as P values (Student's t Test, fold change > 1.5 between groups and < 1.5 within groups; * P < 0.5 were considered statistically significant). Genes were annotated using Panther biological processes. (B) Heat map showing differential signal strength intensities for cell cycle genes between NMuMG and E1KD monosomes and polysomes compared to total, unfractionated mRNA. (C) Left Total RNA isolated from NMuMG and E1KD cells were subjected to qPCR analysis to assess steady state Cdc27 mRNA expression levels normalized to Actb (control). Data are presented as means ± s.e.m., n=3. Right Immunoblot analysis examining protein expression levels of Cdc27, hnRNP E1, and GAPDH (control) in whole cell lysates isolated from asynchronous NMuMG and E1KD cells. (D) Semi-quantitative RT-PCR using gene specific primers to Cdc27 (30 cycles) and Actb (21 cycles; control) on RNA isolated from polysome profiles of asynchronous NMuMG and E1KD cells. (E) Total RNA isolated from HMLE and HMLE-E1KD cells were subjected to qPCR analysis to assess steady state Cdc27 mRNA expression levels normalized to Actb (control). Data are presented as means ± s.e.m., n=3. Right Immunoblot analysis examining protein expression levels of Cdc27, hnRNP E1, and GAPDH (control) in whole cell lysates isolated from asynchronous HMLE and HMLE-E1KD cells. (F) Semi-quantitative RT-PCR using gene specific primers to Cdc27 (32 cycles) and Actb (23 cycles; control) on RNA isolated from polysome profiles of asynchronous HMLE and HMLE-E1KD cells.
HnRNP E1 regulates Cdc27 expression at the translational level in a cell cycle-dependent manner
Since Cdc27 is a component of a cell cycle-regulatory complex, we hypothesized that its expression might be regulated in a cell cycle-dependent manner. Indeed, we found that as G0-synchronized cells are stimulated to re-enter the cell cycle, Cdc27 protein expression is increased in a cell cycle-dependent manner with maximal expression observed between 18-24 hours post stimulation, corresponding to late S and G2/M phases (Fig. 2A). This increased expression of Cdc27 protein is due to translational induction as there was no significant change in total Cdc27 mRNA levels (Fig. 2B). In synchronized E1KD cells, however, Cdc27 expression is constitutively high throughout the cell cycle (Fig. 2A) with no observable change in total mRNA (Fig. 2B), suggesting that hnRNP E1 is involved in post-transcriptional regulation of Cdc27 expression. To determine the efficacy of the synchronization method, progression through the cell cycle was assessed by flow cytometric analyses of propidium iodide staining of total cellular DNA in synchronized NMuMG and E1KD cells (Fig. 2C). Progression through the cell cycle can be visualized by the shift in the population from G1 through S phase. Reconstitution of wild-type hnRNP E1 in E1KD cells (termed KIWT for knock-in wild-type) resulted in a rescue of the cell cycle-dependent regulation of Cdc27 expression (Fig. S1). Polysome profile analyses of G0-synchronized cells show that Cdc27 mRNA shifts from the monosome fractions to the polysome fractions upon mitogenic stimulation in NMuMG cells, while sedimenting constitutively to the polysome fractions in E1KDs, irrespective of cell cycle phase (Fig. 2D). Global changes in mRNA expression were controlled for by comparison of GAPDH mRNA sedimentation in G0-synchronized (0h) and released (18h) cells (Fig. 2D). Further, as analyzed by RNA-immunoprecipitation, interaction between hnRNP E1 and Cdc27 mRNA, as observed in G0-synchronized cells, is attenuated upon mitogenic stimulation (Fig. 2E). As a positive control, interactions between hnRNP E1 and L1CAM mRNA, which has previously shown to be translationally regulated in a cell cycle-dependent manner by hnRNP E1 (15), are also lost upon mitogenic stimulation of G0-synchronized cells (Fig. 2E). Based on our previous results (13,14), the dissociation of hnRNP E1 from 3′-UTRs is mediated through its phosphorylation suggesting that as G0-synchronized cells progress through the cell cycle, kinase activation results in the phosphorylation and dissociation of hnRNP E1 from the Cdc27 mRNA.
Figure 2. HnRNP E1 regulates Cdc27 expression at the translational level in a cell cycle-dependent manner.
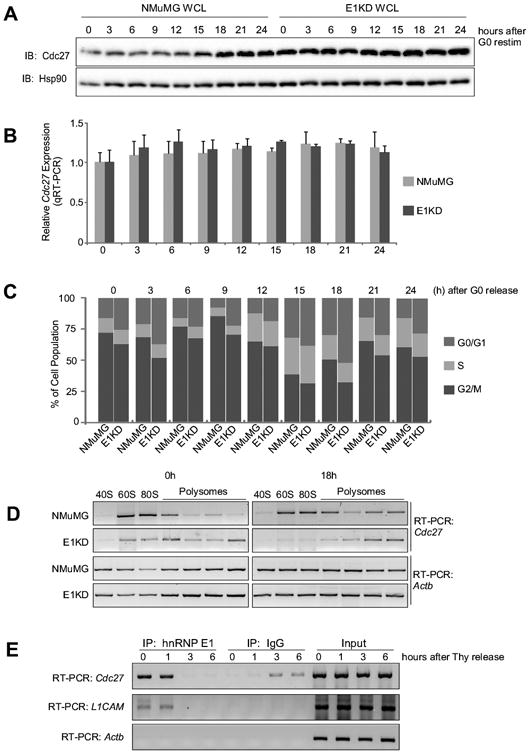
(A) Immunoblot analysis of protein expression of Cdc27 and Hsp90 (control) in NMuMG and E1KD cells that were synchronized by overnight culturing in DMEM + 0.5% FBS, then released into complete medium. (B) Total RNA isolated from NMuMG and E1KD cells released from G0 arrest at the indicated times were subjected to qPCR analysis to assess steady state Cdc27 mRNA expression levels normalized to Actb (control). Data are presented as means ± s.e.m., n=3. (C) Bar graph representing percent cell cycle phase distribution of synchronized populations of NMuMG and E1KD cells by flow cytometric analysis of propidium iodide staining. Cells were synchronized as described in A above. Cells were gated from debris, then gated again on singlets to obtain analyses of populations of single cells. (D) Semi-quantitative RT-PCR analysis using gene specific primers for Cdc27 (30 cycles) and Actb (21 cycles; control) using RNA isolated from NMuMG and E1KD polysome profiling of G0-synchronized (0 h) and released (18 h) cells. (E) Semi-quantitative RT-PCR analysis using gene specific primers for Cdc27 (30 cycles), L1CAM (34 cycles), and Actb (20 cycles) using RNA isolated from α-hnRNP E1 immunoprecipitates. HnRNP E1 was immunopurified from extracts isolated from NMuMG cells that had been synchronized by double thymidine block, washed, and released into complete medium for times indicated in figure.
Pak1 phosphorylates hnRNP E1 in a cell cycle-dependent manner allowing for Cdc27 translation
To identify the kinase mediating the dissociation of hnRNP E1 from the Cdc27 transcript, we first interrogated Akt2 since we have previously shown that TGFβ-induced activation of Akt2 is responsible for serine 43 phosphorylation of hnRNP E1 (13,14). Our data, however, demonstrate that neither TGFβ stimulation (data not shown), nor use of the PI3K inhibitor LY294002 to block Akt activation, had any significant effect on expression of Cdc27 (Fig. 3A). Since the Pak1 kinase has also been shown to phosphorylate hnRNP E1 (15), we investigated its potential role in translational regulation of Cdc27. Treatment of NMuMG cells with the Pak1-specific inhibitor IPA3 led to attenuation of Cdc27 expression (Fig. 3B), suggesting that activation of Pak1 is necessary for Cdc27 expression. In E1KD cells, neither LY294002 (Fig. 3A) nor IPA3 (Fig. 3B) had any effects on Cdc27 expression levels. As shown (Fig. 3C), immunoprecipitation of hnRNP E1 from G1/S-synchronized NMuMG cells followed by immunoblotting for phospho-RXXS*/T* reveals mitogen-induced phosphorylation of hnRNP E1, except in the presence of the Pak1-specific inhibitor, IPA-3. This phospho-specific antibody recognizes phosphorylated serine or threonine three residues downstream from arginine and is capable of recognizing either the predicted Akt2 or Pak1 phosphorylation sites on hnRNP E1. In addition, Cdc27 expression is induced in a manner that temporally coincides with maximal (3 hours post-stimulation) hnRNP E1 phosphorylation (Fig. 3B; upper panel) and activation (i.e. phosphorylation) of Pak1 (Fig 3B; middle panel). The kinetics of hnRNP E1 phosphorylation, and subsequent Cdc27 expression, support the earlier finding that Cdc27 expression is regulated post-transcriptionally. In E1KD cells (Fig. 3B), Cdc27 is not sensitive to the inhibitory effects of IPA-3, even though activated Pak1 is efficiently inhibited. These results suggest that Pak1 is responsible for hnRNP E1 phosphorylation in response to mitogenic stimulation and that activated Pak1 is necessary for Cdc27 expression in the presence of hnRNP E1. Further support for a role of Pak1 in translational regulation of Cdc27 is provided by the RNA-immunoprecipitation analysis (Fig. 3D) demonstrating that the dissociation of hnRNP E1 from the Cdc27 mRNA, observed at 3h post-stimulation of G1/S-synchronized cells, is lost in the presence of the Pak1 inhibitor. This suggests that by preventing Pak1-mediated hnRNP E1 phosphorylation, the interaction between hnRNP E1 and Cdc27 mRNA is stabilized.
Figure 3. Pak1 phosphorylates hnRNP E1 in a cell cycle-dependent manner, allowing for Cdc27 translation.
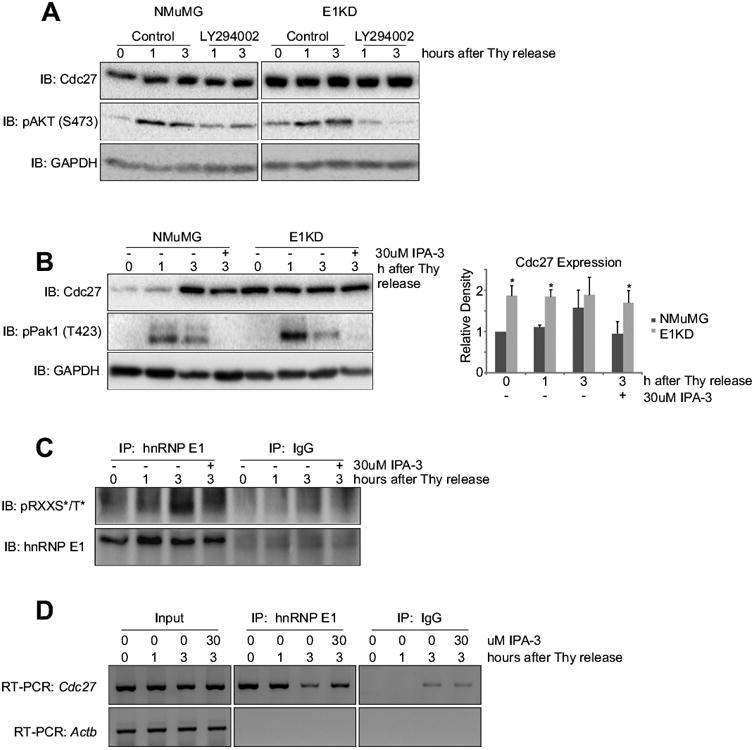
(A) Immunoblot analysis of protein expression in whole cell lysates isolated from NMuMG and E1KD cells synchronized and released ± 10 μM LY294002 for indicated times. (B) Left Immunoblot analysis of protein expression using whole cell lysates isolated from NMuMG and E1KD cells synchronized and released ± 30 μM IPA-3 for indicated times. Right Graph illustrating relative density of bands observed in Cdc27 immunoblot compared with 0h NMuMG sample (n = 3; *P < 0.05) (C) Immunoblot analysis of phosphorylated-RXXS*/T* from α-hnRNP E1 or α-IgG immunoprecipitates isolated from NMuMG cells synchronized by double-thymidine block, then released ± 30 μM IPA-3. PVDF membrane was stripped and re-probed using α-hnRNP E1 antibody. (D) Semi-quantitative RT-PCR analysis using gene specific primers for Cdc27 and Actb (control) using RNA isolated from α-hnRNP E1 immunoprecipitates. HnRNP E1 was immunopurified from NMuMG cells synchronized at G1/S using double-thymidine block and release ± 30 μM IPA-3.
Loss of APC/C-Cdh1 function and Cdh1 expression in hnRNP E1-silenced E1KD cells
Since Cdc27 is a core component of the APC/C, we were interested in assessing the activity of the APC/C in E1KD cells where Cdc27 expression is constitutively high. Immunoblot analysis of APC/C substrates reveals stabilization of polo-like kinase 1 (Plk1) and a decrease in levels of securin and cyclin B in E1KD compared to NMuMG cells (Fig. 4A). Plk1 is a substrate of the APC/C-Cdh1 while cyclin B and securin are substrates of APC/C-Cdc20, suggesting that APC/C-Cdh1 is inactive in E1KD cells, while the APC/C-Cdc20 complex appears to be constitutively active. Since silencing hnRNP E1 expression led to modulated levels of APC/C substrates, we investigated the expression of APC/C co-factors responsible for its activation. Immunoblot analysis indicates that expression of Cdc20 is constitutively high throughout the cell cycle, in contrast to the almost complete loss of Cdh1 expression in E1KD cells (Fig. 4B). This result is consistent with the observation that APC/C-Cdh1 substrates (i.e. Plk1) are stabilized and APC/C-Cdc20 substrates (cyclin B and securin) are lost in E1KD cells.
Figure 4. APC/C-Cdh1 function is lost in E1KDs, as well as expression of Cdh1.
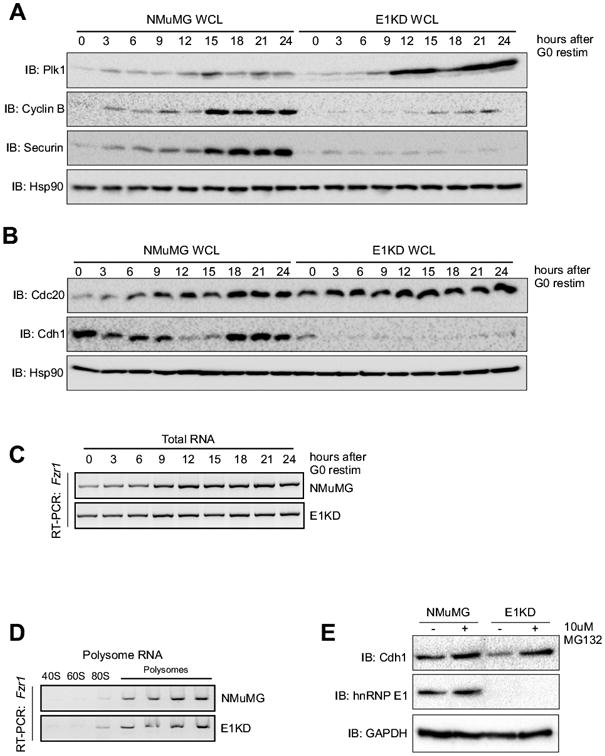
(A) Immunoblot analysis of protein expression of Plk1, cyclin B1, securin, and Hsp90 (control) using whole cell lysates isolated from G0-synchronized and released NMuMG and E1KD cells (B) Immunoblot analysis of protein expression of Cdc20, Cdh1, and Hsp90 (control) using whole cell lysates isolated from G0-synchronized and released NMuMG and E1KD cells. (C) Semi-quantitative RT-PCR analysis using gene specific primers for Fzr1 on total RNA isolated from NMuMG and E1KD cells synchronized as described in A above. (D) Semi-quantitative RT-PCR analysis with gene specific primers for Fzr1 using RNA isolated from polysome profiling of NMuMG and E1KD cytosolic extracts. (E) Immunoblot analysis of protein expression in whole cell extracts prepared form NMuMG and E1KD cells treated ± 10 μM MG132 for 1 hour using α-Cdh1, α-hnRNP E1, and α-GAPDH (control) antibodies.
We chose to further investigate the mechanism underlying the loss in Cdh1 expression, as it is poorly understood. RT-PCR analysis using total RNA from NMuMG and E1KD cells and gene specific primers for Cdh1 (gene name: Fzr1) revealed that the observed loss of Cdh1 protein was not a result of a decrease in Fzr1 mRNA levels (Fig. 4C). Polysome profile analysis was also performed, and the results indicate that Fzr1 mRNA is actively being translated in both NMuMG and E1KD cells (Fig. 4D), despite not being observed via immunoblot in E1KD cells. To determine whether proteasomal degradation was causing the decreased levels of Cdh1, E1KD cells were treated with the 26S proteasome inhibitor, MG132. The results (Fig. 4E) demonstrate that Cdh1 expression is rescued in E1KD cells upon treatment with MG132, indicating that while the Fzr1 gene is being transcribed and translated into Cdh1 protein, upon hnRNP E1 loss it is being rapidly degraded either directly or indirectly through the proteasome.
Cdc27 redirects the APC/C to degrade Cdh1
Cdh1 is normally targeted for degradation during S phase by the SCF/βTRCP ubiquitin ligase complex, as shown in Figure 4B (middle panel) where at 12-15 h post-stimulation of G0-synchronized NMuMG cells, Cdh1 expression is attenuated. However, E1KD cells have low levels of Cdh1 and exhibit aberrant activation of APC/C-Cdc20 activity. To determine whether the APC/C-Cdc20 complex in E1KD cells might be responsible for the observed decreased levels of Cdh1, we employed the allosteric inhibitor of APC/C activity, proTAME (21,22). As shown, (Fig. 5A) in both mouse (E1KD) and human mammary epithelial cells (HMLE E1KD) silenced for hnRNP E1, proTAME rescues Cdh1 expression, suggesting a role for the APC/C in targeting Cdh1. In order to address whether constitutive expression of Cdc27 plays a role in aberrant APC/C-Cdc20 activation and Cdh1 degradation observed in E1KDs, we established a cell line stably expressing Cdc27, termed NMuMG-Cdc27. As shown by immunoblot analysis (Fig. 5B), overexpression of Cdc27 in NMuMG cells leads to a concomitant loss of Cdh1 expression levels, similar to that observed in the E1KD cells. We suggest a role for the APC/C complex in mediating degradation of Cdh1 by in vitro ubiquitination assay using APC/C immunopurified from G1- or mitotic-synchronized NMuMG and E1KD cells and either purified recombinant myc-tagged Cyclin B (Fig. 5C; top panel) or GST-Cdh1 (Fig. 5C; bottom panel) as substrate. Immunoblot analysis (Fig. 5B; top panel) revealed that mitotic-APC/C isolated from NMuMG cells ubiquitinates recombinant myc-tagged cyclin B in vitro more efficiently than G1-APC/C (Fig. 5C; top panel). This result suggests that the mitotic-APC/C immunopurified from NMuMG cells is associated with Cdc20 and the G1-APC/C is associated with Cdh1. Both mitotic- and G1-APC/C complexes isolated from E1KD and NMuMG-Cdc27 cells are able to ubiquitinate myc-tagged cyclin B in vitro with similar efficiencies (Fig. 5C; top panel), suggesting that the APC/C is dysregulated in E1KDs and NMuMG-Cdc27 cells. Simultaneously, we investigated the ability of the APC/C to ubiquitinate recombinant GST-Cdh1 in vitro. Interestingly, we find that the APC/C immunopurified from G1- and mitotic-synchronized NMuMG, E1KD, and NMuMG-Cdc27 cells is capable of ubiquitinating GST-Cdh1 in vitro (Fig. 5C; bottom panel). Others have shown previously that APC/C-Cdh1 can auto-ubiquitinate Cdh1 (23), however APC/C-Cdc20-mediated ubiquitination of Cdh1 has not been previously described. In Figure 5D, we propose a model wherein hnRNP E1 represses translation of Cdc27 mRNA until Pak1-mediated phosphorylation relieves translational silencing, resulting in increased Cdc27 expression. Aberrant mitogenic signaling, as often occurs in tumorigenesis, could result in constitutive phosphorylation of hnRNP E1 and high levels of Cdc27 expression, dysregulation of APC/C activity, and loss of Cdh1 expression.
Figure 5. Cdc27 redirects Cdh1 to alternative degradation by the APC/C.
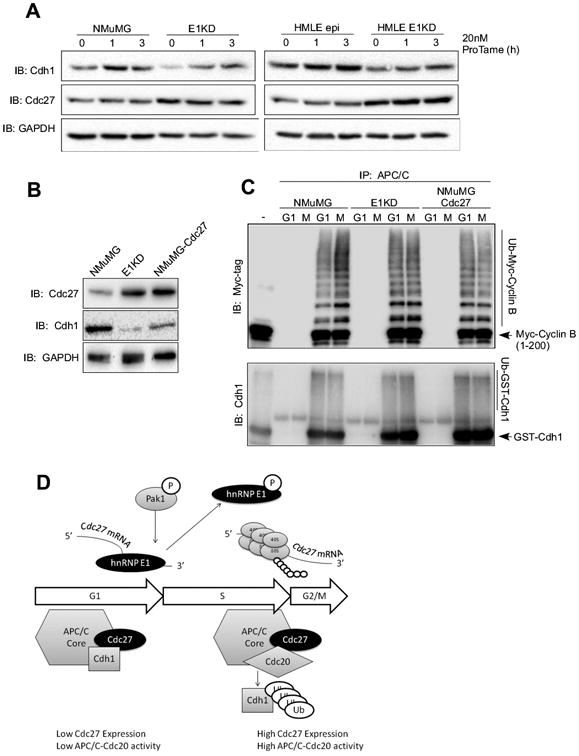
(A) Immunoblot analysis of protein expression using α-Cdh1, α-Cdc27, and α-GAPDH (control) antibodies in whole cell extracts isolated from NMuMG, E1KD, HMLEEpi and HMLEEpi-E1KD cells treated with the APC/C-specific inhibitor, proTAME, at 20 nM for the times indicated in the figure. (B) Immunoblot analysis of protein expression using α-Cdc27, α-Cdh1, and α-GAPDH (control) antibodies in whole cell extracts isolated from NMuMG, E1KD, and NMuMG-Cdc27 cells overexpressing V5-tagged Cdc27 open reading frame. (C) In vitro ubiquitination of recombinant myc-tagged cyclin B1 (top) and GST-tagged Cdh1 (bottom) using APC/C immunopurified from NMuMG, E1KD, and NMuMG-Cdc27 cells synchronized using double-thymidine (G1) and thymidine-nocodazole (M) synchronization. Immunoblot was performed using α-Myc and α-Cdh1 antibodies, respectively. (D) Schematic of proposed mechanism for Pak1/hnRNP E1-mediated regulation of Cdc27 expression during the cell cycle.
Constitutive Cdc27 expression results in mitotic aberrations and aneuploidy
In order to determine the biological impact of aberrant APC/C activity and decreased Cdh1 expression, we performed immunofluorescence confocal microscopy of NMuMG, E1KD, and NMuMG-Cdc27 cells, staining for hnRNP E1, α-tubulin, and chromosomal DNA using DAPI (Fig. 6A). We observed mitotic abnormalities such as metaphase plates with misaligned chromosomes, multipolar spindles, and nondisjunction in E1KD and NMuMG-Cdc27 cells that were absent in the NMuMG control cells (Fig. 6A). In an effort to quantify the effects of abnormal mitoses and due to the relatively low percentage of mitotic cells in each cell line, we chose to perform metaphase spread analyses. A significant proportion (P<0.05) of E1KD and NMuMG-Cdc27 cells contained aneuploid genomes compared to NMuMG controls (Fig. 6B). Taken together, these results suggest that increased Cdc27 expression correlates with improper division of chromosomes and thus results in aneuploidy. We predict that increased Cdc27 expression could be an adaptive mechanism for cancer cells, allowing those cells acquiring favorable phenotypes to survive and ultimately result in disease recurrence.
Figure 6. . Constitutive Cdc27 expression results in mitotic aberrations and aneuploidy in NMuMG and E1KD cells.
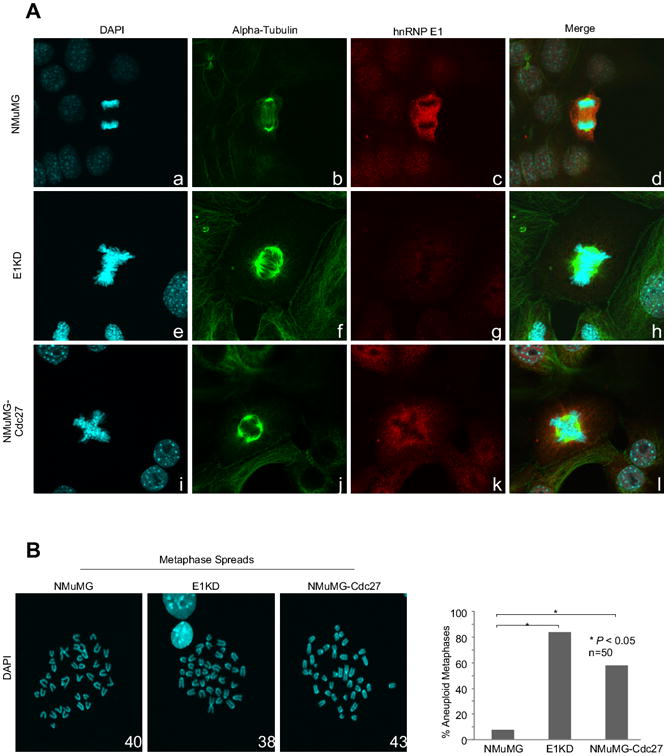
(A) Confocal immunofluorescent microscopy using α-alpha Tubulin, α-hnRNP E1, and DAPI on NMuMG, E1KD, and NMuMG-Cdc27 cells was performed to visualize mitoses. (B) Metaphase spread analyses were performed and percentages of aneuploid metaphases compared to total are represented. Chromosomes were stained with DAPI and counted using ImageJ Cell Count Plugin and 50 metaphases were counted per cell line. (*P<0.05, n=50)
Cdc27 expression is significantly higher in breast cancer patient tumor samples
To correlate our findings with human breast cancer data, we assessed Cdc27 protein expression by immunohistochemistry in human breast tissue microarrays (TMAs) (Fig. 7). Cdc27 expression was significantly higher in tumor tissue compared with normal breast tissue (Fig. 7A). Furthermore, when tumor biopsies were scored and divided into groups based on Cdc27 expression levels, a significant correlation between disease recurrence and Cdc27 expression was observed, with high Cdc27 expression associated with relapse (Figure 7B, Supplementary Table S3; P = 0.046). To investigate the relationship between Cdc27 and Cdh1 expression in human breast cancer, we assessed Cdh1 protein expression by immunohistochemistry using the same human breast TMAs (Fig. 7C). No significant correlation between Cdh1 and Cdc27 was discovered; however, low expression of Cdh1 was observed in 83% of metastatic lymph node biopsies (Supplementary Table S4), while low Cdh1 expression was not observed in the primary tumor when compared to normal tissue. This data suggests that loss of Cdh1 may play a more significant role in later stages of cancer progression. It is important to note that although these data are preliminary due to the small sample size, they are consistent with studies reporting a correlation between low Cdh1 expression and poor prognosis in breast cancer (24).
Figure 7. Cdc27 expression is significantly higher in breast cancer patient tumor samples compared to normal tissue, and high Cdc27 expression predicts disease recurrence.
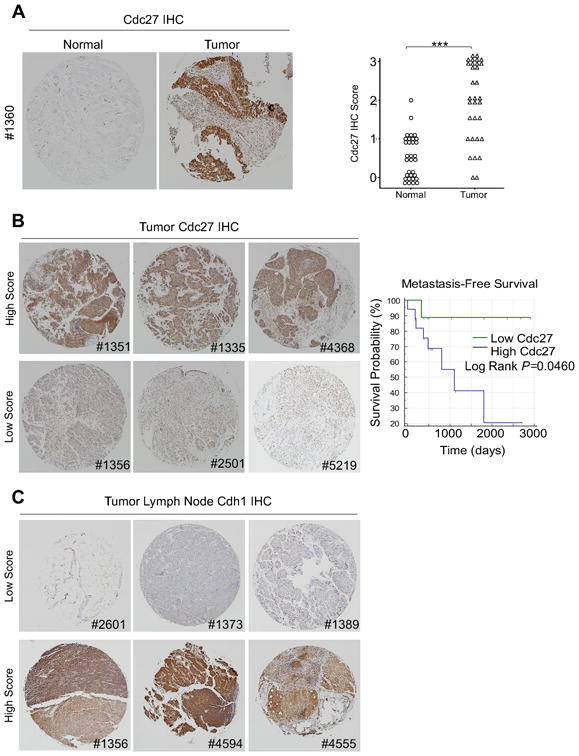
(A) Left Representative images of Cdc27 immunohistochemistry (IHC) performed on normal and tumor breast TMAs. Right Graph showing quantitation of Cdc27 staining in normal and tumor breast tissue biopsies. The intensity and proportion of Cdc27 staining was scored on a scale of 0 (low/negative staining) to 3 (intense staining). Independent scoring by a second investigator was blinded to sample type and clinicopathological data (***P<0.001). (B) Left Representative images of breast tumor specimens which were divided into high Cdc27 scoring biopsies (score = 2 or 3) or low scoring biopsies (score = 0 or 1). Right Kaplan–Meier survival analysis of low and high scoring biopsies (right panel; log rank P = 0.023). (C) Representative images of malignant lymph node specimens which were divided into low Cdh1 scoring biopsies (score = 0 or 1) or high scoring biopsies (score = 2 or 3). All images are × 40 magnifications and images were stitched using Microsoft ICE Image Composite software.
In summary, we have shown that Cdc27 is translationally repressed by the RNA-binding protein hnRNP E1 in a cell cycle-dependent manner. Following mitogenic stimulation, the interaction between Cdc27 mRNA and hnRNP E1 is abolished as a result of Pak1-mediated phosphorylation of hnRNPE1, coinciding with an increase in Cdc27 translation. Constitutive Cdc27 expression, upon hnRNP E1 loss, leads to dysregulation of the APC/C, which in turn aberrantly targets Cdh1 for degradation. We show that overexpression of Cdc27 drives chromosomal instability and induces aneuploidy in normal mouse mammary epithelial cells, and that Cdc27 is overexpressed in a small sample group of breast cancer tissue microarray samples. Further experiments are necessary to delineate the mechanism by which irregular Cdc27 expression induces aberrant APC/C activity. Others have reported an inverse relationship between APC/C-Cdc20 substrate securin and Cdc27 (25), which is consistent with our findings. However, in contrast to our findings, the aforementioned study (25) and another (26) conclude that low Cdc27 correlates with poor survival in breast cancer. While our study did not find a significant correlation between survival and Cdc27 expression, this may be due to the relatively small sample size in the tissue microarray analysis. We predict that aberrant expression of Cdc27, whether up- or down-regulated, will result in improper APC/C activity and contribute to aneuploidy. Future studies will aim to characterize the contribution of Cdc27 expression levels to APC/C activity. These data emphasize the importance of proper regulation of Cdc27 expression in maintaining chromosomal integrity and appropriate cell cycle progression.
Supplementary Material
Acknowledgments
This work was supported by Grants CA055536 and CA154663 from the National Cancer Institute. BVH was supported by funding from the Abney Foundation. GSH was supported by an American Heart Association Pre-doctoral Fellowship 10PRE3870024. This study used the services of the MUSC Center for Oral Health Research (COHR), which is partially supported by the National Institute of General Medicine grant P30GM103331, the MUSC Flow Cytometry Facility which is supported by P30GM103342, the Cell & Molecular Imaging Shared Resource of MUSC supported by P30CA138313, the Biorepository & Tissue Analysis Shared Resource, Hollings Cancer Center, MUSC and the Gene Targeting and Knockout Shared Resource at MUSC.
Note: This study was supported by research grants awarded to P.H. Howe by the National Cancer Institute NIH (grants NIH-CA 555536 and NIH-CA 154663).
Footnotes
Conflict of interest: No potential conflicts of interest were disclosed by authors.
Author contributions: Conception and design: L.A. Link, P.H. Howe
Development of methodology: L.A. Link, B.V. Howley, G.S. Hussey, and P.H. Howe
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): L.A. Link
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): L.A. Link, B.V. Howley
Writing, review, and/or revision of the manuscript: L.A. Link, B.V. Howley, G.S. Hussey, P.H. Howe
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): L.A. Link, B.V. Howley
Study supervision: P.H. Howe.
References
- 1.Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):2776–8. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nature reviews Cancer. 2006;6(5):369–81. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 3.Penas C, Ramachandran V, Ayad NG. The APC/C Ubiquitin Ligase: From Cell Biology to Tumorigenesis. Front Oncol. 2011;1:60. doi: 10.3389/fonc.2011.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278(5337):460–3. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- 5.Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Molecular biology of the cell. 2000;11(5):1555–69. doi: 10.1091/mbc.11.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/C(Cdc20), and APC/C(Cdh1) play distinct roles in connecting mitosis to S phase. The Journal of cell biology. 2013;201(7):1013–26. doi: 10.1083/jcb.201211019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tipton AR, Wang K, Link L, Bellizzi JJ, Huang H, Yen T, et al. BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. The Journal of biological chemistry. 2011;286(24):21173–9. doi: 10.1074/jbc.M111.238543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. The Journal of cell biology. 2001;154(5):925–36. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Listovsky T, Sale JE. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. The Journal of cell biology. 2013;203(1):87–100. doi: 10.1083/jcb.201302060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32(3-4):325–40. doi: 10.1007/s10555-013-9427-7. [DOI] [PubMed] [Google Scholar]
- 11.Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih Ie M, Vogelstein B, et al. The role of chromosomal instability in tumor initiation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):16226–31. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussey GS, Link LA, Brown AS, Howley BV, Chaudhury A, Howe PH. Establishment of a TGFbeta-induced post-transcriptional EMT gene signature. PloS one. 2012;7(12):e52624. doi: 10.1371/journal.pone.0052624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussey GS, Chaudhury A, Dawson AE, Lindner DJ, Knudsen CR, Wilce MC, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Molecular cell. 2011;41(4):419–31. doi: 10.1016/j.molcel.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nature cell biology. 2010;12(3):286–93. doi: 10.1038/ncb2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng Q, Rayala SK, Gururaj AE, Talukder AH, O'Malley BW, Kumar R. Signaling-dependent and coordinated regulation of transcription, splicing, and translation resides in a single coregulator, PCBP1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(14):5866–71. doi: 10.1073/pnas.0701065104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho JJ, Robb GB, Tai SC, Turgeon PJ, Mawji IA, Man HS, et al. Active stabilization of human endothelial nitric oxide synthase mRNA by hnRNP E1 protects against antisense RNA and microRNAs. Molecular and cellular biology. 2013;33(10):2029–46. doi: 10.1128/MCB.01257-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature. 2015;522(7557):450–4. doi: 10.1038/nature14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passmore LA, McCormack EA, Au SW, Paul A, Willison KR, Harper JW, et al. Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. The EMBO journal. 2003;22(4):786–96. doi: 10.1093/emboj/cdg084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4(5):P3. [PubMed] [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Zeng X, King RW. An APC/C inhibitor stabilizes cyclin B1 by prematurely terminating ubiquitination. Nat Chem Biol. 2012;8(4):383–92. doi: 10.1038/nchembio.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell. 2010;18(4):382–95. doi: 10.1016/j.ccr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benmaamar R, Pagano M. Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle. 2005;4(9):1230–2. doi: 10.4161/cc.4.9.2048. [DOI] [PubMed] [Google Scholar]
- 24.Fujita T, Liu W, Doihara H, Date H, Wan Y. Dissection of the APCCdh1-Skp2 cascade in breast cancer. Clin Cancer Res. 2008;14(7):1966–75. doi: 10.1158/1078-0432.CCR-07-1585. [DOI] [PubMed] [Google Scholar]
- 25.Talvinen K, Karra H, Pitkanen R, Ahonen I, Nykanen M, Lintunen M, et al. Low cdc27 and high securin expression predict short survival for breast cancer patients. APMIS. 2013;121(10):945–53. doi: 10.1111/apm.12110. [DOI] [PubMed] [Google Scholar]
- 26.Lee YJ, Lee HJ, Lee JS, Jeoung D, Kang CM, Bae S, et al. A novel function for HSF1-induced mitotic exit failure and genomic instability through direct interaction between HSF1 and Cdc20. Oncogene. 2008;27(21):2999–3009. doi: 10.1038/sj.onc.1210966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.