

Abstract
Background & Aims
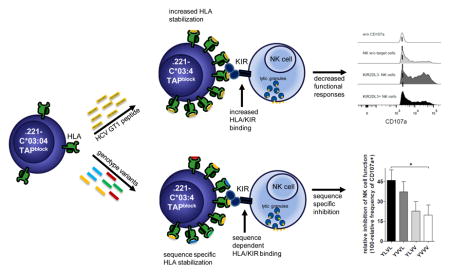
Both NK cells and HLA/KIR interactions have been shown to play an important role in the control, clearance and progression of HCV disease. Here we aimed at elucidating the effects of viral peptides derived from HCV on HLA stabilization, changes in KIR binding and primary NK cell function.
Methods
TAP-deficient 722.221 cells stably transfected with HLA-C*03:04 were used to screen 200 overlapping peptides, covering the NS3 and core protein of HCV genotype 1, for their ability to bind and stabilize HLA-C*03:04. Binding of KIR2DL3 to the HLA-peptide complex was assessed using a KIR2DL3-IgG fusion construct. Primary NK cells were isolated from healthy donors to investigate the effects of identified peptides on KIR2DL3+ NK cell function.
Results
Thirty-one peptides able to stabilize HLA-C*03:04 were identified. One 9mer peptide YIPLVGAPL resulted in significant higher KIR2DL3 binding to HLA-C*03:04+ 722.221 cells and suppression of primary KIR2DL3+ NK cell function. Interestingly this sequence exhibited a high frequency of mutations in different HCV genotypes. These genotype-specific peptides showed lower HLA-C*03:04 stabilization, decreased binding of the inhibitory KIR2DL3 and lower inhibition of NK cell function.
Conclusion
Taken together we show that a viral peptide derived from the core protein of HCV genotype 1 binding to HLA-C*03:04 results in a sequence-dependent engagement of the inhibitory NK cell receptor KIR2DL3, while the large majority of the remaining 30 HLA-C*03:04 binding HCV core peptides did not. These data show that sequence variations within HCV can modulate NK cell function, providing potential pathways for viral escape.
Keywords: HCV, KIR, HLA, NK
Graphical Abstract
Introduction
Natural Killer (NK) cells play a pivotal role in the early defense against virus infections and malignancies [1, 2], as they are part of the innate arm of the human immune system and able to respond rapidly against encountered pathogens without prior need of sensitization. The main NK cell function is the elimination of target cells by directed release of perforin and granzyme or engagement of apoptosis-inducing receptors such as FAS[2]. NK cells can furthermore shape the following antigen-specific immune responses through the production of cytokines and chemokines or by interacting directly with other cells of the immune system, such as T cells, dendritic cells and monocytes [3, 4].
Due to the potent nature of their effector functions, the activation of NK cells is tightly regulated via a plethora of activating and inhibiting receptors and their interaction with respective ligands[5, 6]. One major group of receptors on NK cells are the killer cell immunoglobulin like receptors (KIRs), which interact mainly with HLA class I molecules on the surfaces of other cells[7, 8]. Both activating and inhibiting KIRs have been described. The binding of specific KIRs to their respective HLA ligand can be furthermore modulated by the HLA class I-presented peptide [9]. These interactions have been shown to play an important role in regulating the NK cell response against the human immunodeficiency virus (HIV), and also enable HIV-1 to escape NK cell recognition through the selection of viral sequence variations [10].
In the case of hepatitis virus infections, NK cells play an important role in the clearance of infection and disease progression[1]. A large number of studies have highlighted the importance of NK cells in viral hepatitis, focusing mainly on patients infected with HBV and HCV[11]. KIR-HLA interactions seem of particular importance in HCV infection as suggested by genetic association studies which linked HLA-C genotypes in conjunction with KIR2DL3 to viral clearance [12, 13]. A more recent study further expanded on this, with the finding that KIR2DS3 is negatively associated with viral clearance [14]. The exact mechanism underlying these associations is not fully understood, but probably depends on the strength of the KIR/HLA interaction, which in case of KIR2DL3 and HLA-C alleles of the group 1 (HLA-C1) is rather weak, compared to other inhibition events[12]. HLA-C*03:04 is a common member of the HLA-C1 group, and is expressed at varying frequencies, ranging from 1% in Saudi Arabian populations all the way to 54% in Brazilians (allelfrequencies.net). In Caucasian populations in Germany the range is between 14–21%. It is a ligand for the above mentioned KIR2DL3 and the described role in HIV infection [15] might indicate further involvement in other viral infections.
While numerous HCV-derived CD4+ and CD8+ T cell epitopes have been described, no data are available on the role of HCV peptides presented by HLA class I molecules for KIR binding and KIR+ NK cell function. Our aim was therefore to investigate whether viral peptides derived from HCV core have an effect on the function of primary KIR+ NK cells.
Material and Methods
Cell lines and human peripheral blood mononuclear cells
We used a previously described 721.221 human B-cell line, which has been stably transduced with ICP47, a HSV protein that blocks TAP-dependent loading of MHC class I molecules, and also transfected with HLA-C*03:04. Additionally we used the same cell line without ICP47 TAP-block. These cells were kept in RPMI medium 1640, supplemented with 10% heat-inactivated fetal calf serum, streptomycin and penicillin (R10). A TAP-KO cell line based on the 721.221-C*03:04 cells was generated (221-C*03:04-TAP1-KO) using CRISPR/CAS technology [15](plasmids were supplied by addgene). Cells were selected in R10 supplemented with Puromycin, Blasticidine and Neomycin. Human primary blood mononuclear cells (PBMC) were isolated from healthy donors, using density centrifugation and either used directly or cryopreserved in liquid nitrogen until use. All donors gave informed consent.
HLA stabilization and KIR binding assay
HLA stabilization and KIR binding assays were performed as previously described [16]. In short, cells were washed with FBS free RPMI 1640 (R0) to remove any remaining foreign peptides from culturing in R10. Afterwards 721.221-ICP47-C*03:04, or 221-C*03:04-TAP1-KO cells were pulsed with 100μM of the respective HCV peptide for 20h at 26°C. Previously described peptides that stabilized HLA-C*03:04 expression GAVDPLLAL (GAL) and GAVDPLLKL (GKL) [17], were used as positive controls, whereas culturing in the absence of peptides was used as negative control. After peptide-pulsing, cells were stained using an anti-HLA-ABC antibody (clone W6/32), fixed in 4% paraformaldehyde and analyzed using flow cytometry. KIR binding was analyzed after 20h of HLA stabilization, as described above, with the respective peptides of interest. Cells were stained using KIR2DL3-Fc chimera (R&D), for 1h on ice, secondary staining with anti-human FC antibody was performed for 30min on ice. Afterwards cells were fixed and analyzed using flow cytometry.
NK cell degranulation assay
Primary NK cells were isolated from PBMCs of healthy donors, using Ficoll-Hypaque centrifugation, and rested overnight in R10 supplemented with 1ng/ml IL-15. NK cells were co-incubated with peptide-pulsed 721.221-ICP47-C*03:04 cells at an effector to target (E:T) ratio of 5:1 in the presence of 3μL anti-CD107a in 96well plates. After 1h incubation at 37°C, GolgiPlug (BD) was added followed by 5 additional hours of incubation at 26°C. Cells were subsequently stained using anti-CD3, anti-CD16, anti-CD56 and anti-KIR2DL3 for 30mins at 4°C, fixed using 4% paraformaldehyde in PBS for 30mins and analyzed by flow cytometry. The gating strategy to identify responses in KIR2DL3+ and KIR2DL3- cells is shown in Supplementary Fig. 1.
Data acquisition, analysis and statistics
Flow cytometry was performed on a BD LSRFortessa and BD Canto II (BD Bioscience) and analyzed using FlowJo software v10 (Tree Star, Inc.). Figures were designed and statistical analysis done using GraphPad Prism 5 (GraphPad Software, Inc.). All values in bar graphs represent mean+SEM unless stated otherwise. Association of KIR2DL3 with HCV genotype was performed in GraphPad using the latest metadata [18] and data on KIR distribution in different ethnicities, kindly provided by Mary Carrington.
Results
Several peptides derived from the core and NS3 protein of HCV bind to HLA-C*03:04
The goal of this study was to investigate the effect of HCV-core and NS3-derived epitopes presented by HLA-C*03:04 on binding to KIR2DL3 and modulation of NK cell activation. To identify suitable epitopes that are presented by HLA-C*03:04, we screened a pool of 200 overlapping peptides (OLP) of 15 amino acid length and overlapping by 11 amino acids, spanning both core and NS3 of HCV genotype 1. We identified 31 peptides that stabilized HLA-C*03:04 (Fig. 1) on the surface of 721.221-ICP47-C*03:04 cells. We subsequently focused on those peptides that showed a clear increase in HLA-C*03:04 expression, defined by having a MFI higher than the mean value plus 2 standard deviations of all non-stabilizing peptides for further assessment, resulting in ten 15mer peptides (Fig. 1 and Supplementary Fig. 2).
Fig. 1. HCV core and NS3 derived peptides stabilize HLA-C*03:04.

(A) Histograms of HLA-ABC staining of 721.221-ICP47-C*03:04 cells pulsed with control peptides. The numbers represent the MFI values obtained during the measurement depicted. (B) HLA-C*03:04 expression on HCV core and NS3 peptide-pulsed 721.221-ICP47-C*03:04 cells. HLA-C*03:04 expression is shown as relative mean fluorescence intensity (RFI) as compared to unloaded 721.221-ICP47-C*03:04 cells. Each bar represents mean +/− SEM of 3 independent experiments for each peptide. The black line represents the cutoff set by the no peptide control and the grey line the cutoff for the selected petides. The grey area is underlying the positive and negative controls.
As the optimal length of a peptide binding to HLA class I molecules is between 9–11 amino acids, we attempted to predict the optimal binding sequence for HLA-C*03:04 within these ten 15mer peptides based on previously published data and binding motives (Table 1). These 9mer peptides were compared to the original 15mer peptides for their ability to stabilize HLA-C*03:04 expression (Table 1).
Table 1.
HLA stabilization comparing 15mer and 9mer sequences.
| 15mer | 9mer | ||||
|---|---|---|---|---|---|
| Number | Sequence | HLA Stabilization | Number | Sequence | HLA Stabilization |
| 32 |
TLTCGFADLMGYIPL core125-139 |
2,5 | 32* |
TLTCGFADL core125-133 |
1,0 |
| 34 | LMGYIPLVGAPLGGA core133-147 |
3,0 | 34* |
YIPLVGAPL core136-144 |
3,3 |
| 88 | FIPVENLETTMRSPV NS31195-1209 |
2,4 | 88* |
PVENLETTM NS31197-1205 |
1,0 |
| 104 |
AATLGFGAYMSKAHG NS31259-1273 |
2,7 | 104* |
AATLGFGAY NS31259-1267 |
1,1 |
| 114 | FLADGGCSGGAYDII NS31299-1313 |
2,5 | 114* |
CSGGAYDII NS31305-1313 |
1,1 |
| 119 |
HSTDATSILGIGTVL NS31319-1333 |
2,5 | 119* |
HSTDATSIL NS31319-1327 |
1,6 |
| 120 | ATSILGIGTVLDQAE NS31323-1337 |
2,3 | 120* |
SILGIGTVL NS31325-1333 |
3,3 |
| 142 | GINAVAYYRGLDVSV NS31411-1425 |
1,8 | 142* |
NAVAYYRGL NS31413-1421 |
1,6 |
| 155 | DFSLDPTFTIETITL NS31463-1477 |
3,1 | 155* |
FSLDPTFTI NS31464-1472 |
1,2 |
| 198/199 | **VTKYIMTCM**** NS31639-1653 |
2,4 | 198/199* |
VTKYIMTCM NS31641-1649 |
0,9 |
Two of the newly synthesized 9mer peptides, 34* and 120*, showed a markedly higher stabilization of HLA-C*03:04 compared to the original 15mer, while the other 9mer peptides showed no improved or even reduced HLA stabilization capacity. These two 9mer peptides and the original ten 15mer peptides were subsequently used to investigate their impact on the binding of KIR2DL3 to the HLA-peptide complex. We performed titration experiments to identify the optimal concentration of 34* to be used in later assays (Supplementary Fig. 4). Taken together, we identified 10 novel HCV peptides that bound to HLA-C*03:04.
Binding of KIR2DL3 to HLA-C*03:04 is significantly increased by one HCV core derived epitope which also inhibits KIR2DL3+ NK cell function
To analyze the effects of the twelve selected peptides on binding of KIR2DL3 to HLA-C*03:04+ cells, we loaded 721.221-ICP47-C*03:04 cells with the respective peptides and assessed KIR binding using a KIR2DL3-IgG fusion construct (Fig. 2A). The most dramatic increase in KIR2DL3-binding was observed for the 9mer peptide number 34* (YLVL), spanning position 136–144 of the core protein of HCV genotype 1 (Fig. 2B and Supplementary Fig. 3). The longer corresponding 15mer peptide number 34 (sequence), exhibited the second highest KIR binding. While some of the other peptides also allowed for some KIR2DL3 binding, binding was lower compared to YLVL. Titration experiments performed to determine the optimal concentration of 34* revealed similar concentration dependent kinetics as HLA stabilization (Supplementary Fig. 4).
Fig. 2. Binding of KIR2DL3-Fc to HCV core and NS3 derived peptides presented by HLA-C*03:04.

(A) Representative KIR2DL3-Fc staining on peptide pulsed 721.221-ICP47-C*03:04 cells. Histogram shows no peptide control (white), a non-binding peptide (upper grey), a binding peptide (lower grey) and peptide 34* (black). The numbers represent the MFI values obtained during the measurement depicted. (B) Relative binding of KIR2DL3-Fc to HCV core and NS3 derived peptide-pulsed 721.221-ICP47-C*03:04 cells. KIR2DL3-Fc binding is shown as relative mean fluorescence intensity (RFI) compared to unloaded 721.221-ICP47-C*03:04 cells. Each bar represents mean +/− SEM of 3 independent experiments for each peptide. The grey area is underlying the positive and negative controls.
We next addressed the functional consequence of the increased engagement of KIR2DL3 by YLVL presented by HLA-C*03:04 on KIR2DL3+ NK cell function. For this we performed degranulation assays using purified primary NK cells from healthy donors in combination with YLVL-pulsed 721.221-ICP47-C*03:04 cells as target cells (Fig. 3A). The above observed strong binding of KIR2DL3 to the HLA-C*03:04+ cells pulsed with YLVL also resulted in a significant reduction in primary NK cell response towards the target cells (Fig. 3B). This reduction in NK cell responses, while barely visible for the whole CD56dim NK cell population, was dramatically increased in the KIR2DL3+ NK cell subset. In contrast, primary NK cells lacking KIR2DL3 were much less affected by the peptide-pulsed target cells. Taken together, these data demonstrate that the newly identified HCV peptide YIPLVGAPL inhibits KIR2DL3+ NK cell function by increasing the binding of KIR2DL3 to HLA-C*03:04.
Fig. 3. YLVL inhibits primary NK cell function when presented on HLA-C*03:04 cells.

(A) Representative staining of CD107a on NK cells after co-incubation with peptide pulsed 721.221-ICP47-C*03:04. Control without CD107a-AB (white), NK cells without target cells (light grey), KIR2DL3- (dark grey) and KIR2DL3+ (black) NK cells against YLVL pulsed target cells. The numbers represent the % of CD107a+ NK cells for the depicted measurement. (B) Relative frequency of CD107a+ NK cells when co-cultured with 721.221-ICP47-C*03:04 cells pulsed with YLVL compared to GKL control. Each bar represents mean +/− SEM of 7 independent experiments for each peptide.
Naturally occurring sequence variations of YIPLVGAPL modulate KIR2DL3 binding and NK cell function
The peptides we used for our initial screening assays were based on the HCV genotype 1 sequence. Comparison with other HCV genotypes revealed that YLVL often contained sequence variations in other HCV genotypes. We next assessed whether these sequence variations have a differential effect on HLA-C*03:04 stabilization, KIR2DL3 binding and KIR2DL3+ NK cell function. In other genotypes, the leucine in position 4 and 9 of YIPLVGAPL often changes to valine, resulting in the following peptide variants; YIPVVGAPL, YIPLVGAPV or YIPVVGAPV (Table 2). The variants for HCV genotype 6, 3/4 and 2 in combination with genotype 1 accounted for the four most common variants, which amount to 88.7% of all published sequences for HCV core136–144 (hcv.lanl.gov).
Table 2.
Naturally occurring sequence variations in HCV core136–144.
| Name | Main HCV genotype | Sequence | Frequency of HCV sequences |
|---|---|---|---|
| YLVL | 1 | YIPLVGAPL | 69.5% |
| YVVL | 6 | YIPVVGAPL | 7.7% |
| YLVV | 3+4 | YIPLVGAPV | 6.0% |
| YVVV | 2 | YIPVVGAPV | 5.5% |
We therefore assessed whether these naturally occurring sequence variations might show a differential effect on KIR2DL3 binding and subsequent activation of KIR2DL3+ NK cells. 721.221-ICP47-C*03:04 cells were pulsed with peptides corresponding to the different variations of core136–144 and HLA-C*03:04 stabilization and KIR2DL3 binding were assessed (Fig. 4A+B). The genotype 2 (YVVV) variant peptide of core136–144 showed a markedly decreased ability to stabilize HLA-C*03:04 expression compared to the genotype 1 YLVL peptide, while the other variant peptides exhibited similar stabilization capacities. Overall the genotype 1 derived peptide YLVL showed the highest capability to increase KIR2DL3 binding compared to YLVV and YVVV, but not the YVVL sequence. The different capacity of the four variant peptides to increase binding of KIR2DL3 to peptide pulsed 721.221-ICP47-C*03:04 cells was mirrored in their ability to modulate NK cell function. Functional responses of KIR2DL3+ NK cells were generally lower for those viral variant peptides that showed higher KIR2DL3 binding and HLA stabilization, with YLVL showing the overall highest inhibition followed by YVVL, YLVV and finally YVVV.
Fig. 4. Sequence-specific modulation of KIR2DL3 binding and NK cell function.

(A) HLA-C*03:04 expression by 721.221-ICP47-C*03:04 cells pulsed with genotype variants of YLVL. (B) Relative binding of KIR2DL3-Fc to 721.221-ICP47-C*03:04 cells pulsed with genotype variants of YLVL. (C) Relative induction of KIR2DL3 binding in relation to stabilization of HLA-C*03:04 on 721.221-ICP47-C*03:04 cells pulsed with genotype variants of YLVL. (D) Relative frequency of CD107a+ NK cells out of KIR2DL3+ NK cells when co-cultured with 721.221-ICP47-C*03:04 cells pulsed with YLVL and its genotype variants in comparison to GKL. Each bar represents the mean +/− SEM of 5 independent experiments. The grey area is underlying the positive and negative controls.
Overall we observed a modulation of KIR2DL3+ NK cell function by naturally occurring variants of the based on the ability of these peptide variants to core136-144 peptide stabilize HLA-C*03:04 expression as well as the capacity to increase binding of KIR2DL3 to peptide-pulsed 721.221-ICP47-C*03:04 cells.
Discussion
Understanding the complexity of host-pathogen interactions involving multiple arms of the immune system remains one of the major challenges in immunology. Prompted by observations that KIR-HLA interactions play an important role in the clearance of HCV infection[12, 14] and that NK cells can impose immune selection pressure in several viral infections[1], we investigated the impact of HCV-derived peptides on KIR-HLA interactions and primary NK cell function. We identified several novel HLA-C*03:04-binding epitopes in HCV core and NS3, and show that the HLA-C*03:04-restricted peptide YIPLVGAPL derived from the core protein (position 136–144) of HCV genotype 1 can inhibit KIR2DL3+ NK cell function. We furthermore demonstrate that several naturally occurring sequence variations of YIPLVGAPL in other HCV genotypes exhibit reduced capacities to bind to the inhibitory NK cell receptor KIR2DL3 and a lower capacity to inhibit KIR2DL3+ NK cell function. Taken together, these data provide a novel mechanism by which HCV might be able to evade NK cell-mediated recognition through the presentation of viral epitopes that engage inhibitory NK cell receptors.
HCV genotype 1 accounts for roughly 40–50% of all hepatitis C cases worldwide and remains a major health burden, even in light of the new highly effective DAA treatments[19, 20]. Why 10–50% of all symptomatic patients are able to clear the infection spontaneously, while the rest develops persistent infections, remains unclear[21]. Recent data suggest that host genetic factors contribute importantly to this differential outcome of HCV infection and disease progression. The most prominent host gene polymorphisms associated to date with HCV disease outcome, IL28b[22] and KIR2DL3[12], both represent genes of the innate immune system. The protective effect of certain IL28b polymorphisms might be due to an increased ability of hepatocytes to respond to members of the interferon family [23] leading to an increased antiviral state in potential target cells. The described effect of KIR2DL3 and HLA-C alleles of the HLA-C group 1 (HLA-C1) family that serve as ligands for KIR2DL3, including HLA-C*03:04, on HCV clearance [12] is thought to be due to the comparatively weak interaction between HLA-C1 molecules and the inhibitory KIR2DL3 receptor [24]. This weak inhibitory interaction might result in stronger antiviral NK cell responses in HLA-C1/KIR2DL3+ individuals, and a strengthening of the binding of KIR2DL3 to HLA-C1 molecules might provide a benefit to the virus. Here we show that the HCV core-derived viral epitope YIPLVGAPL can increase binding of inhibitory KIR2DL3 to the respective HLA-C*03:04/peptide complex, leading to a significant inhibition of KIR2DL3+ NK cell function.
One of the limitations of our study is that we used peptides of 15AA length for the initial HLA stabilization screen, we therefore cannot exclude the possibility that we might have missed some peptides which also stabilize HLA-C*03:04. Nevertheless none of the tested 9mer or 15mer peptides showed similar effects on KIR binding compared to YIPLVGAPL and the original 15mer P34. Therefor any peptide that we missed will most likely only be a weak binder of KIR2DL3.
Previous studies have demonstrated that the sequences of peptides presented by HLA class I molecules impact KIR binding and KIR+ NK cell function, including self-epitopes as well as peptides [17] derived from EBV [25, 26] and HIV [16, 27]. However, no data on the ability of HCV peptides to modulate HLA/KIR interactions have been reported to our knowledge to date, despite strong data showing an important role for KIR-HLA interactions in HCV disease outcome [12, 13]. Our data demonstrate that most HLA-C*03:04-restricted HCV peptides identified did not allow for binding of KIR2DL3, but that one peptide, YIPLVGAPL derived from HCV core, induced strong binding of KIR2DL3 to HLA-C*03:04+ target cells and inhibited the function of KIR2DL3+ primary NK cells. The crystal structure of HLA-Cw3 with KIR2DL3 was described several years ago [28], and illustrated that role of residues in the HLA-presented epitope, in particular positions 7 and 8, in the binding to KIR2DL3. Interestingly, while the sequence of the YIPLVGAPL peptide does not follow the described canonical peptide sequence motive for HLA-C*03:04 [29], it nevertheless stabilized HLA-C*03:04 expression on TAP-deficient cell lines and increased the binding of KIR2DL3. This is in line with recent data demonstrating that the binding motives for peptide presented by HLA-C molecules allow for more variability in the peptide sequence [29]. One concern might be the levels of HLA expression on the generated cell lines compared to normal human cells. We did however not find higher levels of HLA, when comparing the generated cell lines pulsed with stabilizing peptides to human PBMCs (Supplementary Fig. 5).
A number of naturally occurring sequence variations of YIPLVGAPL are described; which are primarily seen in other HCV genotypes. These variations are characterized by a switch from leucine to valine at position 4 and/or 9, and showed effects on HLA stabilization, KIR binding and NK cell function in our studies. In particular, the switch from leucine to valine in the C-terminal HLA-binding anchor position 9 of the peptide resulted in reduction of HLA-C*03:04 expression on TAP-deficient cell lines, in line with the critical role of this residue in binding to HLA-C*03:04. The observed reduction in KIR2DL3 binding in the context of these two peptides (YIPLVGAPV, YIPVVGAPV) is most likely a reflection of the reduced expression of its ligand HLA-C*03:04. In contrast, changes in position 4 had little effect on HLA expression, KIR2DL3 binding or the function of KIR2DL3+ NK cells. It remains unknown whether NK cell-mediated immune pressure could induce HCV to adapt to the KIR/HLA genotypes in certain host populations, exploiting the ability of specific sequence variants in HLA class I-presented epitopes to inhibit KIR+ NK cell function, as suggested in the context of HIV infection [10]. When we compared KIR distribution in different ethnicities with the latest metadata analysis on HCV genotype distribution [18], we found that, among HCV infected individuals, carrying the KIR2DL3 gene strongly correlates (R2=0.8317, p=<0.0001) with being infected with HCV genotype 1 (data not shown). Future studies are also required to determine the abundance by which the identified HCV peptides are presented by HLA class I on HCV infected cells in vivo, and whether the function of intrahepatic NK cells is modified by HLA class I presented HCV peptides to the same degree, as a recent publication hinted towards distinct functional features of intrahepatic NK cells [30].
In conclusion we identified several novel HLA-C*03:04-presented epitopes in HCV core and NS3, and provide novel functional evidence demonstrating that one of these HCV-derived peptides presented by HLA-C*03:04 can inhibit primary NK cell function through the engagement of an inhibitory KIR. These data provide first evidence for a novel pathway by which HCV can inhibit recognition by NK cells through the engagement of inhibitory NK cell receptors, providing a possible mechanism for viral escape.
Supplementary Material
Acknowledgments
The authors would like to thank Camille R. Simoneau with help for the generation of TAP-KO cell lines. Furthermore we would like to express our gratitude to all study participants. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 841). This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Abbreviations
- HCV
Hepatitis C virus
- KIR
Killer cell immunoglobulin like receptor
- HLA
human leukocyte antigen
- NK
Natural Killer
- OLP
overlapping peptide
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest
Author Contribution: SL designed and performed the experiments, analyzed the data and wrote the paper; GM performed experiments and wrote the paper; AH designed the experiments and wrote the paper; AC and MZ performed experiments; CK and WGM designed experiments and wrote the paper; MC analyzed the data and wrote the paper; HW analyzed the data and wrote the paper; MA designed the experiments, analyzed the data and wrote the paper.
Author names in bold designate shared co-first authorship
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annual review of immunology. 2013;31:163–94. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nature immunology. 2008;9(5):503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 3.Crome SQ, Lang PA, Lang KS, Ohashi PS. Natural killer cells regulate diverse T cell responses. Trends Immunol. 2013;34(7):342–9. doi: 10.1016/j.it.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Moretta A. The dialogue between human natural killer cells and dendritic cells. Curr Opin Immunol. 2005;17(3):306–11. doi: 10.1016/j.coi.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annual review of immunology. 2013;31:227–58. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nature immunology. 2008;9(5):495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nature reviews Immunology. 2013;13(2):133–44. doi: 10.1038/nri3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamil KM, Khakoo SI. KIR/HLA interactions and pathogen immunity. Journal of biomedicine & biotechnology. 2011;2011:298348. doi: 10.1155/2011/298348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long EO, Rajagopalan S. HLA class I recognition by killer cell Ig-like receptors. Seminars in immunology. 2000;12(2):101–8. doi: 10.1006/smim.2000.0212. [DOI] [PubMed] [Google Scholar]
- 10.Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476(7358):96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nature medicine. 2013;19(7):859–68. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305(5685):872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 13.Knapp S, Warshow U, Hegazy D, Brackenbury L, Guha IN, Fowell A, et al. Consistent beneficial effects of killer cell immunoglobulin-like receptor 2DL3 and group 1 human leukocyte antigen-C following exposure to hepatitis C virus. Hepatology. 2010;51(4):1168–75. doi: 10.1002/hep.23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dring MM, Morrison MH, McSharry BP, Guinan KJ, Hagan R, Irish HCVRC, et al. Innate immune genes synergize to predict increased risk of chronic disease in hepatitis C virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5736–41. doi: 10.1073/pnas.1016358108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods. 2014;11(8):783–4. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Teijlingen NH, Holzemer A, Korner C, Garcia-Beltran WF, Schafer JL, Fadda L, et al. Sequence variations in HIV-1 p24 Gag-derived epitopes can alter binding of KIR2DL2 to HLA-C*03:04 and modulate primary natural killer cell function. Aids. 2014;28(10):1399–408. doi: 10.1097/QAD.0000000000000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405(6786):537–43. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 18.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61(1):77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeuzem S, Asselah T, Angus P, Zarski JP, Larrey D, Mullhaupt B, et al. Faldaprevir (BI 201335), deleobuvir (BI 207127) and ribavirin oral therapy for treatment-naive HCV genotype 1: SOUND-C1 final results. Antiviral therapy. 2013;18(8):1015–9. doi: 10.3851/IMP2567. [DOI] [PubMed] [Google Scholar]
- 20.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Subramanian GM, et al. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014;146(3):736–43. e1. doi: 10.1053/j.gastro.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Wiegand J, Deterding K, Cornberg M, Wedemeyer H. Treatment of acute hepatitis C: the success of monotherapy with (pegylated) interferon alpha. The Journal of antimicrobial chemotherapy. 2008;62(5):860–5. doi: 10.1093/jac/dkn346. [DOI] [PubMed] [Google Scholar]
- 22.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 23.Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2010;30(8):555–64. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parham P. Immunology. NK cells lose their inhibition. Science. 2004;305(5685):786–7. doi: 10.1126/science.1102025. [DOI] [PubMed] [Google Scholar]
- 25.Hill AB, Lee SP, Haurum JS, Murray N, Yao QY, Rowe M, et al. Class I major histocompatibility complex-restricted cytotoxic T lymphocytes specific for Epstein-Barr virus (EBV)-transformed B lymphoblastoid cell lines against which they were raised. The Journal of experimental medicine. 1995;181(6):2221–8. doi: 10.1084/jem.181.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, et al. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. European journal of immunology. 2004;34(6):1673–9. doi: 10.1002/eji.200425089. [DOI] [PubMed] [Google Scholar]
- 27.Fadda L, Korner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS pathogens. 2012;8(7):e1002805. doi: 10.1371/journal.ppat.1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maenaka K, Juji T, Stuart DI, Jones EY. Crystal structure of the human p58 killer cell inhibitory receptor (KIR2DL3) specific for HLA-Cw3-related MHC class I. Structure. 1999;7(4):391–8. doi: 10.1016/s0969-2126(99)80052-5. [DOI] [PubMed] [Google Scholar]
- 29.Rasmussen M, Harndahl M, Stryhn A, Boucherma R, Nielsen LL, Lemonnier FA, et al. Uncovering the peptide-binding specificities of HLA-C: a general strategy to determine the specificity of any MHC class I molecule. Journal of immunology. 2014;193(10):4790–802. doi: 10.4049/jimmunol.1401689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marquardt N, Beziat V, Nystrom S, Hengst J, Ivarsson MA, Kekalainen E, et al. Cutting edge: identification and characterization of human intrahepatic CD49a+ NK cells. Journal of immunology. 2015;194(6):2467–71. doi: 10.4049/jimmunol.1402756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.