

Abstract
BACKGROUND
For a more complete understanding of pharmacodynamic, metabolic, and pathophysiologic effects, protein kinetics, such as production rate and fractional catabolic rate, can offer substantially more information than protein concentration alone. Kinetic experiments with stable isotope tracers typically require laborious sample preparation and are most often used for studying abundant proteins. Here we describe a practical methodology for measuring isotope enrichment into low-abundance proteins that uses an automated procedure and immunoaffinity enrichment (IA) with LC-MS. Low-abundance plasma proteins cholesteryl ester transfer protein (CETP) and proprotein convertase subtilisin/kexin type 9 (PCSK9) were studied as examples.
METHODS
Human participants (n = 39) were infused with [2H3]leucine, and blood samples were collected at multiple time points. Sample preparation and analysis were automated and multiplexed to increase throughput. Proteins were concentrated from plasma by use of IA and digested with trypsin to yield proteotypic peptides that were analyzed by microflow chromatography-mass spectrometry to measure isotope enrichment.
RESULTS
The IA procedure was optimized to provide the greatest signal intensity. Use of a gel-free method increased throughput while increasing the signal. The intra- and interassay CVs were <15% at all isotope enrichment levels studied. More than 1400 samples were analyzed in <3 weeks without the need for instrument stoppages or user interventions.
CONCLUSIONS
The use of automated gel-free methods to multiplex the measurement of isotope enrichment was applied to the low-abundance proteins CETP and PCSK9.
Protein kinetics measurements are often performed to understand the influence of an observed pharmacodynamic, metabolic, or pathophysiologic effect, and can be used to determine whether an observed change in protein concentration is due to the modulation of protein synthesis or protein clearance (1–6). The procedure for determining protein kinetics has been described previously (7–9) but generally involves infusing a participant with an isotopically labeled amino acid, such as leucine, followed by measuring the incorporation and elimination of the labeled amino acid into and from the protein of interest. Traditionally, these analyses require sophisticated and laborious sample preparation techniques that include protein purifications followed by GC-MS on the hydrolyzed protein to measure isotope enrichment (10–12). However, recent advances in LC-MS and its associated techniques have facilitated the analysis of isotope enrichment by removing the requirement for extensive protein purification (13–18). For selectivity, LC-MS can separate and measure unique proteolytic peptide sequences (typically generated through tryptic digestion of the target protein into peptide fragments) rather than analysis of single amino acids (derived from hydrolyzed protein) by GC-MS. As a result, the need to isolate the protein of interest from matrix proteins is reduced, and multiplexed analysis of several proteins can be obtained from a single sample (19–21).
The precision of the isotope enrichment measurement is dictated greatly by the signal intensity of the isotope-labeled peptide, which is often a small percentage of the unlabeled peptide. The use of highly sensitive mass spectrometers and low-flow or nanoflow chromatography can enhance the analytical signal and enable isotope-enrichment measurements for lower-abundance proteins; however, nanoflow chromatography can be difficult to implement for analysis of clinical samples because it has relatively low throughput and requires high levels of expertise. An approach that has been used to increase the analytical signal for protein quantification assays uses immunoaffinity enrichment (IA)6 to concentrate analytes while removing the majority of matrix proteins (22–25). Much work has been done in this area to automate and increase the throughput of IA mass spectrometry assays (26, 27), which is critical for clinical protein kinetics measurements where intensive sampling is required to accurately model synthesis and clearance.
Cholesteryl ester transfer protein (CETP) is a plasma protein that transfers lipids from 1 lipoprotein particle to another and has been a target for treatment of atherosclerosis and cardiovascular disease. Circulating concentrations of CETP are relatively low (approximately 1800 ng/mL) (28). Proprotein convertase subtilisin/kexin type 9 (PCSK9) circulates at concentrations much lower than those of CETP (approximately 90 ng/mL) (29) and regulates the number of LDL receptors at the surface of hepatocytes, thereby affecting the clearance of LDL cholesterol. PCSK9 is a potential target for the treatment of cardiovascular disease and has been shown to be modulated by statins and fibrates (30).
Here, we describe the practical application of a multiplexed IA mass spectrometry technique to measure the kinetics of these 2 low-abundance plasma proteins. IA increases the effective concentration of analyte proteins in the sample, but also removes the majority of plasma proteins, reducing matrix effects (signal suppression) and background. However, the concentration of PCSK9 remains so low it cannot be measured by LC-MS using conventional ultraperformance liquid chromatography (UPLC) technology. To overcome this problem, we used microflow chromatography (approximately 10 μL/min) to achieve sufficient analytical sensitivity while retaining a higher throughput and robustness compared to nanospray chromatography (<1 μL/min).
Materials and Methods
STUDY PROTOCOL
Individuals were enrolled in an ongoing Merck-sponsored phase I study (MK-0859 PN026; NCT00990808) (unpublished data on file; Merck Sharp & Dohme Corp). All protocols and procedures were approved by the Human Investigational Review Boards at the University of Pennsylvania and Columbia University. All subjects provided written informed consent. Participants received a bolus injection of labeled leucine, [5,5,5-2H3]-L-leucine, immediately followed by a constant infusion over a 15-h period. Samples were collected at 0 (prebolus), 20, and 40 min and 1, 2, 4, 6, 8, 10, 12, 14, 15, 15.5, 16, 18, 21, 24, and 48 h after the bolus injection of labeled leucine. At each time point, approximately 15 mL whole blood was collected with heparin as an anticoagulant. Immediately after collection, samples were placed on ice and centrifuged at 4 °C to obtain plasma. Plasma was aliquoted in 1.5-mL volumes into a 3.6-mL internally threaded Nunc cryotube and stored at −80 °C until transfer to the analysis site, where the samples were also stored at −80 °C until analysis.
OPTIMIZATION OF IMMUNOAFFINITY
Immunoaffinity beads covalently coupled to anti-CETP and anti-PCSK9 mAbs (both developed in house) were prepared by use of Dynabeads MyOne Tosylactivated (Life Technologies Corp.) following the manufacturer’s recommended protocol. Prewashing the coupled magnetic beads with an acidic solution (100 mmol/L glycine, pH 2.5) was beneficial for increasing the recovery with anti-PCSK9 antibodies.
Sample processing was completed in 4 days. On day 1, after thawing plasma samples in a room-temperature water bath, samples were vortex-mixed, and 0.5 mL plasma was transferred to a 96-well deep-well plate by use of a Freedom EVO (Tecan Trading) liquid-handling robot. We centrifuged the plates at 2000g for 30 min at 4 °C to remove precipitated protein and debris. We mixed clarified sample supernatants with 55 μL of 10× radioimmunoprecipitation assay (RIPA) lysis buffer (EMD Millipore Corp.), protease inhibitors (EMD Millipore Corp.), and IA beads for both CETP and PCSK9 (0.6 mg each/sample). Samples were incubated overnight (18–20 h) at 4 °C with rotation. On day 2, the magnetic beads in each sample were washed with 500-μL volumes once with RIPA buffer (Technova Corp.) and twice with 2 washes with PBS. Proteins bound to the beads were released during a 20-min incubation with 100 μL of 10% formic acid in water. During each liquid transfer step, we separated magnetic beads from the solution using a custom magnetic device that pulled the beads to the side of each well. Enriched proteins were transferred to a new plate and completely dried by use of a SpeedVac system and stored at −20 °C. On day 3, dried proteins were reconstituted and reduced in 80 μL of 50 mmol/L ammonium bicarbonate buffer (pH 7.4) containing 5 mmol/L dithiothreitol for 30 min at 37 °C with mixing. Free sulfhydryl groups were alkylated with the addition of 10 μL of 80 mmol/L iodoacetamide in 50 mmol/L ammonium bicarbonate buffer and incubated for 1 h at room temperature. Trypsin (0.05 μg in 10 μL of 50 mmol/L ammonium bicarbonate buffer) was added to each sample, and proteins were digested during overnight (18–20 h) incubation at 37 °C. On day 4, samples were acidified with 10 μL of 10% formic acid in water and transferred to a 96-well nonbinding surface polystyrene round-bottom analysis plate (Corning).
OPTIMIZATION OF INSTRUMENT PARAMETERS
Candidate proteolytic peptides were first identified empirically with recombinant protein and then confirmed with in silico tools. Unlabeled peptides and isotope-labeled versions of candidate peptides were synthesized and obtained from New England Peptide, GTVSGT-2H3Leu-IGLEFIR for PCSK9 and ITKPAL-2H3Leu-VLNEHTAK for CETP. We determined multiple reaction monitoring (MRM) transitions for each labeled and unlabeled peptide by direct infusion into a TSQ Vantage QQQ mass spectrometer (Thermo Fisher Scientific). The optimal instrument parameters, including collision energy (CE), were determined empirically.
LIQUID CHROMATOGRAPHY–TANDEM MASS SPECTROMETRY ANALYSIS (ISOTOPE ENRICHMENT)
Isotope enrichment was determined by use of a nano-Acquity UPLC system (Waters) coupled with a TSQ Vantage QQQ mass spectrometer with a Captivespray ESI source (Bruker-Michrom). The analytical column was a 0.3 × 50–mm Magic C18AQ column, 3 μm particle size (Bruker-Michrom), held at 50 °C. Solvent A was 0.1% formic acid in water, and solvent B was acetonitrile. The initial solvent composition was 2% B held at 15 μL/min for 2 min, at which time the flow rate was reduced to 10 μL/min and the solvent composition increased to 50% B over 2.5 min with a linear gradient. The flow rate was then increased to 15 μL/min and the solvent composition increased to 85% B and was held for 0.5 min before being lowered back to 2% B. The injection volume was 20 μL. Including autosampler washes and injection time, the sample-to-sample injection time was <10 min. The transitions monitored for CETP (ITKPALLVLNEHTAK > LLVLNEHTAK) were 550.00 > 653.38 for M0 and 551.00 > 654.88 for M3 with S-lens = 115, CE = 17, and Q1 and Q3 values 0.7 and 0.4. For PCSK9 (GTVSGTLIGLEFIR > LIGLEFIR) the transitions monitored were 731.91 > 960.588 for M0 and 733.41 > 763.586 for M3 with S-lens = 150, CE = 23, and Q1 and Q3 resolution values both 0.4.
QC SAMPLES
We generated QC samples by adding synthetic heavy-labeled ITKPAL-2H3Leu-VLNEHTAK and GTVSGT-2H3Leu-IGLEFIR peptides to a pool of processed samples from a single donor. Three levels of QC samples were prepared (high, medium, and low). The low QC contained only the naturally occurring ratio of isotopes without added peptide. Aliquots (0.5 mL) of this material were stored at −80 °C and thawed to be added to the sample plate after tryptic digestion and before sample analysis.
MRM ACCURACY ASSESSMENT
We analyzed a series of samples created with varying isotope enrichments with synthetic peptide standards with both MRM and selected ion monitoring (SIM) data acquisition settings. For SIM, the instrument settings for CETP (ITKPALLVLNEHTAK) were 550.66 (1.5) amu for 0.2 s with Q1 and Q3 resolution settings both 0.1. For PCSK9 (GTVSGTLIGLEFIR), the instrument settings were 732.41 (1.5) amu for 0.2 s with Q1 and Q3 resolution settings 0.25 and 0.1. Quantification of isotope enrichment with SIM was calculated with the peptide molecular weight ± 100 mDa.
ASSESSMENT OF LIMIT OF QUANTIFICATION
Limit of quantification (LOQ) for isotope enrichment could not be determined in the same manner as the LOQ for quantification, because both isotope enrichment and concentration varied in each sample. In this assay, the LOQ for isotope enrichment was based on signal intensity of the M3 peptide, which was always less intense and therefore more difficult to measure than that of the M0 peptide. An unlabeled peptide solution containing both the CETP and PCSK9 peptide was titrated at decreasing concentrations, and the M3/M0 signal was measured at each concentration (n = 5).
ASSESSMENT OF INTRA- AND INTERASSAY REPRODUCIBILITY
We assessed the intraassay reproducibility of repeated measurements with the QC samples by analyzing 6 sets on a single day with a single sample preparation. We assessed interassay reproducibility by analyzing each QC sample on 6 separate days with 6 separate sample preparations.
MEASUREMENTS OF CETP AND PCSK9 CONCENTRATIONS BY IMMUNOASSAYS
We measured PCSK9 concentration using a Meso Scale Discovery assay developed in house. Briefly, after thawing and vortex-mixing, samples and controls were diluted 1:8 in assay buffer and added to the plate in duplicate (50 μL/well). Calibrators were added undiluted onto the plate. Samples, calibrators, and controls were incubated and followed by a biotin-linked secondary antibody that bound streptavidin-linked electrochemiluminescent ruthenium label, Meso Scale Discovery Sulfo-Tag™. A read buffer containing a coreactant was then introduced to the well. Plates were read on a MSD Sector 6000 instrument. CETP concentration was measured at PPD (Richmond, VA) by ELISA assay.
KINETICS CALCULATIONS
The isotope enrichment (M3/M0 measured − M3/M0 background) was divided by the number of leucines in the fragment peptides (3 for CETP and 2 for PCSK9) and was converted to enrichment: (M3/M0/n)/{1 + [(M3/M0/n)/100]}. The fractional catabolic rate (FCR) for PCSK9 was calculated with WinSAAM version 3.0.7 by fitting the isotope enrichment to a compartmental model consisting of 3 sequential compartments representing a hepatic precursor leading to a synthetic delay followed by plasma PCSK9 that was directly cleared. The hepatic precursor was represented by the plasma [2H3]leucine enrichment, which was measured as previously described (31). The PCSK9 pool size was calculated as the product of the mean plasma PCSK9 concentration, measured at 3 time points during the metabolic study, and the plasma volume, assumed to be 4.5% of body weight. The production rate for PCSK9 was calculated as the product of the FCR and the plasma PCSK9 pool size. For CETP, enrichments were plotted over time, and the slopes of the enrichment curves were calculated.
Results
SELECTION OF PEPTIDES AND MRM TRANSITIONS
Candidate peptide analytes for kinetic measurements must contain at least a single leucine residue, have a sequence unique to the target protein in the human plasma proteome, and have properties that are amenable to LC/MS. Peptides from CETP and PCSK9 identified previously (16) met these criteria and were selected for this assay. Additional peptides were investigated but were found to be inferior on the basis of our selection criteria. The peptides measured in this assay were ITKPALLVLNEHTAK for CETP and GTVSGTLIGLEFIR for PCSK9, and the MRMs were selected on the basis of greatest signal-to-noise ratio (S/N). Because of the low concentration of isotope-labeled leucine as a precursor, the statistical chance of a peptide to have incorporated multiple leucines was small enough to ignore for these calculations. The amino acid tracer used for these studies was 5,5,5-2H3 leucine, which lacked sufficient mass to resolve M3 from the natural isotopes of unlabeled peptides (M0). Therefore, there was substantial background for M3 (approximately 4% for PCSK9 and approximately 9% for CETP) resulting from the endogenous isotopes that could not be separated by chromatography or low-resolution mass spectrometry. Background was different for the CETP and PCSK9 peptides owing to the different sequences and chemical formulas.
OPTIMIZATION OF IMMUNOAFFINITY
Increasing sample processing throughput is critical when analyzing large sample sets. A 2-fold reduction in processing and analysis time was gained by combining anti-CETP and anti-PCSK9 coupled IA beads and multiplexing sample processing and LC/MS. Plasma tested with individual or combined purifications showed no loss of recovery of either analyte when beads were combined. Compared with in-gel digestion, in-solution digestion resulted in a >5-fold increase in peptide signals while avoiding the technical difficulties involved with cutting and handling gel pieces. This gain in analytical sensitivity allowed us to use microflow liquid chromatography flow rates rather than nanoflow, which also resulted in increased throughput and practical usability.
The final optimized IA binding and wash conditions were determined by testing 1- and 0.5-mL plasma volumes under several wash conditions. Each condition was tested with plasma from a set of 20 donors. Each condition was evaluated by comparing the precision [standard deviation (σ)] of the M3/M0 isotope ratio measurements. The combination of a lower sample volume (0.5 mL) and 1 wash with a more stringent buffer (RIPA) resulted in the highest precision [σ(CETP) = 0.114; σ(PCSK9) = 0.457] compared with 0.5 mL plasma with PBS + 1% Triton X-100 [σ(CETP) = 0.181; σ(PCSK9) = 0.547] and 1 mL plasma with RIPA wash [σ(CETP) = 0.311; σ(PCSK9) = 0.651].
SELECTION OF MRM QUADRUPOLE RESOLUTION SETTINGS
We observed that decreasing the quadrupole resolution settings effectively increased S/N for the M3 PCSK9 peptide. Reducing resolution settings decreased absolute signal intensity, but the added selectivity effectively decreased the background preferentially for the target peptide, thus increasing S/N. For a typical sample, S/N for PCSK9 M3 was 3:1 with unit resolution settings. Resolution settings of 0.4 for both Q1 and Q3 increased S/N to 10:1 while reducing overall signal intensity only <2-fold.
ACCURACY OF MRM MEASUREMENT
To verify the accuracy of the MRM measurement, both MRM and SIM data acquisitions were applied to a set of synthetic peptide samples. It is important to note that these different types of analyses effectively measure isotope enrichment differently. The isotope pattern of the parent peptides was recorded with SIM data acquisition. With MRM acquisition, independent measurements of unlabeled and labeled fragments were recorded. As demonstrated in Fig. 1, isotope enrichment measurements were identical for the 2 acquisition settings for both peptides as determined by correlation plots with slopes for both peptides near 1 (1.02 for both CETP and PCSK9 enrichment curves). For both proteins, the y axis intercept was greater than zero due to the contribution from the natural isotopes of the unlabeled peptide.
Fig. 1.
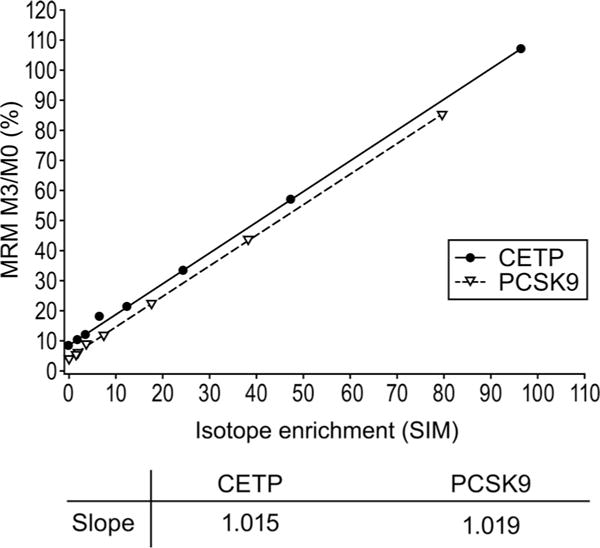
Comparison of SIM and MRM values for identical samples.
LIMIT OF QUANTIFICATION
Because the signal from the labeled peptide (M3) was always much lower than that of the unlabeled peptide (M0), it was practical to consider only the labeled peptide when evaluating LOQ for these measurements. Because this assay was not designed to quantify proteins, LOQ was based solely on signal intensity of the M3 peptide required to achieve a precise ratio measurement. As demonstrated in Table 1, baseline measurements at high concentrations were accurate and consistent (with low CVs) until the peak height of M3 decreased to 1 E + 04, where the CVs approached or exceeded 20%. As a result, samples for which the M3 peak was <1 E + 04 could not be reliably measured by this assay.
Table 1.
Effect of M3 peak height of CETP and PCSK9 proteolytic peptides on the ability to calculate isotope enrichment.
| M3 peak height | Calculated M3/M0 | Error, % | CV, % |
|---|---|---|---|
| CETP | |||
| 5.60E+05 | 0.085 | 1 | 3 |
| 2.70E+05 | 0.081 | 6 | 2 |
| 1.30E+05 | 0.084 | 3 | 6 |
| 8.90E+04 | 0.081 | 6 | 3 |
| 4.40E+04 | 0.078 | 9 | 8 |
| 1.80E+04 | 0.074 | 14 | 8 |
| 8.90E+03 | 0.059 | 32 | 19 |
| 5.00E+03 | 0.049 | 43 | 15 |
| 3.40E+03 | 0.061 | 30 | 39 |
| 2.40E+03 | 0.063 | 27 | 36 |
| PCSK9 | |||
| 1.90E+05 | 0.042 | 2 | 4 |
| 1.20E+05 | 0.042 | 2 | 3 |
| 5.70E+04 | 0.040 | 3 | 3 |
| 3.70E+04 | 0.040 | 3 | 6 |
| 1.90E+04 | 0.036 | 12 | 11 |
| 8.50E+03 | 0.032 | 22 | 10 |
| 4.40E+03 | 0.029 | 30 | 22 |
| 3.00E+03 | 0.023 | 44 | 49 |
| 1.50E+03 | 0.022 | 46 | 29 |
| 7.50E+02 | 0.019 | 54 | 41 |
ASSESSMENT OF INTRA- AND INTERDAY REPRODUCIBILITY
With the spiked peptide QC samples, the intraassay variability (CV) ranged from 1.6% to 2.2% for CETP and 3.6% to 8.2% for PCSK9. The observed interassay variability ranged from 2.3% to 3.4% for CETP and 3.0% to 9.0% for PCSK9 (Table 2). For both intra- and interassay reproducibility, the PCSK9 measurement was more variable than the CETP measurement. This was likely due to a large difference in signal intensity. However, in both cases, the reproducibility was sufficient to allow measurements of <1% isotope enrichment (on the basis of standard deviation).
Table 2.
Interassay reproducibility values (M3/M0) for CETP and PCSK9 QC samples through assay validation.
| Peptide | Low | Medium | High |
|---|---|---|---|
| CETP (ITKPALLVLNEHTAK) | |||
| Mean | 0.092 | 0.193 | 0.292 |
| SD | 0.0032 | 0.0043 | 0.0100 |
| CV, % | 3.4 | 2.3 | 3.4 |
| Samples, n | 6 | 6 | 4 |
| PCSK9 (GTVSGTLIGLEFIR) | |||
| Mean | 0.040 | 0.197 | 0.363 |
| SD | 0.0036 | 0.0060 | 0.0016 |
| CV, % | 9.0 | 3.0 | 4.4 |
| Samples, n | 6 | 6 | 4 |
ISOTOPE ENRICHMENT MEASUREMENTS
A total of 78 isotope enrichment curves were calculated for the 39 participants’ samples for each protein from 2 separate periods. This required the analysis of >1400 samples and QCs. To ensure consistent data across the entire sample set and reduce sample preparation and instrument resource time, samples were processed in parallel and the mass spectrometer was operated continuously. Sample processing was conducted with 72 samples (2 participants each having two 18-sample periods) on each 96-well plate, 4 plates at a time. Preparation of all samples was completed in 9 days. LC/MS analysis was conducted continuously, with each sample plate requiring <12 h. All samples were analyzed over the course of 10 days with no user interventions (including column changes or instrument cleanings). A small increase in liquid chromatography column pressure was observed, but with no corresponding reduction in instrument stability or data quality. Retention times and peak shape were relatively stable over the course of the experiment. QC samples that were acquired over the experiment (n = 20) also demonstrate the reproducibility and robustness of this method. CVs for each QC level (Table 3) were in line with interassay reproducibility experiments. Fig. 2 shows a typical time course for isotope enrichment for CETP and PCSK9. Incorporation of label into newly synthesized protein was easily observed by the increased M3/M0 ratios, with a corresponding decrease as the label was removed from circulation and newly synthesized protein no longer contained the isotope-labeled leucine.
Table 3.
Interassay reproducibility values (M3/M0) for CETP and PCSK9 QC samples during sample analysis.
| Peptide | Low | Medium | High |
|---|---|---|---|
| CETP (ITKPALLVLNEHTAK) | |||
| Mean | 0.085 | 0.184 | 0.289 |
| SD | 0.0029 | 0.0057 | 0.0064 |
| CV, % | 3.4 | 3.1 | 2.2 |
| Samples, n | 20 | 20 | 20 |
| PCSK9 (GTVSGTLIGLEFIR) | |||
| Mean | 0.041 | 0.203 | 0.373 |
| SD | 0.0050 | 0.0186 | 0.0234 |
| CV, % | 12.1 | 9.2 | 6.3 |
| Samples, n | 20 | 20 | 20 |
Fig. 2. Typical isotope-enrichment curves for peptides ITKPALLVLNEHTAK (CETP) and GTVSGTLIGLEFIR (PCSK9) for a clinical period.
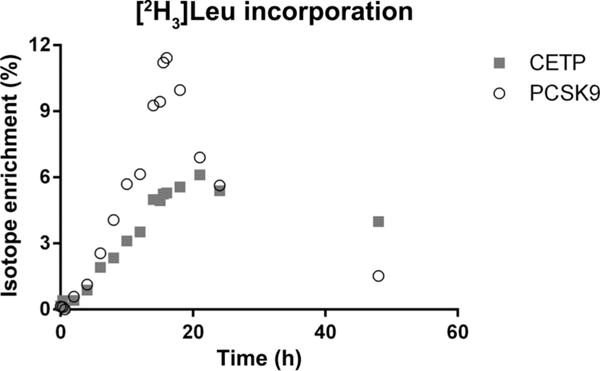
Background from natural isotopes of M0 was subtracted to calculate isotope enrichment.
KINETIC PARAMETERS FOR CETP AND PCSK9
By use of immunoaffinity-based methods to measure pool size and mass spectrometry to measure isotope enrichment, the pool size, fractional catabolic rate (FCR), and production rate were determined for all participants, with mean values from a single period presented in Table 4. Individual participant values are included in Supplemental Table 1, which accompanies the online version of this article at http://www.clinchem.org/content/vol60/issue9. Participants were on daily doses of either 20 mg atorvastatin (n = 29) or placebo (n = 10) for approximately 4 weeks. These values agree well with the only other report of CETP and PCSK9 kinetics (n = 3) (16). Differences between the 20 mg atorvastatin and placebo groups were not significant (P > 0.05) for all parameters except CETP production rate (P = 0.047) by 2-sided t-test assuming equal variance.
Table 4.
Pool size, FCR, and production rate calculated for CETP and PCSK9.a
| Pool size, μg | FCR, pools/day | Production rate, μg · kg−1 · day−1 | |
|---|---|---|---|
| CETP | |||
| Atorvastatin | 7090.54 (2334.4) | 0.48 (0.09) | 37.28 (10.5) |
| Placebo | 7688.97 (3055.3) | 0.51 (0.19) | 45.25 (10.9) |
| P | 0.523 | 0.416 | 0.047 |
| PCSK9 | |||
| Atorvastatin | 976.48 (385.8) | 1.92 (0.05) | 21.7 (10.1) |
| Placebo | 778.50 (265.7) | 1.92 (0.87) | 18.8 (10.8) |
| P | 0.143 | 0.997 | 0.446 |
Data are mean (SD). P values compare atorvastatin- and placebo-treated groups.
Discussion
Previously, protein kinetics measurements have been performed on relatively highly abundant proteins by either GC or LC-MS, with LC-MS increasingly used due to selectivity advantages. Recent technical advances have enabled the measurement of protein kinetics for plasma proteins that had previously been unmeasurable. These advances include the use of antibody-coated magnetic beads, which are used to enrich samples for the proteins of interest, the use of liquid handlers to efficiently and accurately process hundreds of samples, and highly sensitive LC-MS platforms capable of measuring low-abundance analytes with the throughput required to support clinical protocols. The use of LC-MS to measure low-abundance protein kinetics has been demonstrated previously, but in limited quantity (16). The use of gel-free isolations markedly increases applicability by increasing throughput while increasing sample recovery. This, in turn, allows the use of LC-MS platforms that are more practical in the clinical setting than most nanospray platforms. Furthermore, gel-free methods enable one to multiplex the analysis of multiple proteins in a single sample preparation and LC-MS run. Because kinetic measurements require a large number of time points, the ability to automate and multiplex these measurements significantly reduces the overall cost of analysis and makes these measurements practical for clinical studies.
As protein enrichment capabilities continue to improve along with commercially available mass spectrometers, we expect that the kinetics of even less-abundant proteins and proteins with very slow synthesis rates may be investigated by use of this methodology. As the calculation of synthesis and clearance rates requires the measurement of protein concentration, mass spectrometry–based methods have the potential to measure isotope enrichment and protein concentration simultaneously. Future experiments will likely take advantage of the mass spectrometer’s ability to quantify both endogenous protein and isotope enrichment.
Supplementary Material
Acknowledgments
The authors thank Amy Johnson-Levonas and Kathleen Newcomb for editorial assistance in the preparation of this manuscript and Yang Liu for assistance with statistical analysis, Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ.
Research Funding: Merck & Co. We conducted studies of the effects of anacetrapib, a CETP inhibitor, on lipoprotein kinetics. The study was funded by Merck and some of the samples were used for this ancillary work.
Footnotes
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: M.E. Lassman, T. McAvoy, A.Y.H. Lee, D. Chappell, O. Wong, H. Zhou, D.E. Gutstein, and O.F. Laterza, Merck & Co.
Consultant or Advisory Role: H.N. Ginsberg, member of advisory boards for Merck & Co.; D.J. Rader, Merck & Co.
Stock Ownership: M.E. Lassman, T. McAvoy, A.Y.H. Lee, D. Chappell, H. Zhou, D.E. Gutstein, and O.F. Laterza, Merck & Co.
Honoraria: G. Reyes-Soffer, H.N. Ginsberg, and J.S. Millar, Merck & Co.
Expert Testimony: None declared.
Patents: None declared.
Other Remuneration: G. Reyes-Soffer, Merck & Co.
Role of Sponsor: No sponsor was declared.
Nonstandard abbreviations: IA, immunoaffinity enrichment; CETP, cholesteryl ester transfer protein; PCSK9, proprotein convertase subtilisin/kexin type 9; UPLC, ultraperformance liquid chromatography; RIPA, radioimmunoprecipitation assay; MRM, multiple reaction monitoring; CE, collision energy; SIM, selected ion monitoring; LOQ, limit of quantification; FCR, fractional clearance rate; S/N, signal-to-noise ratio.
References
- 1.Barrett PH, Chan DC, Watts GF. Thematic review series: patient-oriented research. Design and analysis of lipoprotein tracer kinetics studies in humans. J Lipid Res. 2006;47:1607–19. doi: 10.1194/jlr.R600017-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Boren J, Taskinen MR, Adiels M. Kinetic studies to investigate lipoprotein metabolism. J Intern Med. 2012;271:166–73. doi: 10.1111/j.1365-2796.2011.02497.x. [DOI] [PubMed] [Google Scholar]
- 3.Claydon AJ, Beynon R. Proteome dynamics: revisiting turnover with a global perspective. Mol Cell Proteomics. 2012;11:1551–65. doi: 10.1074/mcp.O112.022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Matthan NR, Lichtenstein AH, et al. Effects of different doses of atorvastatin on human apolipoprotein B-100, B-48, and A-I metabolism. J Lipid Res. 2007;48:1746–53. doi: 10.1194/jlr.M700067-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Millar JS, Brousseau ME, Diffenderfer MR, Barrett PH, Welty FK, Cohn JS, et al. Effects of the cholesteryl ester transfer protein inhibitor torcetrapib on VLDL apolipoprotein E metabolism. J Lipid Res. 2008;49:543–9. doi: 10.1194/jlr.M700268-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Parhofer KG, Barrett PH. Thematic review series: patient-oriented research. What we have learned about VLDL and LDL metabolism from human kinetics studies. J Lipid Res. 2006;47:1620–30. doi: 10.1194/jlr.R600013-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parhofer KG, Hugh P, Barrett R, Bier DM, Schonfeld G. Determination of kinetic parameters of apolipoprotein B metabolism using amino acids labeled with stable isotopes. J Lipid Res. 1991;32:1311–23. [PubMed] [Google Scholar]
- 9.Watts GF, Ooi EM, Chan DC. Therapeutic regulation of apoB100 metabolism in insulin resistance in vivo. Pharmacol Ther. 2009;123:281–91. doi: 10.1016/j.pharmthera.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Cohn JS, Wagner DA, Cohn SD, Millar JS, Schaefer EJ. Measurement of very low density and low density lipoprotein apolipoprotein (Apo) B-100 and high density lipoprotein Apo A-I production in human subjects using deuterated leucine. Effect of fasting and feeding. J Clin Invest. 1990;85:804–11. doi: 10.1172/JCI114507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frischmann ME, Ikewaki K, Trenkwalder E, Lamina C, Dieplinger B, Soufi M, et al. In vivo stable-isotope kinetic study suggests intracellular assembly of lipoprotein (a) Atherosclerosis. 2012;225:322–7. doi: 10.1016/j.atherosclerosis.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 12.Thongtang N, Diffenderfer MR, Ooi EM, Asztalos BF, Dolnikowski GG, Lamon-Fava S, Schaefer EJ. Linkage between C-reactive protein and triglyceride-rich lipoprotein metabolism. Metabolism. 2013;62:369–75. doi: 10.1016/j.metabol.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasumov T, Ilchenko S, Li L, Rachdaoui N, Sadygov RG, Willard B, McCullough AJ, Previs S. Measuring protein dynthesis using metabolic 2H labeling, high-resolution mass spectrometry, and an algorithm. Anal Biochem. 2011;412:47–55. doi: 10.1016/j.ab.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Willard B, Rachdaoui N, Kirwin JP, Sadygov RG, Stanley WC, et al. Plasma proteome dynamic: analysis of lipoproteins and acute phase response proteins with 2H2O metabolic labeling. Mol Cell Proteomics. 2012;12:1–16. doi: 10.1074/mcp.M111.014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilechenko S, Previs SF, Rachdaoui N, Willard B, McCullough AJ, Kasumov T. An improved measurement of isotopic ratios by high resolution mass spectrometry. J Am Soc Mass Spectrom. 2013;24:309–12. doi: 10.1007/s13361-012-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee AY, Yates NA, Ichetovkin M, Deyanova E, Southwick K, Fisher TS, et al. Measurement of fractional synthetic rates of multiple protein analytes by triple quadrupole mass spectrometry. Clin Chem. 2012;58:619–27. doi: 10.1373/clinchem.2011.172429. [DOI] [PubMed] [Google Scholar]
- 17.Tomazela DM, Patterson BW, Hanson E, Spence KL, Kanion TB, Salinger DH, et al. Measurement of human surfactant protein-B turnover in vivo from tracheal aspirates using targeted proteomics. Anal Chem. 2010;82:2561–7. doi: 10.1021/ac1001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Castro-Perez J, Lassman ME, Thomas T, Li W, McLaughlin TM, et al. Measurement of apo(a) kinetics in humans a microfluidic device with tandem mass spectrometry. Rapid Comm Mass Spectrom. 2013;27:1294–302. doi: 10.1002/rcm.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh EJ, Shulman NJ, Dai DF, Vincow ES, Karunadharma PP, Pallanck L, et al. Topograph, a software platform for precursor enrichment corrected global protein turnover measurements. Mol Cell Proteomics. 2012;11:1468–74. doi: 10.1074/mcp.O112.017699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price JC, Holmes WE, Li KW, Floreani NA, Neese RA, Turner SM, Hellerstein MK. Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Anal Biochem. 2012;420:73–83. doi: 10.1016/j.ab.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Li W, Wang SP, Mendoza V, Rosa R, Hubert J, et al. Quantifying apoprotein synthesis in rodents: coupling LC-MS/MS analyses with the administration of labeled water. J Lipid Res. 2012;53:1223–31. doi: 10.1194/jlr.D021295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ackermann BL, Berna MJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev Proteomics. 2007;4:175–86. doi: 10.1586/14789450.4.2.175. [DOI] [PubMed] [Google Scholar]
- 23.Becker JO, Hoofnagle AN. Replacing immunoassays with tryptic digestion-peptide immunoaffinity enrichment and LC-MS/MS. Bioanalysis. 2012;4:281–90. doi: 10.4155/bio.11.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dufield DR, Radabaugh MR. Online immunoaffinity LC/MS/MS. A general method to increase sensitivity and specificity: how do you do it and what do you need? Methods. 2012;56:236–45. doi: 10.1016/j.ymeth.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, et al. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal Chem. 2003;75:4818–26. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 26.Razavi M, Frick LE, LaMarr WA, Pope ME, Miller CA, Anderson NL, Pearson TW. High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J Proteome Res. 2012;11:5642–9. doi: 10.1021/pr300652v. [DOI] [PubMed] [Google Scholar]
- 27.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics. 2010;9:184–96. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brousseau ME, Schaefer EJ, Wolfe ML, Bloedon LT, Digenio AG, Clark RW, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004;350:1505–15. doi: 10.1056/NEJMoa031766. [DOI] [PubMed] [Google Scholar]
- 29.Dubuc G, Tremblay M, Pare G, Jacques H, Hamelin J, Benjannet S, et al. A new method for measurement of total plasma PCSK9: clinical applications. J Lipid Res. 2010;51:140–9. doi: 10.1194/jlr.M900273-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, et al. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;7:22. doi: 10.1186/1476-511X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millar JS, Maugeais C, Ikewaki K, Kolansky DM, Barrett PH, Budreck EC, et al. Complete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 production. Arterioscler Thromb Vasc Biol. 2005;25:560–5. doi: 10.1161/01.ATV.0000155323.18856.a2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.