

Abstract
Chagas disease, also called American trypanosomiasis, is a parasitic disease caused by Trypanosoma cruzi (T. cruzi). Recent findings have underscored the abundance of the causative organism, (T. cruzi), especially in the southern tier states of the US and the risk burden for the rural farming communities there. Due to a lack of safe and effective drugs, there is an urgent need for novel therapeutic options for treating Chagas disease. We report here our first scientific effort to pursue a novel drug design for treating Chagas disease via the targeting of T. cruzi tubulin. First, the anti T. cruzi tubulin activities of five naphthoquinone derivatives were determined and correlated to their anti-trypanosomal activities. The correlation between the ligand activities against the T. cruzi organism and their tubulin inhibitory activities was very strong with a Pearson's r value of 0.88 ( P value < 0.05), indicating that this class of compounds could inhibit the activity of the trypanosome organism via T. cruzi tubulin polymerization inhibition. Subsequent molecular modeling studies were carried out to understand the mechanisms of the anti-tubulin activities, wherein, the homology model of T. cruzi tubulin dimer was generated and the putative binding site of naphthoquinone derivatives was predicted. The correlation coefficient for ligand anti-tubulin activities and their binding energies at the putative pocket was found to be r = 0.79, a high correlation efficiency that was not replicated in contiguous candidate pockets. The homology model of T. cruzi tubulin and the identification of its putative binding site lay a solid ground for further structure based drug design, including molecular docking and pharmacophore analysis. This study presents a new opportunity for designing potent and selective drugs for Chagas disease.
Keywords: Chagas disease, Trypanosoma cruzi, Naphthoquinone Derivatives, Tubulin Polymerization Inhibition, Homology Modeling, Binding Site Prediction, Structure Based Drug Design (SBDD)
Graphical abstract

1. INTRODUCTION
Chagas disease, also called American trypanosomiasis, which can affect both animals and humans, is caused by the protozoan parasite Trypanosoma cruzi. The disease is endemic in South America; however, due to population mobility, it is also significant in the US [1]. Recent reports indicate that of the Chagas disease cases tested on blood donors in Texas, 36% were acquired locally. Furthermore, 73% of tested triatomine insects from 11 counties in Texas were found to be positive for T. cruzi [2]. A separate study reported a T. cruzi infection rate of 8.8% in sheltered dogs in Texas [3]. Other studies have corroborated this abundance of T. cruzi in vectors and animal carriers in Texas [4], and the high correlation between human and dog infection (r2 = 0.955) [5]. These findings point to a high reservoir of T. cruzi in the Southern tier states of the US [6], and a likely underestimation of the prevalence of this disease in the US, including this region in particular.
Chagas disease is ranked as the third most widely spread tropical disease following malaria and schistosomiasis [7]. However, unlike malaria and schistosomiasis, both of which have safe and effective treatments, the drugs for Chagas disease, benznidazole and nifurtimox, are neither FDA approved nor safe or effective [8]. Chagas disease has two distinct clinical phases, an acute and a chronic phase. The acute phase occurs in the first two months of infection [8] and is diagnosed by a positive parasitological and molecular based test for the parasite T. cruzi. Once detected it can be cured by use of both of the drugs. After the acute infection, the disease enters into an asymptomatic chronic phase. The chronic phase is diagnosed by serological based immunoassay techniques and characterized by undetectable levels of the parasite. Of the chronic cases, 30% will develop the pathological conditions of the disease. The efficacy of the two drugs’ markedly diminish with the length of the chronic infection, more so in adults than in children seventeen years and younger [9]. In addition, these treatments cause adverse side effects in 40% of the infected which necessitate the physicians’ cessation of treatment [8]. Combined with the fact that the diagnosis of Chagas disease usually occurs years or even decades post onset of infection, it is crucial that safe and effective drugs be developed that can enable treatment at both the acute and chronic stages of the disease.
Naphthoquinone natural products comprise diverse structures known to possess a number of useful biological activities including: anti-viral, anti-fungal [10], anti-neoplastic [11], anti-hypoxic [12], anti-ischemic [13], anti-platelet, anti-inflammatory, and anti-allergenic activities [13] and have been investigated for their anti-trypanosomal activities [14]. A recent study showed that imido-substituted 1, 4-naphthoquinones are more potent anti-trypanosomal agents than the clinically used Nifurtimox by the arrest of T. cruzi cell proliferation at the G2/M phase [15]. Since the cell cycle arrest pointed to a tubulin disruptive process, we hypothesized that the anti-trypanosomal activities occurred via T. cruzi tubulin polymerization inhibition, an idea that provided the impetus to pursue a novel drug design for Chagas disease via the targeting of T. cruzi tubulin.
Microtubules are ubiquitous cellular polymers made up of two chains of tubulin dimers (α/β) that exhibit a characteristic dynamic instability. The dynamic instability is a state of perpetual stochastic reversions among periods of growth, shortening and pauses that imbue the microtubules with their cellular functionalities. For example, the spindle apparatus formed by microtubules is responsible for the separation of replicated chromosomes with high integrity into two daughter cells during mitosis. Hence, microtubules have been used as a validated target for anticancer chemotherapy [16]. By extension, targeting the parasite tubulin is a theoretically viable approach to the treatment of parasitic diseases. Indeed, the anti-protozoal activities of benzimidazoles have been correlated to β-tubulin sequences. The work of Chan et al. [18] sought to harness the anti-tubulin properties of certain herbicides to target trypanosomatid protozoans and the parasite Leishmania mexicana [19], while George et al. [20] followed the same approach for the treatment of the African trypanosomes. Targeting tubulin is one of the viable and promising techniques for anti-parasite drug design.
The research reported here is focused on bioassay and molecular modeling studies of tubulin polymerization inhibition by naphthoquinone derivatives.
2. METHODOLOGY
2.1 Bioassay
The anti T. cruzi tubulin activities of five newly synthesized ligands were determined by using biological analyses as stated below. The biologically determined anti T. cruzi tubulin activities were then correlated with their respective anti-trypanosomal activities.
2.1.1 Tubulin purification
The method of MacRae and Gull [21] was used for the purification of tubulin from T. cruzi. Briefly, PBS washed parasites were resuspended in 7 ml of PEME buffer (100mM PIPES, pH 6.9, 2mM EGTA, 1mM MgSO4, and 0.1 mM EDTA) supplemented with 4M glycerol, 0.1 mM GTP, and 50 μg/ml leupeptin. The resuspended parasites were sonicated 4 × 30 s, with 2 min cooling intervals between bursts (BRANSONIC 50/60 Hz, 80 watts) and centrifuged twice at 40, 000 g for 30 minutes at 4 °C (BECKMAN induction drive centrifuge model J2-21M). The resulting cell- free supernatant fraction was loaded into DEASE-Sephadex column (Sigma, DFF 100 DEAE-Sepharose) equilibrated with PEME buffer supplemented with 0.2 M KCl, 0.1 mM GTP, and 12.5 μ g/ml leupeptin. The column was washed with the equilibrium buffer after adding the sample and tubulin was eluted with PEME buffer with 0.6 M KCl, 0.1 mM GTP, and 12.5 μg/ml leupeptin. The peak eluted fractions were pooled and concentrated to 800-900 μl utilizing an Amicon Ultra-5 centrifugal filter device (MILLIPORE UFC9 030 08). Protein concentrations were determined using a BCA Protein Assay Kit (Pierce, 23225) according to the manufacturer's instructions using Bovine Serum Albumin (BSA) as a standard.
2.1.2 Polymerization Assay
T. cruzi tubulin polymerization assays were performed according to the procedures of Yakovich et al. [22]. Polymerization reactions were conducted in buffer containing 0.1M PIPES (pH 6.9), 1mM EGTA, 5mM MgCl2, 1.2 mg/ml purified T. cruzi tubulin, and different concentrations of the 3-chloro,2- imido-substituted 1,4- naphthoquinones (IMNDQ) (0.6-40 μM in 50% DMSO). Components of the reaction mixture were added to a 96-well microplate in a final volume of 50 μl, incubated at room temperature for 15 minutes, and then placed at 4 °C for 5 minutes. Polymerization was initiated by adding 1mM GTP, and assayed photometrically by measuring the absorbance at 405 nm using a microplate reader at 30 °C.
2.1.3 Statistical Analysis
All experiments were done in triplicate and the means and standard errors (S.E.) were determined. Data were analyzed by one way ANOVA using excel with the Analysis ToolPak add-in activated. P < 0.05 was considered significant.
2.2 Molecular Modeling
The host terminal computer used was equipped with Microsoft Windows 7 Professional OS and an Intel Xeon 3.40 GHz dual processor with 64.0 GB physical memory. All molecular modeling studies were performed with Molecular Operating Environment software (MOE 2013.0802) [23]. Default parameters were used unless specified otherwise.
2.2.1 Homology Modeling
The protein sequences of T. cruzi were mined from UniProtKB. The atom coordinates of the template protein 1JFF were downloaded from the RCSB Protein Data Bank. The homology modeling protocol entailed sequence alignment and matching model sequences with template sequences. The modeling was done with parameters selected to disable C-terminal and N-terminal outgap modeling, as well as the inclusion of selected ligand environments for induced fit and automatic detection of disulfide bonds. Protonate 3D was applied prior to energy minimization of the final model. Homology model validation was carried out by Phi-Psi plot of the model.
2.2.2 Binding sites discovery
The Site Finder algorithm built into MOE was used to calculate the possible binding sites in the model. Site Finder implements a geometric method where relative positions and accessibility of the receptor atoms are taken into account along with their chemical classifications. It is based upon finding alpha shapes, which generally identify regions of tight atomic packing; filtering alpha shapes that are exposed; classifying alpha shapes as hydrophobic or hydrophilic; clustering alpha spheres and rating the sites according to their probability for ligand binding [24].
2.2.3 Putative binding site prediction
The putative active site is defined as the binding pocket in which the binding energies of tested ligands correlate with the inhibition activities of the ligand in assay experiments. In order to identify the putative pocket for the IMDNQ (3-chloro,2- imido-substituted 1,4-naphthoquinones) ligands, five compounds: IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10 and IMDNQ11 were docked into the three pockets, respectively. The docking procedure was followed as recommended by MOE analysis using forcefield Amber:12 EHT. The receptor was prepared by modeling missing residues and protonated using Protonate3D. Tethered energy minimization was also carried out. To perform docking studies, ligands were converted into an MDB file format and the energy was minimized. The differing ligand conformations were generated by rotation of all rotatable bonds. Docking was done using the Triangle Matcher protocol. The forcefields used to refine poses were London dG for rescoring and GBVI/WSA dG for rescoring [23]. Duplicate poses were removed and 30 poses with lower energy were retained as best poses. When required, pharmacophore constraints were included to guide placement or to filter poses. The best poses were used in the evaluation of energy stabilization and ligand binding mode for the further calculation of correlation efficiency.
3. RESULTS AND DISCUSSION
3.1 Bioassay
As discussed in the introduction, certain naphthoquinones exhibit anti-trypanosomal activities. For example, the naturally occurring naphthoquinone lapachol and some of its derivatives show trypanocidal activity against T. cruzi [25]. Additionally, some naphthofuranquinones were found to be active against epimastigote and trypomastigote forms of T. cruzi [26]. Similarly, 2, 3-diphenyl-1, 4-naphthoquinone (DPNQ), was found to be effective against T. cruzi [14]. Previously, some novel imido-1, 4-naphthoquinone derivatives [15], were synthesized and investigated for anti-trypanosomal activities. Some of them showed remarkable anti-trypanosomal activity at G2/M of the cell cycle [15]. Since the cell cycle arrest pointed to tubulin disruptive processes, experiments were designed to evaluate activities of five selected imido-1, 4-naphthoquinone ligands against T. cruzi tubulin assembly (Table 1). The anti T. cruzi tubulin activities of the ligands IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10 and IMDNQ11 were determined and correlated with their anti-trypanosomal activities. As shown in Table 1, the previously determined anti-trypanosomal activities gave IC50s of 2.77, 4.83, 0.70, 2.23 and 6.10 μM, respectively. Similarly, their activities against T. cruzi tubulin polymerization gave IC50s of 6, 12, 3.2, 8, and >40 μM, respectively. Compound IMDNQ11 was the least potent and least selective. IMDNQ11 was included in the tubulin assembly inhibition experiments for comparative purposes. The most potent IMDNQ, IMDNQ3, has an anti-trypanosomal IC50 of 0.70 μM and was also determined to be the most active in inhibiting T. cruzi polymerization with an IC50 of 3.2 μM. Similarly, the least potent derivative, IMDNQ11 with an anti-trypanosomal IC50 of 6.10 μM exhibited the least anti-tubulin activity with an IC50 of >40 μM. IMDNQ1, IMDNQ2 and IMDNQ10 showed anti-tubulin IC50 values of 6, 12, and 8 μM, respectively. Fig. 1 shows that a correlation was established between anti-trypanosomal activities of the imido-1, 4-naphthoquinone derivatives and their activities toward T. cruzi tubulin inhibition, with a correlation coefficient of r = 0.88, P value = 0.55. The results show strong evidence that this class of compounds inhibits the trypanosome organism via T. cruzi tubulin polymerization inhibition.
Table 1.
Anti-trypanosomal and anti Trypanosoma cruzi tubulin activities of five imido-1, 4-naphthoquinone derivatives. Nifurtimox is included as an anti-trypanosomal activity reference.
| Molecule | Structure | Anti-trypanosome IC50 (μM) | Anti T. cruzi tubulin IC50 (μM) |
|---|---|---|---|
| Nifurtimox |
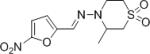
|
10.67 | N/A |
| IMDNQ1 |
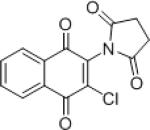
|
2.77 | 6 |
| IMDNQ2 |
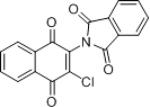
|
4.83 | 12 |
| IMDNQ3 |
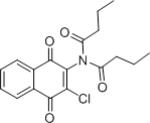
|
0.70 | 3.2 |
| IMDNQ10 |
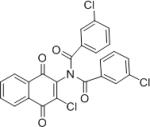
|
2.23 | 8 |
| IMDNQ11 |
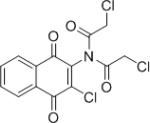
|
6.10 | >40 |
Figure 1.

The correlation coefficient of Anti-trypanosomal Log (IC50) and Anti Trypanosoma cruzi tubulin Log (IC50) is r =0.88.
3.2 Molecular modeling
The study of molecular modeling is indispensable in understanding the mechanism of inhibition of T. cruzi tubulin and the facilitation of designing lead compounds (drugs) for treating Chagas disease. To understand how the 2-imido-1, 4- naphthoquinone derivatives with anti-trypanosomal activities [27,28] inhibited T. cruzi tubulin, the 3D structure of the T. cruzi tubulin dimer was homology modeled, and subsequently, the modeled dimer was used in molecular docking studies to examine the nature of the receptor-ligand binding mode.
3.2.1 Sequence determination
In order to carry out homology modeling, the sequences of the target protein chains of T. cruzi tubulin (α chain and β chain) are required, as well as the 3D structure of the template homologue. Protein sequences of T. cruzi were mined from UniProtKB (www.uniprot.org), and are identified by their accession numbers. Fig. 2 shows the sequence alignment of alpha chains and beta chains from different database entries of T. cruzi tubulin. The amino acid lengths of the alpha chains, Q27352TBA is labeled as 1α; Q26973TBA labeled as 2α and Q8T9X5TBA labeled as 3α are 451, 425 and 451, respectively. The lengths of the beta chains, P08562 labeled as 1β; Q4DQP2 labeled as 2β and Q8STF3 labeled as 3β are all composed of 442 amino acids. Chain alignments gave a pairwise residue identity of 99.3% between 1α and 2α; 99.6% between 1α and 3α; and 98.8% between 2α and 3α. Residue variation of the alpha chains were found at positions 376 (CYS to SER) and 425 (LEU to TRP) for 2α and positions 215 (ARG to ALA) and 270 (SER to THR) for 3α. Similarly, the beta chains are highly conserved, with pairwise alignment giving values of residue conservation between 99.1 and 99.8%. The beta chains have residue variations at 274 on 1β, where SER replaces THR, and at 288 and 289, where SER and VAL are replaced by GLU and LEU, respectively, and finally, at 306 (ALA to ARG). Additionally, 3β has residue variation at 103 (LYS to GLN). All the variations identified in the α and β chains were either conserved or remotely located from the putative active site such that their effect(s) on ligand binding would remain largely insignificant. Consequently, our model of a 1α1β dimer was held as a representative dimer for the nine possible dimer combinations.
Figure 2.

Sequence alignment of Trypanosoma cruzi tubulin.
3.2.2 Homology modeling
The Bos taurus tubulin structure from the RCSB Protein Data Bank ID 1JFF is a homologue of T. cruzi tubulin, with 80% residue similarity and a well resolved 3D structure [29]. In addition it possesses a straight tubulin dimer structure similar to a functional microtubule unit, therefore, it is a good choice for a 3D template for modeling the target T. cruzi tubulin dimer. The sequence alignment of T. cruzi tubulin and Bos taurus tubulin gave a pairwise residue identity of 83.7% and 84.8% for the α and β chains, respectively. This high residue similarity attests to the appropriateness of the template for the generation of a good model because residue similarity is a good predictor of structure similarity [30].
Homology modeling was carried out using the aforementioned protocol in methodology. The resulting homology model of the target protein shows retained ligands in respective binding sites. The forcefield Amber12:EHT modules implemented in MOE with parameters enabled for bonded, van der Waals, electrostatic and restraints terms were loaded and used to prepare the template structure by modeling the missing residues on the alpha chain residues at 34-60, 440-450 and beta chain 427-445. This was followed by automatic protonation and tethered minimization with a deviation constraint of 0.5 Å for all atoms. Insertions and deletions were modeled from fragments of high resolution chains from the Protein Data Bank which superposes well onto anchor residues on either side of the insertion area [31]. Loops were modeled with their original substrates/ligands, GTP, GDP and taxol, retained to maintain the model environment. Ten intermediate models were generated and the average backbone coordinates of the models were determined. The root mean square deviation, RMSD, of each intermediate model to the average positions for all the intermediate models was used as criteria for selecting the working model. Hence, the final homology model was the one with the smallest RMSD following the defined criteria.
Fig. 3 shows the homology model of the T. cruzi tubulin dimer with the ligands GDP, GTP and taxol in the respective binding sites which have been retained for induced fit.
Figure 3.

Homology model of Trypanosoma cruzi tubulin dimer with substrates/ligands GDP (highlighted in green), GTP (highlighted in yellow) and taxol (highlighted in red) retained in their original respective binding sites.
Finally, the Phi-Psi dihedral angle plot was used to validate our final model. The Phi-Psi dihedral angle plot provides an independent method to evaluate the conformational quality of the protein structure and offers a visual representation of any dihedral angle outliers in the model structure [23]. The plots use a MOE reference database of a selected 129,858 residues derived from 556 protein structures. A Z-Score threshold of 4 (99.99%) was applied in which the favored regions are rendered green and the allowed but disfavored regions are rendered yellow while the outliers are rendered red. Fig. 4 is the result of the evaluation of the dihedral angles of the homology model and shows that the Phi-Psi plot cluster of the homology model is of high quality with minimal outliers.
Figure 4.

Model validation by Phi-Psi Plot showing that the Phi-Psi plot cluster of the model is of high quality with minimal outliers.
3.2.3 Pocket Discovery
The homology model of this study serves as a means to uncover candidate pockets for the binding of IMDNQ ligands to the target protein. Pocket discovery, carried out by the Site Finder algorithm in the MOE module, revealed three pockets between the tubulin α and β chains, as shown in Fig. 5. Pocket 1 is located on one side of the inter-dimer region, while pockets 2 and 3 are located at the other end diametrically opposite to the first pocket. Pockets 2 and 3 are partially overlapping and, therefore, upon docking produced similar energy profiles. All the candidate pockets lie between the chains and interact with residues of both chains.
Figure 5.

The modeled structure of the Trypanosoma cruzi tubulin dimer showing candidate binding pockets in between chains. Pocket 1 is highlighted in yellow, pocket 2 in green, and pocket 3 in red. Pocket 1 is the putative binding pocket for IMDNQ ligands.
In order to identify the putative pocket for the IMDNQ ligands, the three pockets were docked with the ligands IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10 and IMDNQ11. Again these ligands have known T. cruzi tubulin inhibition IC50s of 8, 12, 3.2, and >40 μM, respectively. The binding energy profiles for the ligands in the three pockets are shown in Fig. 6A. Correlations were calculated between the binding energies of the ligands in each of the pockets and the natural log of IC50, with pocket 1 showing a good correlation as shown in Fig. 6B. The putative active site is defined as the binding pocket which gives energy stabilizations that correlate with inhibition activities in assay experiments (Table 2). The calculations showed that the correlation coefficient between receptor-ligand stabilization energy and tubulin polymerization inhibition (log IC50) of pocket 1 is 0.79 while the other two contiguous binding pockets showed no correlation between binding energy and activity. Hence, pocket 1 was designated as the putative binding pocket for IMDNQ ligands bound in the T. cruzi tubulin. The high correlation coefficient of 0.79 at the putative binding pocket is remarkable and provides strong evidence that the site is involved in the inhibition of T. cruzi tubulin polymerization and, hence, the corollary inhibition of the T. cruzi organism.
Figure 6.

A. Energy profiles for ligands 1-5, representing IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10, and IMDNQ11 in Pockets 1, Pocket 2 and Pocket 3. B. Binding energy associated with Anti- Trypanosoma cruzi tubulin Ln (IC50) for the putative pocket (pocket 1).
Table 2.
Activities and Binding Energies of the validation ligands at the putative site.
| Molecule | Anti-Tubulin IC50 (μM) | Binding Energy in putative site (kcal/mole) |
|---|---|---|
| IMDNQ1 | 6 | 6.23 |
| IMDNQ2 | 12 | 6.21 |
| IMDNQ3 | 3.2 | 7.01 |
| IMDNQ10 | 8 | 6.74 |
| IMDNQ11 | >40 | 6.01 |
As shown in the close up view of the predicted putative binding site (Fig. 7), the loop of the sequences from α96 to α103 (highlighted in the purple ribbon) is located inside the pocket 1, right next to the bound GTP molecule. Therefore it has been shown that within this loop, residue 101 interacts with the GTP pocket on the N site of the α-chain while residues 97, 98, 100 and 102 interact with the IMDNQ molecules. This means that the putative binding site of the IMDNQ ligands (in the yellow shadow) share the loop on the alpha chain with the N site for GTP.
Figure 7.

The close up view of pocket 1, the predicted putative binding site. The loop of residues from 96 to 103 on the α chain is shown in the purple ribbon and the loop of residues from 251 to 263 on the β chain is shown in the green ribbon. The GTP molecule at the N site is also included as a light pink stick model.
Additionally, the loop of residues 251-263 on the β chain (highlighted as the green ribbon) in the IMDNQ binding site overlaps with the colchicine binding site (buried deep at the midsection of the inter-dimer region), shifted into the β chain side on the loop 248-255. This is very important and interesting information for the design of highly potent but low toxic drugs for the potential treatment of Chagas disease. As previously discussed, tubulin dynamics is a promising target for new chemotherapeutic agents and the colchicine binding site is one of the most important pockets for potential human tubulin polymerization destabilizers [32]. In a comparison of the T. cruzi tubulin model produced in this study with the Bos taurus tubulin protein structure (complexed with colchicine) obtained from RCSB PDB ID 1SAO [33], it is revealed that pocket 2 of this study's model coincides with the B. taurus colchicine binding site. Hence, the predicted putative binding site of naphthoquinone based ligands is different from the colchicine binding site which is implicated in the inhibition of human tubulin polymerization. An effective drug would typically attack the parasite and remain harmless to the host, therefore, an ideal approach to drug design would be to identify target proteins that are either present only in the parasite, or alternatively, have three dimensional structures which are sufficiently different from the corresponding host proteins. Therefore, the revelation of the differences between the putative binding site for the naphthoquinone based ligands and the colchicine binding site of the human tubulin polymerization inhibitors is a very important finding that presents a novel opportunity for ligand selectivity enhancement and reduction of toxicities of future drugs for Chagas disease.
The major interactions exhibited by IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10 and IMDNQ11 binding in the putative pocket consisted mainly of the donor-acceptor interactions of Arg156 on the β-chain and His406 on the α-chain with the imido side groups of the ligands, and the hydrophobic pi-pi interactions of the fused pi rings of the naphthoquinone moiety and the pi ring of the Trp407 residue on the α-chain. For example, Figure 8 shows the major interactions found in the binding mode of IMDNQ3 are two hydrogen bonds between one amide group of the ligand and the Arg156 hydrogen; one hydrogen bond between another amide group and the His406 hydrogen; and a pi-pi interaction between the naphthoquinone pi ring and the pi ring of Trp407. The optimization of the aforementioned major interactions and the general fit of the ligands in the binding pocket seem to explain the observed greater stabilization of the most active ligand IMDNQ3. Further investigation of these interactions will be carried out and reported in a subsequent publication.
Figure 8.

The interactions between the ligand IMNDQ3 and the putative binding pocket. The hydrogen bonds formed between amide groups of the ligand with the guanylyl hydrogens of Arg156 and the hydrogen of the imidazole group of His406 are highlighted as green lines. The pi-pi interactions between the pi rings of naphthoquinone moiety on the ligand and the pi ring of the indole group of Tryp407 is highlighted as a blue line.
4. CONCLUSIONS
The activities of five previously synthesized 3-chloro, 2-imido-1, 4-naphthoquinone ligands, namely, IMDNQ1, IMDNQ2, IMDNQ3, IMDNQ10 and IMDNQ11, against T. cruzi tubulin assembly were examined and determined. It was found that IMDNQ3 possessed the most potent anti-trypanosomal activity with an IC50 of 0.70 μM. IMDNQ3 was also determined to be the most active compound in inhibiting T. cruzi polymerization with an IC50 of 3.2 μM. Similarly, the least potent derivative, IMDNQ11, has an anti-trypanosomal IC50 of 6.10 μM and it exhibited the least anti-tubulin activity with an IC50 of >40 μM. A correlation of r = 0.88 ( P value < 0.05) was established between anti-trypanosomal activities of the imido-1, 4-naphthoquinone derivatives and their activity toward T. cruzi tubulin inhibition. These findings provide strong evidence that this class of compounds inhibit the trypanosome organism via T. cruzi tubulin polymerization inhibition.
In order to understand the mechanism of tubulin inhibition and use it for drug design, a homology model of the T. cruzi tubulin dimer was created and the putative binding site of this class of naphthoquinone derivatives was predicted. The correlation coefficient for ligand anti-tubulin activities and their binding energies at the putative pocket were evaluated and found to be strong (r = 0.79), a high correlation efficiency not replicated in other contiguous candidate pockets. Moreover, the relative locations of the IMDNQ binding site in T. cruzi tubulin and the colchicine binding site in human tubulin were described and compared, thereby providing important information useful in the design of drug selectivity.
In conclusion, the identification of the binding pocket is important because it enables elucidation of pharmacophore features which allows for the characterization of receptor-ligand complexes in the putative binding pocket. Additionally, the formulation of a pharmacophore query can be employed to design potent drug candidates or to search databases of ligands in order to identify candidate molecules for treatment of Chagas disease. The creation of a homology model of T. cruzi tubulin and the discovery of the putative binding site of the naphthoquinone based ligands provides a basis for pharmacophore feature analysis and selectivity enhancement of the drug candidates. This study opens a novel approach to the design of high potent, but low toxic drugs for the treatment of Chagas disease.
ACKNOWLEDGMENT
This work was supported by HUMAA (Howard University Medicine Alumni Association) Endowed Founder's Chair in Basic Science to Yayin Fang, grant #G12 MD007597 from NIMHD, NIH to the RCMI program at Howard University, grant #1208880 from NSF under the Howard University ADVANCE Institutional Transformation (HU ADVANCE-IT) Grant, grant #CHE-1126533 from NSF-MRI program and grant SF#0401723 from NSF under WBHRLSAMP Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bern C, Kjos S, Yabsley MJ, Montgomery SP. Clin. Microbiol. Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia NM, Hotez PJ, Murray KO. Parasites and Vectors. 2014;7:311. doi: 10.1186/1756-3305-7-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tenney TD, Curtis-Robles R, Snowden KF, Hamer SA. Emerg. Infect. Dis. 2014;20:1323–1326. doi: 10.3201/eid2008.131843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kjos S, Snowden KF, Olson KJ. Vector Borne Zoonotic Dis. 2009;9:41–50. doi: 10.1089/vbz.2008.0026. [DOI] [PubMed] [Google Scholar]
- 5.Estrada-Franco JG, Bhatia V, Diaz-Albiter H, Ochoa-Garcia L, Barbabosa A, Vazquez-Chagoyan JC, Martinez-Perez MA, Guzman-Bracho C, Garg N. Emerg. Infect. Dis. 2006;12:624–630. doi: 10.3201/eid1204.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bern C, Montgomery SP. Clin. Infect. Dis. 2009;49:52–54. doi: 10.1086/605091. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization Seventeenth programme report UNDP/TDR. 2005.
- 8.World Health Organization Chagas disease (american trypanosomiasis) 2014.
- 9.Sguassero Y, Cuesta CB, Roberts KN, Hicks E, Comandé D, Ciapponi A, Sosa-Estani S. PLoS One. 2015;10 doi: 10.1371/journal.pone.0139363. doi:10.1371/journal.pone.0139363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tandon VK, Singh RV, Tandon VK, Yadav D. Bioorg. Med. Chem. Lett. 2004;14:2901–2904. doi: 10.1016/j.bmcl.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 11.Gordaliza M, Miguel del Corral J, Angeles Castro M, Mar Mahiques M, García-Grávalos M, San Feliciano A. Bioorg. Med. Chem. Lett. 1996;6:1859–1864. doi: 10.1016/s0968-0896(97)10007-4. [DOI] [PubMed] [Google Scholar]
- 12.Kartoflitskaya AP, Stepanyuk GI, Yushkova VV, Marintsova NG, Novikov VP. Pharm. Chem. J. 1997;31:291–292. [Google Scholar]
- 13.Machado FS, Dutra WO, Esper L, Gollob K, Teixeira MM, Factor SM, Weiss LM, Nagajyothi F, Tanowitz HB, Garg NJ. Semin. Immunopathol. 2012;34:753–770. doi: 10.1007/s00281-012-0351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos EI, Garza KM, Krauth-Siegel RL, Bader J, Martinez LE, Maldonado RA. J. Parasitology. 2009;95:461–466. doi: 10.1645/GE-1686.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khraiwesh MH, Lee CM, Brandy Y, Akinboye ES, Berhe S, Gittens G, Abbas MM, Ampy FR, Ashraf M, Bakare O. Arch. Pharm. Res. 2012;35:27–33. doi: 10.1007/s12272-012-0103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumontet C, Jordan MA. Nat. Rev. Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katiyar SK, Gordon VR, McLaughlin GL. Antimicrob. Agents Chemother. 1994;38:2086–2090. doi: 10.1128/aac.38.9.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan MM, Grogl M, Chen CC, Bienen EJ, Fong D. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5657–5661. doi: 10.1073/pnas.90.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan MM, Tzeng J, Emge TJ, Ho CT, Fong D. Antimicrob. Agents Chemother. 1993;37:1909–1913. doi: 10.1128/aac.37.9.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George TG, Endeshaw MM, Morgan RE, Mahasenan KV, DelfÃn DA, Mukherjee MS, Yakovich AJ, Fotie J, Li C, Werbovetz KA. Bioorg. Med. Chem. 2007;15:6071–6079. doi: 10.1016/j.bmc.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacRae TH, Gull K. Biochem. J. 1990;265:87–93. doi: 10.1042/bj2650087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakovich AJ, Ragone FL, Alfonzo JD, Sackett DL, Werbovetz KA. Exp. Parasitol. 2006;114:289–296. doi: 10.1016/j.exppara.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chemical Computing Group Inc. 2013.
- 24.Soga S, Shirai H, Kobori M, Hirayama N. J. Chem. Inf. Model. 2007;47:400–406. doi: 10.1021/ci6002202. [DOI] [PubMed] [Google Scholar]
- 25.Salas C, Tapia RA, Ciudad K, Armstrong V, Orellana M, Kemmerling U, Ferreira J, Maya JD, Morello A. Biorg. Med. Chem. 2008;16:668–674. doi: 10.1016/j.bmc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 26.Silva RSF, Costa EM, Trindade RLT, Teixeira DV, Pinto Mde C, Santos GL, Malta VR, De Simone CA, Pinto AV, de Castro SL. Eur. J. Med. Chem. 2006;41:526–530. doi: 10.1016/j.ejmech.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Bakare O, Shendel CL, Peng H, Zalkow LH, Burgess E. Bioorg. Med. Chem. 2003;11:3165–3170. doi: 10.1016/s0968-0896(03)00267-0. [DOI] [PubMed] [Google Scholar]
- 28.Berhe S, Kanaan Y, Copeland RL, Jr., Wright DA, Zalkow LH, Bakare O. Lett. in Drug Des. Discov. 2008;5:485–488. [Google Scholar]
- 29.Lowe J, Li H, Downing KH, Nogales E. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 30.Chothia C, Lesk AM. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman HM, Westbrook J, Fend Z, Feng Z, Gilliland G. Nucleic Acid Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Y, Chen J, Xiao M, Li W, Miller DD. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]