

Abstract
The innate inflammatory response to Francisella tularensis (Ft) in macrophages is significantly governed by the expression of type I interferons (IFN). Previously, the proteolytic processing and maturation of pro-IL-1β protein was shown to depend upon type I IFN expression. We show in this report that paracrine type I IFN can profoundly enhance innate recognition and TLR-dependent transcriptional responses to Ft infection upstream of its role in inflammasome regulation in both primary human monocyte-derived macrophages and primary murine peritoneal macrophages, but not murine bone marrow-derived macrophages. This type I IFN-enhanced response is synergistic with TLR2 transcriptional responses, partially TLR2-independent, but strictly MyD88-dependent.
Keywords: IFN-β, Francisella tularensis LVS, macrophage, TLR2, type I Interferon
Introduction
Francisella tularensis (Ft) is a virulent human pathogen and the causative agent of the human disease tularemia. Ft is an aerobic Gram-negative coccobacillus, with a broad host range capable of infecting humans by a number of routes1. Cutaneous infection with Ft following a vector bite causes the ulcero-glandular form of the disease1–3. Alternatively, inhalation of fewer than 10 organisms can cause a pneumonic form of the disease, clinically associated with high rates of mortality1–3. The Ft Live Vaccine Strain (LVS) is attenuated in humans by an unknown mechanism, and was originally derived from a Type B strain (holarctica subspecies) that was previously used as a vaccine strain in the Soviet Union. However, Ft LVS remains fully virulent in mice by certain routes of infection and, as such, LVS has been extensively used in studies investigating immunological responses to Ft infection in the human and mouse models4.
As a pathogen, much of the success of Ft can be attributed to its capacity to avoid early detection by the host innate immune system. In humans, tularemia is characterized by an initial 3 to 5 day period in which the bacteria is immunologically silent, eliciting little to no inflammatory sequelae5. The failure to elicit significant inflammation early is likely due to the multiple molecular strategies employed by Ft to avoid innate detection within host cells such as macrophages and dendritic cells. While both humoral and cellular responses contribute to immunity to Ft, a significant portion of the Ft life cycle, including its replication, occurs in mononuclear phagocytes, and in this niche, Ft is largely protected from many aspects of the humoral defense6 placing an increased importance on cell-intrinsic innate detection.
The Toll Like Receptors (TLRs) represent an important sentinel system for bacterial infections, and work by multiple groups has shown that Ft subverts and evades detection by these receptors in a variety of ways. TLR4 is critical to the recognition of most Gram-negative pathogens, and recognition depends upon the productive interaction of a complex of cell surface TLR4/MD2 with bacterial lipopolysaccharide (LPS), the predominant outer membrane component of all Gram-negative bacteria7. Although Ft produces an LPS, Ft LPS does not behave as a TLR4 agonist and TLR4-deficient mice do not display enhanced susceptibility to Ft8, 9. The molecular basis for the failure of TLR4 to recognize Ft LPS likely resides in the structural peculiarities of Ft LPS, as it displays elongated acyl chains and is tetra-acylated10. Additionally, the Ft LPS lacks a critical phosphate moiety found in LPS from Gram-negative bacteria that activate TLR410. In addition to its ability to evade TLR4, Ft does not express flagellin, rendering Ft invisible to detection through TLR511.
Ft is, however, recognized during in vitro and in vivo infection of macrophages by TLR2, but the relevant TLR2 ligand expressed in vivo remains to be clarified12, 13. However, even the recognition of Ft by TLR2 is significantly less robust than observed for the interaction between peptidoglycan expressed by Gram-positive organisms and TLR2. Unlike the Gram-positive pathogens Staphylococcus and Streptococcus, heat-killed Ft is not recognized by TLR2, suggesting an unstable or rapidly turned over ligand12. TLR2 recognition and signaling also occurs in the phagosome following phagocytosis of the organism and Ft escapes rapidly from the phagosome, thus limiting TLR2 recognition14. It is not clear at present whether other phagosomal/endosomal TLRs participate in Ft recognition in vivo.
After phagosomal escape, Ft replicates in the cytosol where it is detected by at least two interrelated pattern recognition systems. Ft nucleic acids are recognized by the STING/MITA/MPYS receptor system that leads to synthesis and secretion of type I interferon (IFN)15, 16. Secreted Type I IFN, in turn, stimulates the macrophage through IFNAR signaling to increase cytosolic expression of the AIM2 Nod-like receptor (NLR) receptor that, upon recognition of Ft DNA, initiates assembly of the inflammasome and, subsequent to inflammasome activation, the proteolytic processing of IL-1β and IL-1817–19.
What effect type I IFN has in governing additional cell-intrinsic innate responses to Ft beyond inflammasome assembly is not known. We demonstrate here that IFN-β can dramatically enhance recognition of Ft and pro-inflammatory transcriptional responses in a manner that is partially TLR2-independent.
Materials and Methods
Cells and Mice
Primary thioglycollate-elicited peritoneal macrophages (TEPM) were prepared as described previously20. Briefly, 3 ml of 3% sterile thioglycollate (Remel) was injected i.p into 6–8 week old, wild-type (WT) C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) and, four days later, macrophages were harvested by peritoneal lavage with sterile saline. Human monocytes were isolated from whole blood by counter flow centrifugal elutriation from PBMC that were obtained from blood of healthy human volunteers at the Department of Transfusion Medicine, National Institutes of Health (kindly provided by Larry Wahl, NIDCR, NIH). Elutriated monocytes were plated for 2 h in serum-free medium (DMEM + 1% P/S + 1% l-glutamine). Human AB serum (Gemini Bio-Products, 2.5%) was added after 2 h. Cells were left to differentiate to human monocyte-derived macrophages (HMDM) for 7 days at 37° C in 5% CO2 in the absence of growth factors. The murine RAW 264.7 macrophage cell line (ATCC, Manassas, VA) was cultured in RPMI 1630 (Bio-Whittaker) supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. Murine bone marrow-derived murine macrophages (BMDM) were prepared as previously reported21.
Reagents
Antibodies against total p38, phospho-p38, total p65, and phospho-p65 were purchased from Cell Signaling (Danvers, MA). Anti-TLR2 antibodies used in Flow Cytometry analysis were purchased from Biolegend (San Diego, CA). S-[2,3-Bis(palmitoyloxy)-(2-RS)-propyl]- N-palmitoyl-(R)-Cys-Ser-Lys4-OH (Pam3Cys) was purchased from InVivogen (San Diego, CA). Purified, mammalian-expressed murine and human recombinant IFN-β (rIFN-β) was purchased from PBL (Piscataway, NJ).
Generation of Ft LVS stocks
Frozen aliquots of Ft LVS (American Type Culture Collection 29684) were prepared as described22. All Ft LVS were grown in Mueller-Hinton broth (MHB; BD Microbiology Systems, Franklin Lakes, NJ) supplemented with 2% IsoVitaleX (BD Biosciences, Franklin Lakes, NJ), 1.0% glucose, and 0.25% Ferric PPi (Sigma-Aldrich, St. Louis, MO); Mueller-Hinton agar (MHA) with 5% sheep blood (Teknova, Hollister, CA) was used as solid culture medium.
In vitro Ft LVS infection and quantitation of Ft LVS in macrophages
For experiments involving infection of murine TEPM, the RAW 264.7 cell line, or HMDMs, with Ft LVS, cells were primed by pre-treatment with PBS or recombinant IFN-β (100 U/ml) for 4 hours and subsequently infected with Ft LVS for 2 h, at an MOI of ~10 or ~50, as indicated in figure legends. The MOI for each experiment was determined retroactively by colony counts on MHA. After washing twice with medium, infected cells were incubated for 40 min in medium containing 50 μg/ml gentamicin to kill extracellular bacteria. Cells were washed three times with medium, and then the cells were re-incubated with medium only, or with medium supplemented with rIFN-β (100 U/ml). The addition of bacteria was defined as the zero time point.
For experiments involving determination of bacterial colony counts, at the indicated time points, cells were washed twice with ice-cold sterile saline (Baxter, Deerfield, IL) before being lysed in 1 ml of ice-cold 0.02% SDS (Bio-RAD) in sterile saline. Lysates were serially diluted and plated on MHA plates. Confirmation of the intracellular replication of Ft in macrophages utilizing Real Time PCR was accomplished as previously reported8. For experiments involving mRNA expression, macrophages were lysed at the indicated times by direct addition of Tri-Pure reagent (Roche, Indianapolis, IN) to dish.
Quantitative Real Time-PCR (qRT-PCR)
Total mRNA was isolated from peritoneal macrophages using TRIZOL (Invitrogen Carlsbad, CA) reagent according to manufacturer’s instructions. A total of 1 μg of RNA was utilized in oligo (dT) cDNA synthesis (Bio-Rad RT system, Hercules, CA). qRT-PCR was carried out using a an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Waltham, MA) utilizing SYBR Green reagent (Applied Biosystems) and transcript-specific primers (see8 for sequences). mRNA expression profiles were normalized to levels of housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) in each sample and the fold-change in expression was calculated by the 2−ΔΔCt method23.
Quantitation of Secreted Cytokines
Cytokine/chemokine levels in macrophage culture supernatants were analyzed by ELISA in the Cytokine Core Laboratory (University of Maryland, School of Medicine).
Western Blot Analysis
Whole cell lysates from primary murine macrophages were obtained by the addition of lysis buffer (20 mM HEPES, 1.0% TRITON X-100, 0.1% SDS, 150 mM NaCl, 10 mM NaF, 1 mM PMSF) and subsequent incubation at 4° C. Cell lysates were separated by electrophoresis in a denaturing SDS-PAGE gel and subsequent transfer to PVDF membrane. Blots were incubated overnight in relevant primary antibodies at 4° C and washed 3X with PBS, and then incubated with appropriate HRP-conjugated secondary antibody (Jackson Immunochemicals, West Grove, PA). Blots were developed following incubation in ECL PLUS Western Blotting Detection Reagent (Amersham Bioscience, Piscataway, NJ).
FACS Analysis
TEPM from WT C57BL/6 mice were cultured without or with recombinant murine IFN-β for 6 hours. Cells were then harvested, washed, and suspended in fluorescence-activated cell sorting (FACS) buffer and stained with phycoerythrin-labeled anti-mouse TLR2 or mouse IgG1 isotype control antibody (eBiocience, San Diego, CA) for 30 min on ice. The cells were washed, suspended in FACS buffer, and immediately analyzed using a FACSCalibur apparatus (BD Biosciences, Sunnyvale, CA) as described previously12.
Results
Type I Interferon Increases Macrophage Resistance to Ft
Innate immune signaling by Ft infection of macrophages has been shown to involve TLR2 leading to pro-inflammatory cytokine/chemokine production12, 24, the AIM2 inflammasome leading to IL-1β and IL-18 processing17, 18, STING, leading to IFN-β induction15, 16, and the autocrine/paracrine effects of these cytokines and type I IFNs on the infected macrophage. To assess the role that type I IFNs play in the regulation of the innate immune responses to Ft, we initially assessed the paracrine effects of IFN-β on Ft infectivity and bacterial cell killing in TEPM. TEPM from 6–8 week old WT C57BL/6J mice were primed with media alone or containing 100 U/ml of murine rIFN-β for 4 h prior to infection with a low MOI (aiming for MOI ~10, average of actual MOI = 9) of Ft. At 6 and 24 h post-infection, remaining macrophages were lysed and recoverable bacteria plated to determine colony counts (Figure 1A). Priming of macrophages with IFN-β had no effect on the initial infectivity in TEPM as essentially identical numbers of bacteria were recovered from the media-treated and IFN-β-primed macrophages (Figure 1A). At 24 h post-infection, however, we confirmed our previous observations of increasing numbers of Ft in media-treated macrophages, while macrophages primed with IFN-β suppressed the infection potently (Figure 1A)25. Equivalent initial uptake of Ft in media- and IFN-β-treated cell populations, even at an increased MOI of 50, was confirmed by RT-PCR analysis of the Ft LVS 16S rRNA levels in infected macrophages at 6 h post-infection, an assay that we have previously shown to correlate directly with viable bacteria (Figure 1B)8.
Figure 1. Type I Interferon does not enhance bacterial uptake but does increase macrophage killing of Ft.
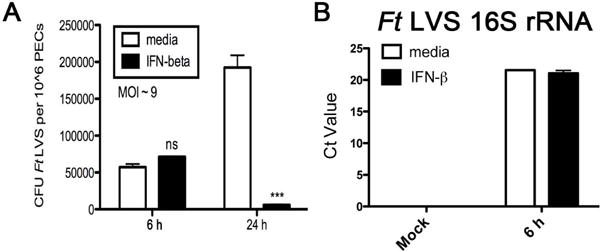
TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h prior to infection with Ft LVS at MOI = 10. (A) Recoverable colony counts of Ft LVS harvested from macrophages 6 or 24 hours p.i. (B) Ft LVS 16S rRNA levels in macrophages measured by RT-PCR 6 h p.i at MOI = 10 or 50. Results are represented as mean ± SEM from 3 experiments.
IFN-β Increases Macrophage Inflammatory Transcriptional Responses to Ft
Since exogenous rIFN-β treatment reduced the growth of Ft in TEPM dramatically (Figure 1), we hypothesized that IFN-β induced by Ft infection may confer enhanced recognition and/or a more robust early innate transcriptional responses to Ft. To test this hypothesis, TEPM from C57BL/6J mice were primed with media alone or rIFN-β for 4 h prior to infection with Ft LVS. At 6 h following infection, cytokine induction in TEPM was analyzed by qRT-PCR. Exposure to rIFN-β dramatically enhanced early induction of pro-inflammatory cytokines IL-1β, IL-6, and MIP2 mRNA (Figure 2A). Measurement of additional cytokines (e.g., TNF-α and KC) revealed an identical pattern of IFN-β-mediated enhancement (data not shown). To confirm that the observed effects of IFN-β on Ft-induced cytokine mRNA induction was not merely the result in a shift in the kinetics of induction, we assayed the levels of secreted cytokine protein by ELISA in the Ft-infected macrophage supernatants at 24 h post-infection (Figure 2B). In agreement with the data obtained at the mRNA level, secreted levels of IL-1β, IL-6, and KC were significantly elevated in the IFN-β-treated macrophages (Figure 2B). We also confirmed the ability of IFN-β to enhance inflammatory responses to Ft at the elevated MOI of 50 (Figure 2C). These data indicate that type I IFN can significantly enhance early innate steady-state mRNA responses to Ft.
Figure 2. IFN-β dramatically enhances early innate cytokine induction in response to Ft infection in macrophages.
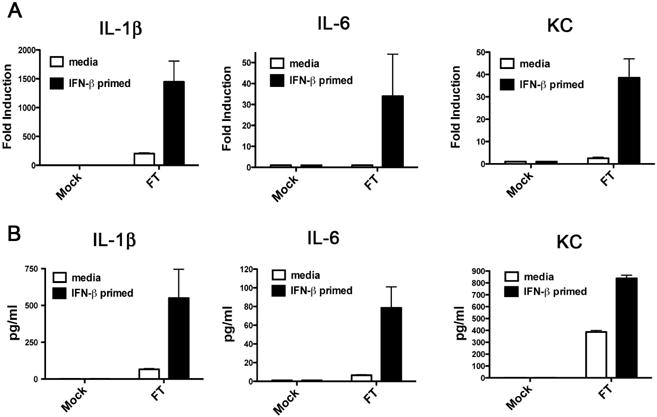
TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h prior to infection with Ft LVS at MOI of 10. (A) Levels of the indicated cytokine mRNA at 6 h p.i. as measured by RT-PCR. (B) Levels of indicated cytokine protein in supernatants at 24 h p.i. as measured by ELISA. (C) Macrophages were treated as in (A) and infected at an MOI of 50 prior to measurement of IL-6 mRNA. Results are represented as mean ± SEM from 3 experiments.
IFN-β Increases Inflammatory responses to Ft in human macrophages
As an enhanced innate transcriptional response to Ft conferred by type I IFN has not been described previously, we sought to demonstrate this effect in multiple macrophage and macrophage-like cell types. Murine BMDM are a widely used in vitro murine macrophage model, and we derived BMDM populations from C57BL/6J mice and performed an identical rIFN-β priming and Ft infection experiment as carried out with TEPM. Unexpectedly, murine BMDM did not display enhanced transcriptional responses to Ft at 6 hours (Figure 3A). A failure of rIFN-β to up-regulate inflammatory cytokine production in the BMDM was independently confirmed by cytokine protein analysis using ELISA (data not shown). To determine if the priming effects of rIFN-β were unique to the TEPM, we utilized the RAW 264.7 murine macrophage-like cell line. rIFN-β priming and Ft infection of RAW 264.7 cells revealed a similar pattern of up-regulated cytokine mRNA as observed in the TEPM, indicating that the priming effects of IFN-β are not an artifact of thioglycollate elicitation (Figure 3B). We additionally sought to investigate whether the observed relationship between Ft and IFN-β priming was species-specific. To accomplish this, we obtained elutriated human monocytes that were differentiated into HMDM in vitro over a 7 day period in the absence of exogenous growth factors, as described in the Methods. Primary HMDM were primed with media alone or human rIFN-β for 4 h, followed by infection with Ft for 6 h, followed by analysis of cytokine mRNA levels by qRT-PCR. Consistent with the data obtained with the TEPM and RAW 264.7 cell line, primary HMDM displayed remarkably elevated cytokine mRNA responses to Ft infection following IFN-β exposure (Figure 3C). Equivalent initial uptake of Ft in each cell type was confirmed by 16s rRNA analysis at 6 hours post-infection (Figure 3D).
Figure 3. IFN-β regulates innate cytokine induction in RAW 264.7 cells and HMDM, but not murine BMDM.
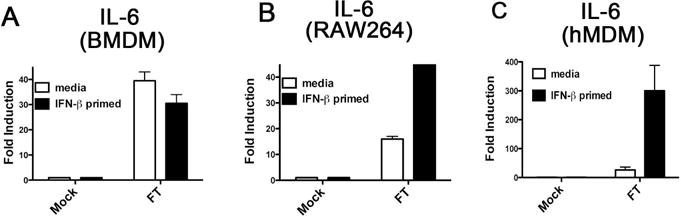
(A) Murine BMDM were differentiated in 12 well plates and treated with media alone or 100 U/ml rIFN-β for 4 h. Subsequent to treatment, BMDM were infected with Ft LVS at an MOI of 13 for 6 h. Total RNA was harvested and used for RT-PCR analysis. (B) RAW 264.7 cells were plated in 12 well plates and treated with media alone or 100 U/ml rIFN-β for 4 h. Subsequent to treatment, RAW 264.7 cells were infected with Ft LVS at an MOI of 13 for 6 h. Total RNA was harvested and used in RT-PCR analysis. (C) HMDM were derived as described in Methods and plated in 6 well plates and treated with media alone or 100 U/ml human rIFN-β for 4 h. Subsequent to treatment, HMDM were infected with Ft LVS at an MOI of 13 for 6 h. Total RNA was harvested and used in RT PCR analysis. (D) RNA from samples treated as in (A–C) were analyzed for Ft 16S rRNA by RT PCR. Results are represented as mean ± SEM from 3 experiments.
IFN-β Increases Innate Immune Signaling in response to Ft
As previous literature has demonstrated an absolute dependence upon TLR2 for innate recognition of Ft by macrophages12, 24, we began to elucidate the mechanism of IFN-β enhanced responses by examining common TLR-regulated signal transduction pathways in TEPM during Ft infection, without or with prior exposure to rIFN-β. It is well-documented that TLR2 ligation induces concomitant activation of both the MAPK and NF-κB signal transduction pathways that, in turn, lead to transcriptional induction of the majority of the pro-inflammatory cytokines26. We therefore reasoned that if rIFN-β were enhancing TLR responses generally, we would observe enhanced Ft-induced signal transduction following IFN-β exposure. We performed Western blot analysis directed against the phosphorylated form of the p38 MAPK (P-p38), as well as against the Ser-536 phosphorylated form of the p65 NF-κB (P-p65) transcription factor. At 6 h post-infection, we observed dramatically enhanced phosphorylation of both p38 MAPK and the NF-κB p65 proteins in macrophages that had been exposed to rIFN-β (Figure 4). These data suggest that IFN-β may be acting to enhance TLR2-dependent signaling.
Figure 4. rIFN-β enhances innate signal transduction in response to Ft LVS infection of murine peritoneal macrophages.
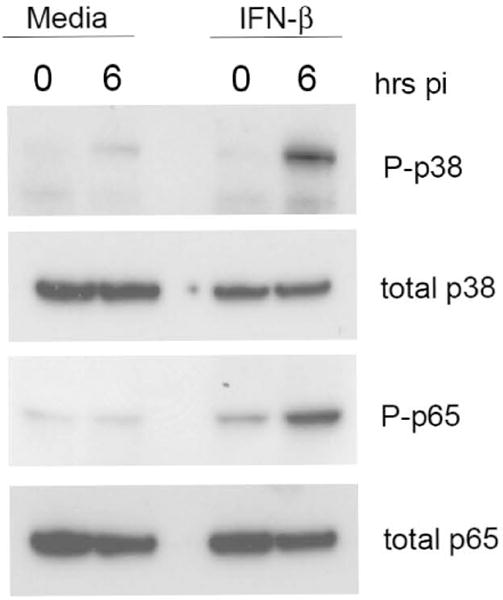
TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h prior to infection with Ft LVS at MOI of 10. Six h post-infection, total cell lysates were harvested for Western blot and probed with antibodies against the indicated proteins.
IFN-β does not generally enhance TLR2 responses
In particular, TLR2/6 has been shown to be both necessary and sufficient for innate recognition of Ft as well as for inflammatory cytokine production during Ft infection in vitro24. Our preceding experiments, therefore, led us to speculate that Type I IFN may be augmenting TLR2-dependent responses to Ft. We initially assayed the potential for rIFN-β to modulate levels of TLR2 mRNA. Following 6 h of IFN-β exposure, TEPM failed to exhibit any appreciable difference in the steady-state levels of TLR2 mRNA by qRT-PCR (Figure 5A). As it remained formally possible that IFN-β might be modulating the cell surface expression of TLR2 independently of any effects on TLR2 transcription, we assessed TLR2 cell surface staining by flow cytometry. Following 6 h of priming with murine rIFN-β, TEPM were fixed and TLR2 expression measured by flow cytometry. In agreement with our data showing no effect of IFN-β on TLR2 steady-state mRNA, we failed to observe a significant increase in surface TLR2 expression following rIFN-β treatment (Figure 5B). Quantitation of the mean fluorescent intensities of TLR2 staining from the experiment in Figure 5B are presented in Figure 5C and confirmed that IFN-β-priming failed to affect TLR2 expression.
Figure 5. rIFN-β does not enhance TLR2 expression or cytokine gene induction in response to purified TLR2 ligands.
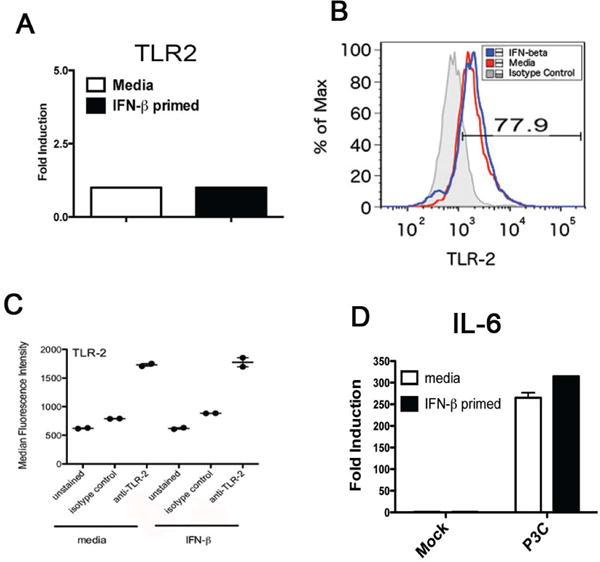
TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h. (A) Total RNA from media and rIFN-β-treated macrophages was harvested and used in RT-PCR analysis of steady-state TLR2 transcript levels. (B) Media and rIFN-β-treated macrophages were fixed and stained for TLR2 surface expression by flow cytometry. (C) Quantitation of mean fluorescence intensity from samples analyzed in (B). (D and E) Media- and rIFN-β pre-treated macrophages were stimulated for 3 h with the indicated dose of TLR2 ligand P3C (D) or P2C (E). Following stimulation total RNA was harvested and used for RT PCR analysis of IL-6 or TNFα transcript levels. Results are represented as mean ± SEM from 3 experiments. (F) Media and rIFN-β-treated macrophages were stimulated with 250 ng/ml P3C for indicated times and cell lysates probed utilizing indicated antibodies.
Type I IFN is known to regulate the abundance of select innate signal transduction elements and transcription factors27. It was therefore possible that IFN-β may modulate the strength of signaling induced by TLR2 without affecting receptor levels directly. To exclude this possibility formally, C57BL/6J TEPM were pre-treated with media or rIFN-β for 4 h prior to stimulation with purified synthetic TLR2 ligands for an additional 3 h. The tri-acylated lipopeptide Pam3Cys (P3C) and di-acylated lipopeptide Pam2Cys (P2C) are widely utilized and extensively characterized ligand for the TLR2/1 and TLR2/6 receptor heterodimers, respectively28, and were used to stimulate macrophages. Pam3Cys and Pam2Cys treatment elicited robust cytokine transcriptional induction in the TEPM, but did not exhibit enhanced cytokine induction following IFN-β exposure (Figure 5D and 5E). Finally, as we have shown that IFN-β exposure leads to increased innate immune signaling following Ft infection (Figure 4), we examined MAPK signaling in response to P3C without or with IFN-β exposure. We observed no effect of IFN-β on P3C-induced p38 activation (Figure 5F), in agreement with the gene induction data.
IFN-β Enhanced Responses to Ft are MyD88-Dependent and Partially TLR2-Independent
As we eliminated a general enhancement of TLR2-dependent responses as a mechanism for increased innate recognition of responsiveness to Ft in IFN-β-treated macrophages, we sought to define more precisely requirements for IFN-β priming using a genetic approach. TEPM were obtained from WT C57BL/6J and background-matched MyD88−/− mice, and were treated either with media alone or murine rIFN-β for 4 h, followed by Ft infection for 6 h. We observed a strict MyD88 dependence for induction of MIP2 and TNF-α mRNA in both the media- and rIFN-β-primed conditions (Figure 6A). These data indicate that IFN-β-enhanced Ft responses are mediated via a MyD88-dependent mechanism.
Figure 6. rIFN-β-mediated enhanced recognition of Ft LVS is fully MyD88-dependent, but partially TLR2-independent.
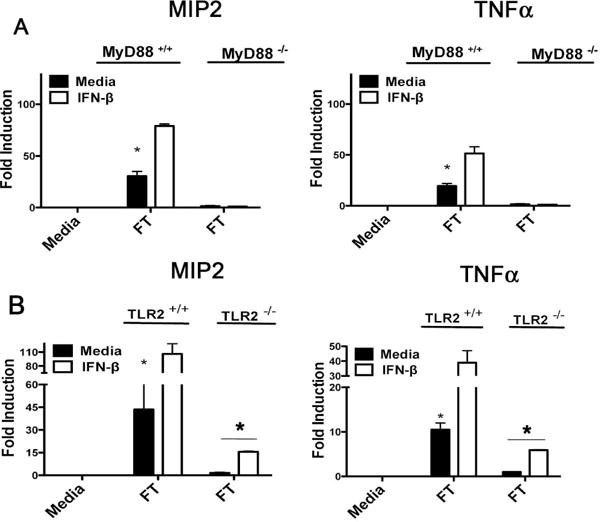
(A) WT C57BL6/J and MyD88−/− TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h prior to infection wit Ft LVS at MOI of 10 for 6 h. Total RNA was harvested from control and infected cells and used in RT-PCR analysis of the indicate cytokines. (B) WT C57BL6/J and TLR2−/− TEPM were treated either with media alone or 100 U/ml rIFN-β for 4 h prior to infection wit Ft LVS for 6 h. Total RNA was harvested from control and infected cells and used in RT-PCR analysis of the indicated cytokine genes. Results are represented as mean ± SEM from 3 experiments.
To distinguish the TLR2-dependent contributions to Ft-mediated cytokine induction from the unknown IFN-β-regulated pathway, we obtained TEPM from WT and TLR2−/− mice. Ft infection of TLR2−/− macrophages, revealed a complete loss of cytokine induction in media-treated TLR2−/− macrophages, in agreement with the previous literature12, 24. Surprisingly, we observed a partial restoration of Ft-dependent cytokine induction in TLR2−/− macrophages following exposure to IFN-β (Figure 6B). However, as the total enhancement of cytokine induction in the presence of both type I IFN and TLR2 expression was significantly beyond the additive effects of either one alone (Figure 6), we propose that the IFN-β governed pathway works synergistically with TLR2 expression to drive transcription from inflammatory promoters.
These data indicate that IFN-β can enhance innate responses to Ft even in the absence of TLR2 expression, and likely regulates the sensitivity of a TLR2-independent pathway for the detection of Ft.
Discussion
Ft remains a relevant and dangerous human pathogen due to its extreme virulence at low infectious doses. In the absence of an effective approved vaccine, a greater understanding of the interaction between Ft and the host innate immune system is vital. Prior work has shown that the success of Ft as a pathogen is intimately related to the capacity of the organism to remain immunologically “silent” during the critical initial days following infection, effectively defeating the surveillance of multiple innate immune sensing systems29. To date, only TLR2 has been shown to be necessary and sufficient for initiation of an inflammatory cytokine response to Ft in macrophages. It should be noted, however, that the vast majority of these studies have been carried out in in vitro macrophage models of infection that may not reflect the full cytokine milieu found in vivo. Notably, several in vivo models of Ft infection revealed that despite an absolute requirement for TLR2 to recognize Ft in cell culture, TLR2-deficient animals did not display the predicted extreme susceptibility that is observed in Ft-infected MyD88−/− mice9. These observations lead to speculation of possible secondary Ft-sensing systems that operate in vivo. Our current work suggests the possibility that the induced production of type I IFN could potentially expand the repertoire of innate receptors capable of responding to Ft and thus provide for a partial in vivo redundancy, as well as synergy, with TLR2. Such an expansion/enhancement of TLR-dependent inflammation would only occur after an initial round of in vivo replication, once Ft had escaped the phagosome and triggered induction of type I IFN from the cytosol25. It is also notable that in vivo, Francisella novicida pathology after lung infection is closely associated with a “cytokine storm” similar to that associated with sepsis30, 31. Type I IFN-mediated enhancement of recognition of Ft could potentially contribute to the amplification of hypercytokinemia and, thus, morbidity and mortality. A model such as this is in concurrence with recent data demonstrating that the IFNAR−/− mice display significantly reduced mortality following intranasal infection with Francisella novicida32.
An unanswered question emerging from our study is the precise molecular mechanism of the increased recognition and signal transduction following Ft infection of IFN-β-treated macrophages. We show that following IFN-β exposure, Ft-driven cytokine production remains exclusively dependent upon expression of the adaptor protein, MyD88 (Figure 6A). This suggests the possibility that type I IFN is licensing recognition of Ft by additional MyD88-dependent Pattern Recognition Receptors (PRRs). While a role for IL-1-mediated feedback through the MyD88-dependent IL-1R cannot be formally excluded in the IFN-β-enhanced response, this appears unlikely as the enhanced cytokine transcription is seen within the first 4–6 h following infection of macrophages and prior to AIM2 inflammasome assembly and IL-1β processing25. One possible model is that IFN-β enhances recognition of Ft by endosomal TLRs, possibly as a result of the capacity of IFN-β to up-regulate transcription of the endosomal nucleic acid receptors TLR3 and TLR933. IFN-β may also enhance bacteriolysis and presentation of nucleic acid ligands to endosomal TLRs, either through increased phagosomal maturation and acidification, or via the induced expression of proteins such as the small interferon inducible GTPases (GBPs) that have been reported to play a role in disruption of intracellular bacteria34, 35. While we have provided data for representative cytokines as read-outs for each experiment, we did not observe a selectivity in the enhanced expression of any individual cytokine that would suggest a particular innate signal transduction cascade is being augmented.
An additional consideration related to mechanism is the possibility that distinct cell types may rely on differing pattern recognition systems to respond to Ft. Our work has been carried out in macrophages, a dedicated innate immune cell type, however it has been shown that Ft can infect many cell types, including different types of macrophages, in the pulmonary environment6, 36. Whether, for example, structural cells display the same relationship with type I interferon and Ft will remain to be investigated.
Another interesting point related to molecular mechanism is our failure to observe IFN-β-mediated enhancement in the murine BMDMs. While BMDMs are the preferred macrophage model for many researchers, the use of high doses of recombinant cytokines or conditioned media to derive the BMDM ex vivo may influence the steady-levels of some IFN-responsive genes37, and therefore, the differentiation state of the macrophage, presumably by inducing type I IFNs that act in an autocrine/paracrine manner38, 39. Regardless, our demonstration that type I IFN significantly enhances cytokine responses to Ft in thioglycollate-elicited primary macrophages, the RAW 264.7 macrophage cell line, as well as primary HMDM, clearly indicates that the observed effects of IFN-β are likely not an artifact of thioglycollate elicitation. Further work is ongoing to characterize the relevant differences in these cell types; however, we observed no global difference in responses to purified TLR2 or TLR4 ligands in TEPM vs. BMDM (data not shown).
In summary, this work highlights the importance of the emerging complex relationship between type I IFNs, long believed to be relevant only to viral infections, and select bacterial pathogens. In particular, it suggests that type I IFNs may play a role in tuning the sensitivity of innate PRR systems to bacterial infections in vivo.
Acknowledgments
This work was supported in part by NIH R56 AI-18797 (SNV) and NIH T32 AI095190.
Abbreviations
- Ft
Francisella tularensis
- LVS
Live Vaccine Strain
- LPS
Lipopolysaccharide
- IFN-β
Interferon-beta
- IFNAR
Interferon-α/β Receptor
- TEPM
Thioglycollate-elicited peritoneal macrophages
- BMDM
bone marrow derived macrophages
- MDM
monocyte-derived macrophages
- STING
stimulator of interferon genes
Footnotes
Disclosures
The authors declare no conflicts of interest
Authorship
K.R., S.N.V., and D.J.P. carried out study design ; K.R. and D.J.P performed experiments; K.R., S.N.V. and D.J.P prepared the manuscript.
References
- 1.McLendon MK, Apicella MA, Allen LA. Francisella tularensis: taxonomy, genetics, and Immunopathogenesis of a potential agent of biowarfare. Annual review of microbiology. 2006;60:167–85. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. Tularemia vaccine study. II. Respiratory challenge. Archives of internal medicine. 1961;107:702–14. doi: 10.1001/archinte.1961.03620050068007. [DOI] [PubMed] [Google Scholar]
- 3.Saslaw S, Eigelsbach HT, Wilson HE, Prior JA, Carhart S. Tularemia vaccine study. I. Intracutaneous challenge. Archives of internal medicine. 1961;107:689–701. doi: 10.1001/archinte.1961.03620050055006. [DOI] [PubMed] [Google Scholar]
- 4.Sjostedt A. Virulence determinants and protective antigens of Francisella tularensis. Current opinion in microbiology. 2003;6:66–71. doi: 10.1016/s1369-5274(03)00002-x. [DOI] [PubMed] [Google Scholar]
- 5.Sjostedt A. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Annals of the New York Academy of Sciences. 2007;1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 6.Hall JD, Woolard MD, Gunn BM, et al. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infection and immunity. 2008;76:5843–52. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. Journal of endotoxin research. 2006;12:133–50. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 8.Cole LE, Elkins KL, Michalek SM, et al. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. Journal of immunology. 2006;176:6888–99. doi: 10.4049/jimmunol.176.11.6888. [DOI] [PubMed] [Google Scholar]
- 9.Collazo CM, Sher A, Meierovics AI, Elkins KL. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes and infection/Institut Pasteur. 2006;8:779–90. doi: 10.1016/j.micinf.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Gunn JS, Ernst RK. The structure and function of Francisella lipopolysaccharide. Annals of the New York Academy of Sciences. 2007;1105:202–18. doi: 10.1196/annals.1409.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. Journal of leukocyte biology. 2006;80:766–73. doi: 10.1189/jlb.0406294. [DOI] [PubMed] [Google Scholar]
- 12.Cole LE, Shirey KA, Barry E, et al. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infection and immunity. 2007;75:4127–37. doi: 10.1128/IAI.01868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infection and immunity. 2006;74:3657–62. doi: 10.1128/IAI.02030-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole LE, Laird MH, Seekatz A, et al. Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. Journal of leukocyte biology. 2010;87:275–81. doi: 10.1189/jlb.0909619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin L, Hill KK, Filak H, et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. Journal of immunology. 2011;187:2595–601. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. Journal of immunology. 2015;194:3236–45. doi: 10.4049/jimmunol.1402764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature immunology. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones JW, Kayagaki N, Broz P, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9771–6. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang TJ, Basu S, Zhang L, et al. Bacillus anthracis spores and lethal toxin induce IL-1beta via functionally distinct signaling pathways. European journal of immunology. 2008;38:1574–84. doi: 10.1002/eji.200838141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobrovolskaia MA, Medvedev AE, Thomas KE, et al. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. Journal of immunology. 2003;170:508–19. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 21.Pennini ME, Perkins DJ, Salazar AM, Lipsky M, Vogel SN. Complete dependence on IRAK4 kinase activity in TLR2, but not TLR4, signaling pathways underlies decreased cytokine production and increased susceptibility to Streptococcus pneumoniae infection in IRAK4 kinase-inactive mice. Journal of immunology. 2013;190:307–16. doi: 10.4049/jimmunol.1201644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elkins KL, Winegar RK, Nacy CA, Fortier AH. Introduction of Francisella tularensis at skin sites induces resistance to infection and generation of protective immunity. Microbial pathogenesis. 1992;13:417–21. doi: 10.1016/0882-4010(92)90085-3. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infection and immunity. 2006;74:2809–16. doi: 10.1128/IAI.74.5.2809-2816.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole LE, Santiago A, Barry E, et al. Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. Journal of immunology. 2008;180:6885–91. doi: 10.4049/jimmunol.180.10.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Schoggins JW, Wilson SJ, Panis M, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 472:481–5. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mistry P, Laird MH, Schwarz RS, et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:5455–60. doi: 10.1073/pnas.1422576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones CL, Napier BA, Sampson TR, Llewellyn AC, Schroeder MR, Weiss DS. Subversion of host recognition and defense systems by Francisella spp. Microbiology and molecular biology reviews: MMBR. 2012;76:383–404. doi: 10.1128/MMBR.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infection and immunity. 2008;76:3001–10. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma J, Li Q, Mishra BB, Pena C, Teale JM. Lethal pulmonary infection with Francisella novicida is associated with severe sepsis. Journal of leukocyte biology. 2009;86:491–504. doi: 10.1189/jlb.1208728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henry T, Kirimanjeswara GS, Ruby T, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. Journal of immunology. 2010;184:3755–67. doi: 10.4049/jimmunol.0902065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins DJ, Rajaiah R, Tennant SM, et al. Salmonella Typhimurium Co-Opts the Host Type I IFN System To Restrict Macrophage Innate Immune Transcriptional Responses Selectively. Journal of immunology. 2015;195:2461–71. doi: 10.4049/jimmunol.1500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meunier E, Wallet P, Dreier RF, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nature immunology. 2015;16:476–84. doi: 10.1038/ni.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pilla DM, Hagar JA, Haldar AK, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6046–51. doi: 10.1073/pnas.1321700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts LM, Tuladhar S, Steele SP, et al. Identification of early interactions between Francisella and the host. Infection and immunity. 2014;82:2504–10. doi: 10.1128/IAI.01654-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. Journal of leukocyte biology. 2009;86:411–21. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- 38.Moore RN, Pitruzzello FJ, Robinson RM, Rouse BT. Interferon produced endogenously in response to CSF-1 augments the functional differentiation of progeny macrophages. Journal of leukocyte biology. 1985;37:659–64. doi: 10.1002/jlb.37.5.659. [DOI] [PubMed] [Google Scholar]
- 39.Warren MK, Vogel SN. Bone marrow-derived macrophages: development and regulation of differentiation markers by colony-stimulating factor and interferons. Journal of immunology. 1985;134:982–9. [PubMed] [Google Scholar]