

Abstract
Nonalcoholic fatty liver disease represents a wide spectrum of conditions and is currently the most common form of chronic liver disease affecting both adults and children in the United States and many other parts of the world. Great effort has been focused on the development of novel therapies for those patients with the more advanced forms of the disease, in particular those with nonalcoholic steatohepatitis (NASH) and liver fibrosis that can be associated with significant morbidity and mortality. In this review, the authors focus on the role of cell death and sterile inflammatory pathways as well as the self-perpetuating deleterious cycle they may trigger as novel therapeutic targets for the treatment of fibrotic NASH.
Keywords: liver disease, cell death, danger-associated molecular patterns, pattern recognition receptors, inflammation, therapy, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis
Nonalcoholic fatty liver disease (NAFLD) has become one of the most common causes of chronic liver disease worldwide. Estimates of NAFLD prevalence based on “cryptogenic” abnormal liver function, autopsy samples, and findings from ultrasonography and magnetic resonance spectroscopy are 3% to 37%, with the usual figure quoted at approximately 30% (reviewed elsewhere[1] [2]). The spectrum of NAFLD includes isolated steatosis and nonalcoholic steatohepatitis (NASH). Although patients with isolated steatosis appear to have a benign nonprogressive clinical course, those with NASH, characterized by steatosis along with hepatocellular injury, inflammation, and varying degrees of fibrosis[3] may have a potentially serious condition.[4] [5] Among these patients, those with liver fibrosis (stage 2 or higher) appear to be the ones at higher risk of overall and liver-related morbidity and mortality.[6] With NAFLD becoming increasingly common in the developed world over the last decade, NASH demonstrated the greatest increase as a cause of chronic liver disease among new liver-transplant waitlist registrations, increasing almost twofold and becoming the second leading etiology of liver disease among new liver transplant waitlist registrations in 2013.[7]
The clinical importance of NAFLD and the current lack of effective medications to stop or reverse disease progression in patients with NASH have sparked great interest and intense investigation to identify relevant pathophysiologic mechanisms that can be the target for the development of novel therapies. The current and most accepted concept outlining the pathogenesis of NAFLD involves multiple “hits.”[8] These hits are characterized by the occurrence of parallel and sequential events that are the result of a complex interaction between environmental factors, host genetics, and gut microflora and involve both intrahepatic and extrahepatic pathways.[9] [10] This interaction might promote isolated steatosis, innate immune activation, inflammation, cell death, or fibrosis with progressive liver damage.[8] Current pharmacotherapy efforts toward NASH can be largely divided into those with a predominant metabolic, antisteatotic effect such as insulin sensitizers and nuclear receptor modulators, and those with a direct anti-inflammatory, hepatoprotective effect. In this review, we focus on the latter. We present new insights into the relevance of various cell death pathways, sterile inflammation, and the crosstalk between them as key mechanisms in NASH pathobiology and progression, as well as discuss the evolving therapies that are either being tested or have significant potential for the treatment of NASH in patients affected with the more severe forms of this condition.
Increased Cell Death and Activation of Sterile Inflammatory Pathways as a Key Self-Perpetuating Loop Involved in Liver Injury and Fibrosis in NASH
Although several of the early triggers of hepatic steatosis can be traced to events that occur outside the liver in distant organs such as the gut, adipose tissue, and muscle among others, excessive hepatocyte cell death by apoptosis, necrosis, and other forms of cell death (see below) followed by the release of danger or stressed signals by these hepatocytes, and activation of sterile inflammatory pathways can initiate an intrahepatic, self-perpetuating noxious loop that results in chronic injury and fibrosis as an intrinsic response to this damage that can eventually progress to excessive scarring and liver failure ([Fig. 1]).[11] [12]
Fig. 1.
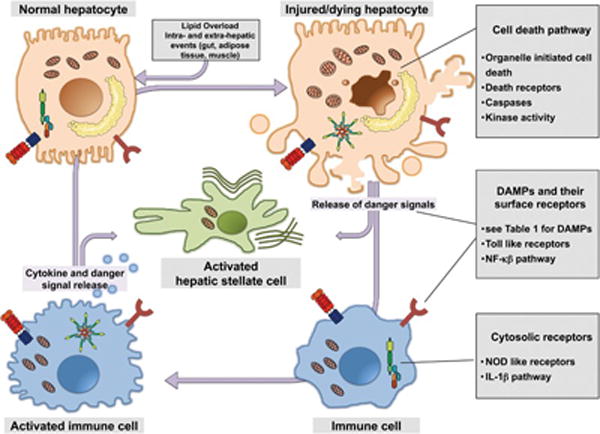
The intrahepatic self-perpetuating noxious loop in nonalcoholic steatohepatitis (NASH). Lipid overloading of the liver may result from both intra- and extrahepatic events in distant organs such as the gut, adipose tissue, and muscle, among others. Accumulation of toxic lipids in hepatocytes may induce cellular stress triggers and lead to hepatocyte cell death by various mechanisms, including apoptosis and necrosis followed by the release of intracellular molecules that act as danger signals to communicate stress to neighboring cells. These signals impart danger-associated molecular patterns (DAMPs), and activate sterile inflammatory pathways in cells of the innate immune system, which in turn release cytokines and additional danger signals. The central consequence of this chronic injury is the activation of hepatic stellate cells that can eventually progress to fibrotic NASH.
Because the original description that caspase activation and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) positive cells are characteristic pathologic features in the liver of NASH patients,[13] growing data have demonstrated that hepatocyte cell death is a key process involved in NASH pathogenesis.[14] [15] Sustained hepatocyte cell death has also been implicated in the development of hepatic fibrosis.[16] [17] In addition to the classical modes of cell death, such as apoptosis and necrosis (oncosis), other forms of hepatic cell death have been more recently described in preclinical models and patients with NASH, including autophagic cell death, pyroptosis, and necroptosis.[18] [19] [20] Apoptosis, a highly organized and genetically controlled process, is the most investigated and best defined form of programmed cell death in NASH. Apoptosis is initiated by either membrane receptors (extrinsic pathway) or intracellular stress leading to organelle dysfunction (intrinsic pathway). Both pathways tend to converge in the activation of effector caspases 3 and 7, which execute the final apoptotic changes.[21] Necrosis or oncosis is an accidental form of cell death, with the fatal consequence being cellular oxygen deprivation, whereby the generation of reactive oxygen species (ROS) leads to mitochondrial dysfunction and a drop in ATP level below the threshold required to maintain cellular integrity.[22] [23] The latter induces membrane rupture with the release of cellular contents.[24] [25] Although ROS production and mitochondrial dysfunction is a central feature of NASH, necrotic cell death is a rare histopathological feature of the disease. Necroptosis is induced by the same death receptors that activate the extrinsic apoptotic pathway, namely tumor necrosis factor receptor-1 (TNF-R1) and Fas.[26] Upon interaction of receptor protein kinases 1 and 3 (RIP1 and RIP3) and a deficiency or absence of caspase 8, cell death, which morphologically resembles necrosis, occurs.[27] Controversy exists on the potential role of this form of cell death in NASH. Pyroptosis is a novel, caspase 1-dependent form of programmed cell death that has been recently shown to occur in vivo during liver injury and that shares features of apoptosis such as DNA fragmentation and necrosis such as plasma membrane permeabilizaton.[20] [28] It is dependent on inflammasome-mediated caspase 1 activation and results in the formation of discretely sized ion-permeable pores in the plasma membrane, which leads to water influx and cell swelling.[29] Its potential role in NASH has yet to be explored.
Dying hepatocytes in which a particular molecular cell death pathway is activated are capable of releasing stress signaling molecules called damage-associated molecular patterns (DAMPs) that can act on neighboring cells, including other hepatocytes as well as nonparenchymal cells of the liver such as immune cells—mainly liver macrophages or Kupffer cells, hepatic stellate cells, and sinusoidal endothelial cells—triggering a variety of responses that initiate an homeostatic, wound-healing response to repair tissue injury. However, the persistence of these signals can induce an exuberant response that can result in tissue inflammation and excessive scarring. Recent advances have occurred in the identification of several DAMPs that play a role in tissue injury during NASH development ([Table 1]). A central consequence of the release of DAMPs is the activation of a sterile inflammatory response via their interaction with immune cells that can result in a full inflammatory response in the absence of infection. Damage-associated molecular patterns are recognized by immune cells via pattern-recognition receptors (PRR). Two key families of PRR have been growingly involved in NASH pathobiology and will be the focus of this review, including membrane-bound toll-like receptors (TLRs) and the cytosolic complex termed the inflammasome.
Table 1.
DAMPs that may be involved in the development of nonalcoholic steatohepatitis
| DAMP | Receptors | Therapy |
|---|---|---|
| ATP | P2X7 | Receptor antagonists, apyrase |
| Cytochrome c | Not known | γ-tocotrienol |
| CPS-1 | Not known | Not known |
| Defensins | TLR4, CCR6 | Antagonists, antibodies |
| Fatty acids | TLR4, NLRP3 | Antagonists, antibodies |
| Ceramides | TLR4, NLRP3 | Antagonists, antibodies |
| Cholesterol crystals | NLRP3 | Antagonists, antibodies |
| HMGB1 | TLR4, RAGE, NLRP3 | Neutralizing antibodies |
| HSP | TLR4, CD14, CD91 | Anti-HSP antibodies |
| Hyaluronic acid | TLR2, TLR4 | Antagonists, hyaluronidase |
| Mitochondrial DNA | TLR9, NLRP3,TLR9 | Antagonists, DNAses |
| Nuclear DNA | TLR9 | TLR9 antagonists, DNAses |
| N-formulated peptides | FPR and FPRL1 | Antibodies |
| S100 proteins | RAGE | Blocking antibodies |
| Uric acid | Nonreceptor | Xanthine oxidase inhibitors |
Abbreviations: ATP, adenosine triphosphate; CPS-1, carbamoyl phosphate synthetase 1; DAMPs, damage-associated molecular patterns; FPR, formyl-peptide receptor; HMGB, high-mobility group box 1 protein; HSP, heat-shock protein; NLRP, NOD-like receptor protein; RAGE, receptor for advanced glycation endproducts; TLR, toll-like receptor.
Targeting Cell-Death Pathways
Organelle-Initiated Cell Death
There are several mitochondrial-, lysosomal-, and endoplasmic reticulum stress-related pathways that could potentially be targeted to attenuate apoptosis. It may be possible to decrease lysosomal effects by targeting the action of cathepsin B. We have already described the proteolytic effects of cathepsin B, including the activation of cell-signaling pathways ultimately leading to cell death. In a preclinical study using a mouse model of the effects of NASH, administration of a cathepsin B inhibitor (R-3020) to mice fed a high-carbohydrate diet led to decreased lysosomal permeabilization and decreased steatosis and liver injury.[22] Both genetic and pharmacological inhibition of cathepsin B decreased hepatocyte apoptosis and histologic evidence of liver injury in a mouse model of cholestasis.[30] More recently, the reversible cathepsin B inhibitor VBY-376 demonstrated potent activity in a mouse model of liver fibrosis. Furthermore, a phase I study to evaluate the safety and pharmacokinetics of VBY-376 in humans was conducted and no serious adverse events were observed.[31] Wu et al using 18β-glycyrrhetinic acid, a biologically active metabolite of licorice root extract, demonstrated that it prevented free fatty acid-induced lipid accumulation and hepatocyte apoptosis both in vitro in a human liver cell line and in vivo in a rat NAFLD model.[32] The proposed mechanism involved stabilizing the lysosomal membrane, inhibiting cathepsin B expression and enzyme activity, and reducing mitochondrial cytochrome c release.
Death Receptors
Mediators of the extrinsic pathway are also potential therapeutic targets for patients with NASH. Blocking the activation of death receptors, such as TNF receptor 1 (TNF-R1), Fas, and tumor necrosis factor-related apoptosis-inducing ligand receptors 1 and 2 (TRAIL-R1/2), and their associated signaling cascades may lead to decrease cell death, inflammation, and fibrosis in patients with NASH. There has been active research on components of the TNF-α and TNF-R1 cascade, including the evaluation of several TNF-α inhibitors as potential therapies for NASH. Pentoxifylline, a weak TNF-α inhibitor, has been studied in several open-label trials.[33] [34] Preliminary results did demonstrate improvement in transaminases and liver histology; however, significant side effects were documented as well, including severe nausea and other gastrointestinal symptoms. More recently, a double-blind, randomized, placebo-controlled trial demonstrated the efficacy of pentoxifylline in improving alanine transaminase (ALT) and histological features of NASH, including hepatocyte apoptosis, without significant side effects.[35] Etanercept, a fusion protein that acts as a “decoy receptor” for TNF-α as well as the various anti-TNF-α monoclonal antibodies currently available for clinical use, have been proposed as potential therapy for the treatment of NASH. However, caution must be taken as a randomized placebo-controlled trial has demonstrated that etanercept was associated with a significantly higher mortality rate in patients with moderate-to-severe acute alcoholic hepatitis (Model for End-Stage Liver Disease Score≥15).[36] Future therapies may also target interrupting the Fas–FasL interaction and formation of the death-inducing signaling complex (DISC). Zou et al performed preclinical studies utilizing Tyr–Leu–Gly–Ala (YLGA) peptides, short strands of peptides corresponding to a portion of FasL that bind readily to Fas. YLGA tetramers were administered to ob/ob mice daily for up to 4 weeks. Mice injected with YLGA peptides had significant reductions in hepatic apoptosis, hepatic inflammation, and ALT levels compared with control animals.[37]
Caspases
Given the key role that caspases play in the intracellular cascade leading to apoptosis, several groups have developed caspase inhibitors as a means to decrease apoptosis. The majority of inhibitors that have been developed are pan-caspase inhibitors. The first pan-caspase inhibitor to enter clinical trials was IDN-6556; it has been studied extensively.[38] When administered to bile-duct–ligated (BDL) mice as a model for hepatocyte apoptosis and liver fibrosis, apoptosis, bile infarcts, chemokine activation, and serum ALT levels were reduced.[30] Similar to the other caspase inhibitors described in this section, IDN-6556 not only inhibits proapoptotic caspases, but also proinflammatory caspases, in particular caspase 1, whose activation is a key consequence of the NLRP3 inflammasome response and will be reviewed below in the section devoted to sterile inflammatory cascades. Thus, part of the effects of these drugs may be related to inhibition of this alternative pathway.[39]
The pan-caspase inhibitor VX-166 was used in a mouse model of NASH (genetically obese male db/db mice fed the methionine–choline-deficient [MCD] diet). VX-166 improved liver fibrosis; however, it failed to improve ALT levels or the net liver injury, as assessed by the NAFLD activity score.[40] The mechanism for decreased fibrosis was thought to be related to the fact that phagocytosis of apoptotic hepatocytes activates HSCs and VX-166 inhibited HSC activation by apoptotic bodies. More recently, Anstee et al evaluated VX-166 in mice models of steatosis (high-fat diet) and steatohepatitis (db/db mice on MCD diet) and demonstrated that VX-166 did not reduce steatosis, but reduced histological inflammation, apoptosis, ALT levels, and oxidative stress, particularly in the MCD model.[41]
The use of caspase inhibitors in human liver disease is being explored. A multicenter, placebo-controlled trial used IDN-6556 in 105 patients with chronic liver diseases, mainly chronic hepatitis C (n = 80), and a few NASH patients (n = 5).[42] IDN-6556, given for 14 days, significantly lowered aminotransferase activity in both hepatitis C virus and NASH patients and appeared to be well tolerated. In a follow-up, larger, double-blind, randomized, placebo-controlled, parallel-dose study using the same compound also named Emricasan (Conatus Pharmaceuticals Inc.), 204 chronic hepatitis C patients were treated with placebo or Emricasan, twice daily for up to 12 weeks. They demonstrated that Emricasan significantly reduced serum aspartate transaminase (AST) and ALT levels and was well tolerated over the study period.[43] More recently, Emricasan was found to decrease liver injury, as well as fibrosis in a murine model of NASH.[44] Another caspase inhibitor, GS-9450, was evaluated in a phase II randomized, double-blind, placebo-controlled study in adult patients with NASH.[45] In this study, patients (n = 124, principally male, mean age 45 years, with body mass index > 30 kg/m2) with biopsy-proven NASH were randomized to receive 1, 5, 10, or 40 mg GS-9450, or placebo once daily for 4 weeks.[45]
After 4 weeks of treatment, patients in the 40-mg treatment group experienced the greatest reduction in ALT and AST levels. At week 4, linear regression of ALT versus GS-9450 dose was highly significant (p < 0.0001), with 35% achieving ALT levels within the normal range (7–56 U/l) compared with 0% at study baseline, and 48% achieving normal levels of AST (5–40 U/l) compared with 20% at baseline. Placebo treatment showed no meaningful change for ALT or AST. In addition, the on-treatment measure of CK18 fragments declined in the 10- and 40-mg dose groups (median baseline and week 4 values were 540 and 445 U/l in the 10-mg group and 562 and 386 U/l in the 40-mg group, respectively). One of the concerns of chronic administration of an inhibitor of apoptosis is the theoretical potential for tumorigenesis. A recent study directly evaluated the potential effects of prolonged administration of the caspase inhibitor, Emricasan, for 26 weeks on carcinogenesis by using a humanized mouse model. Continuous exposure to this drug did not result in increased gastrointestinal or liver tumors, or any other tumors, therefore suggesting that exposure to Emricasan was not carcinogenic.[46]
Kinase Activity
The mitogen-activated protein (MAP) kinase cascades are among the most extensively studied of the signaling systems that transmit stimuli from outside the cell to the nucleus. MAPKs play important roles in a variety of cell processes by controlling transcriptional or translational regulation.[47] Three major MAP kinase cascades have been well characterized in mammals, converging on ERKs, c-Jun N-terminal kinases (JNKs), and p38 MAP kinases; each consists of three classes of serine/threonine kinases, MAP kinase, MAP kinase kinase (MAPKK, also referred to as MEK), and MAPKK kinase (MAPKKK, also referred as MAP3K). In hepatocytes, prolonged phosphorylation of JNK, and especially activation of JNK2, leads to the activation of caspase-8 and the mitochondrial death pathway, which is known to be pivotal in the development of NAFLD.[48] Mice lacking the antiapoptotic caspase-8 homolog cellular FLICE-inhibitory protein developed c-Jun N-terminal kinase- (JNK-) dependent liver injury 21 days after streptozotocin treatment, resulting in loss of insulin.[49] Substitution of insulin and inhibition of JNK using the SP600125 compound in vivo or the genetic deletion of JNK2 in all tissues abolished the injurious effect.[49] Hepatocyte-free cholesterol, which accumulates in NASH, but not in simple steatosis, increases LDH leakage, apoptosis and necrosis associated with JNK1 activation (c-Jun phosphorylation), mitochondrial membrane pore transition, cytochrome c release, oxidative stress (GSSG:GSH ratio) and ATP depletion. A study from the Farrell group demonstrated that Jnk1−/− hepatocytes are refractory to free-cholesterol-induced lipotoxicity and JNK inhibitors (1–2 μM CC-401, CC-930) blocked hepatocellular apoptosis and necrosis.[50] The mixed lineage kinases (MLKs) are a family of serine/threonine protein kinases that function in a phospho-relay module to control the activity of specific MAPKs. Members of the family include MLK1, MLK2, MLK3, dual leucine zipper-bearing kinase, and leucine zipper-bearing kinase.[51] MLK3 is one of the MAP3K that mediates JNK activation in the liver.[52] Loss of MLK3 in mice is protective against diet-induced NASH through attenuation of JNK activation.[53] Inhibitors of MLK3 showed great promise for the treatment of Parkinson’s disease in preclinical models and are a potential therapeutic target for the treatment of human NASH.[54] Apoptosis signal–regulating kinase 1 (ASK1), a 160-kDa serine/threonine protein kinase, is another member of the MAP3K family; it activates both p38 and JNK pathways.[55] ASK1 is preferentially activated in response to various types of stress such as oxidative stress and plays pivotal roles in a wide variety of cellular responses.[56] ASK1-deficient mice exhibit reduced diet-induced hepatic steatosis and fibrosis.[57] ASK1 has also been reported to be involved in liver injury induced by acetaminophen or troglitazone, a first-generation thiazolidinedione insulin sensitizer.[58] [59] In animal models of kidney disease, the ASK1 inhibitor GS-4997 improved kidney histopathologic scores and functional readouts such as direct measurements of the glomerular filtration rate, serum creatinine, and proteinuria.[60] Based on this results, phase 2 trials have been initiated in patients with NASH, pulmonary arterial hypertension, and diabetic kidney disease.[61] [62] [63] Receptor-interacting protein kinases are a group of threonine/serine protein kinases with a relatively conserved kinase domain, but distinct nonkinase regions.[64] Necroptosis is a recently described caspase 8-independent method of cell death that denotes organized cellular necrosis and requires the coactivation of RIP1 and RIP3 kinases. RIP1 is necessary in APAP-induced liver injury and RIP1 inhibition via nectrostatin-1 (Nec-1) reduced hepatotoxicity in APAP-induced acute liver injury.[65] [66] Further, a recent report suggested that selective inhibition of RIP3 using the anticancer drug dabrafenib alleviates APAP injury.[67] The Luedde group demonstrated that RIP3 is upregulated in human NASH and in a dietary mouse model of steatohepatitis. RIP3 mediated liver injury, inflammation, the induction of hepatic progenitor cells/activated cholangiocytes, and liver fibrosis.[68] By screening conventional small-molecule libraries, three RIP3 inhibitors have been identified—GSK840, GSK843, and GSK872—which selectively inhibit RIP3 kinase-dependent necroptosis.[69] Highlighting the fact that diverse modes of acute liver injury have differing requirements for RIP1 and RIP3, a recent study showed that in ConA-induced autoimmune hepatitis, RIP3 deletion was protective, whereas RIP1 inhibition exacerbated disease, while conversely, in acetaminophen-mediated liver injury, blockade of either RIP1 or RIP3 was protective.[70] Future studies are warranted to test applicability and efficacy of the many kinase inhibitors, already available and currently under development, in patients with NASH.
Targeting Danger Signals and Their Surface Receptors
Danger signals are intracellular molecules that are released into the extracellular environment during cellular stress and death, and activate innate immune cells resulting in an inflammatory response.[71] [72] As this occurs in the absence of pathogens it is frequently referred to as a sterile inflammatory response, and the molecules are generically referred to as damage-associated molecular patterns (DAMPs), although many do not have a patterned structure.[73] [Table 1] shows the best characterized DAMPs; many more are likely involved. This deceptively simple concept results in a very complex biology. For example, the nuclear protein HMGB1 upon release can bind one of several receptors, including RAGE (receptor for advanced glycation endproducts), TLR4, and CD24—each resulting in different functions.[74] [75] [76] [77] The consequence of HMGB1-receptor binding is also dependent on the oxidation state of one of three cysteine residues. Fully reduced HMGB1 binds to CXCR4 and stimulates immune cell infiltration, partially reduced HMGB1 activates immune cells to produce cytokines and chemokines via TLR4, and fully oxidized HMGB1 is devoid of immunostimulatory activity.[78]
The concept of DAMPs driving inflammation after tissue injury has been confirmed after diverse forms of injury to a wide range of organs. In the liver, a role for DAMPs has been shown in experimental injury initiated by ischemia reperfusion, acetaminophen, and alcoholic hepatitis.[79] [80] [81] [82] In addition to causing liver injury, the size of the liver and the ability of liver injury to release significant amounts of DAMPs into the circulation opens up the possibility that liver-derived DAMPs may be responsible for systemic inflammation and organ failure. It is interesting to note that many of the receptors activated by DAMPs are also activated by pathogen-associated molecular patterns (PAMPs), and include molecules in the TLR family.[83] Others such as P2 × 7 appear uniquely responsive to DAMPs.[84]
The presence of a large number of DAMPs and receptors suggests significant redundancy, which theoretically makes them unattractive candidates for therapy. However, experimental data suggest otherwise. Interference with individual DAMP pathways results in significant reduction in liver injury in a variety of models. Much of the evidence for a role of DAMPs in NASH comes from receptor-deficient mice, but this is only partially informative because many receptors have DAMPs and PAMPs as ligands. For example, mice deficient in TLR4 and TLR9 have reduced steatosis and inflammation in experimental models of NASH.[85] [86] The long-term nature of NASH models makes it challenging to test therapeutic agents, and there is very little information on direct targeting of DAMP pathways in NASH. The clinical development of antagonists of TLR4 and TLR9 for sepsis and systemic lupus erythematosus has resulted in agents that are safe, and are candidate therapeutic agents for human NASH. Eritoran (Eisai Co.) is a TLR4 antagonist that has in vivo efficacy in blocking TLR4, and protects against liver ischemia reperfusion injury.[87] IRS954 is an oligomer-based TLR9 antagonist that has undergone phase 1b clinical trials in systemic lupus erythematosus, and is a candidate for therapy in NASH.[88] In high-fat diet (HFD) models of NASH IRS954, given concurrently with HFD and 6 weeks after starting a HFD, resulted in a significant reduction in steatosis, and inflammation.[89] The reduction in steatosis is interesting, and was also seen in the original data from MCD-induced NASH in TLR9-deficient mice.[85] This suggests that either blocking TLR9 on hepatocytes or switching off the inflammatory response in liver immune cells results in reduced steatosis. This could be due to the removal of the ability of proinflammatory cytokines such as IL-1β to induce hepatocyte steatosis. The normal phenotype of TLR9-deficient mice and their lack of increased susceptibility to infections are reassuring for anti-TLR9 therapy being low risk strategy. A different approach is to use natural agents such as γ-tocotrienol, which has several effects including antioxidant, and stimulating autophagy as well as reducing cytochrome c signaling.
Overall targeting DAMP pathways in NASH is a very attractive and underdeveloped field. Antibody-based approaches have been used successfully in many types of tissue inflammation and are predicted to be effective in NASH. Inhibition of Hsp90 (heat shock protein 90) by an antibody or small molecule approach as a treatment strategy could be useful in the treatment of inflammatory diseases, including rheumatoid arthritis.[90] Inhibitors of Hsp90 blocks activation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway, leading to a loss of cytokine production. Following Hsp90 inhibition, RIP and IκB kinase, which are members of the NF-κB signaling pathway, are degraded, and activation of MAP kinases can be blocked.[91] Interleukin-1 receptor–associated kinase is also a client of Hsp90, and consequently, inhibition of Hsp also has the ability to diminish innate immunity responses via TLR signaling. SGT1, a component of the inflammasome, has also recently been reported to be degraded following Hsp inhibition. A small molecule inhibitor of Hsp90 (SNX-7081) blocks NF-κB translocation to the nucleus and production of interleukin-1β (IL-1β) and TNFα, both of which are known to be important in NASH.[92] In models of arthritis, SNX-7081 resulted in less tissue inflammation and less overall weight loss.
The advantages of targeting DAMPs are that they are specific to tissue damage-associated inflammation, and that this is not expected to comprise the response to pathogen-derived signals. Furthermore, DAMPs also stimulate fibrogenic responses, and there may be independent antifibrotic effects of DAMP-based therapy.
Targeting Cytosolic Receptors
In addition to cell-surface receptors, the innate immune responses to tissue injury caused by pathogens, cellular stress or environmental insults may be initiated by the cytosolic nucleotide-binding domain and leucin-rich repeat receptors (NLRs), retinoic acid-inducible gene (RIG) like helicase (RLH) receptors and absent-in-melanoma- (AIM-) like receptors.[93] The following paragraph focuses on the role of NLRs in the development and treatment of NASH.
Some NLRs form inflammasomes, which act as scaffolding protein within the complex, caspase-1 and in some cases an adaptor protein.[93] To date, the NLR family comprises 14 NLR genes identified in human and 20 in mouse. Within the liver, inflammasomes are expressed in both parenchymal and nonparenchymal cells and serve as key regulators of inflammation and cell fate.[94] [95] The most studied inflammasome, NLRP3, assembles a complex comprised of the adaptor protein apoptosis-associated speck-like protein (ASC) and the serine protease caspase-1. The NLRP3 inflammasome responds to cellular danger signals by activating caspase-1, releasing IL-1β and IL-18, as well as initiating a novel pathway triggering programmed cell death termed pyroptosis.[96] [97] [98] Activation of the NLRP3 inflammasome in mice results in severe liver inflammation, fibrosis, and hepatocyte pyroptotic cell death.[20] [99] In murine models of NASH, NLRP3 activation is required for the fibrotic response, suggesting that targeting this complex may be a rational strategy to block or reverse the development of fibrotic NASH.[76] Among the various cytokines participating in chronic hepatic inflammation, IL-1β plays a special role. IL-1β was found to promote hepatic stellate cell proliferation, activation, and transdifferentiation into a myofibroblast phenotype.[100] The therapeutic strategy to reduce NLRP3 inflammasome activity can be separated in compounds that affect NLRP3 inflammasome assembly and activation, caspase 1 activation, and IL-1 and IL-18 pathways.
The IL-1 family consists of two IL-1 receptors (IL-1R)—IL-1RI and IL-1RII—and the IL-1R antagonist (IL-1Ra), which does not have agonistic activity and does not trigger downstream signaling.[101] Therefore, anti-IL-1 therapies are based on either recombinant IL-1Ra—anakinra; or monoclonal anti-IL-1β antibodies—canakinumab; or an IL-1 trap—rilonacept. A comprehensive study by Petrasek et al demonstrated that IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice.[102] Based on these results, a phase II study was initiated to test the efficacy of anakinra, pentoxifylline, and zinc compared with methylprednisolone in severe acute alcoholic hepatitis.[103] Programmed cell death, the hallmark of NASH contributes to liver injury and fibrosis, is executed by numerous caspases. As outlined above, several studies provide strong evidence that treatment with pan-caspase inhibitors reduce liver injury and fibrosis in murine models of NASH as well as in patients.[40] [41] [44] [45] To date, only very few drugs have been described targeting the inflammasome directly; no study has evaluated applicability and efficacy of inflammasome blockade in models of NASH. A study by Coll et al identified CRID3—a member of the class of diarylsulfonylurea containing compounds called cytokine release inhibitory drugs (CRIDs)—as a novel inhibitor of the NLRP3 and AIM2 inflammasomes.[104] Changes in the preparation of CRID3 led to the development of MCC950, a potent, selective, small-molecule inhibitor of NLRP3. MCC950 blocked canonical and noncanonical NLRP3 activation at nanomolar concentrations.[105] MCC950 specifically inhibited activation of NLRP3, but not the AIM2, NLRC4, or NLRP1 inflammasomes. MCC950 reduced interleukin-1b (IL-1b) production in vivo and attenuated the severity of experimental autoimmune encephalomyelitis (EAE), a disease model of multiple sclerosis.[105] Furthermore, MCC950 treatment rescued neonatal lethality in a mouse model of CAPS and was active in ex vivo samples from individuals with Muckle–Wells syndrome.[105] A recent study from our group using two murine models of NASH suggested that MCC950 is a potential novel therapy for this disease.[106] Future studies are warranted to decipher the role of targeting cytosolic receptors in NASH treatment.
Acknowledgments
This work was funded by NIH grants R01 DK082451 and U01 AA022489 to Ariel E. Feldstein. Alexander Wree is supported by the START-Program of the Faculty of Medicine, RWTH Aachen.
Abbreviations
- AIM
absent in melanoma like receptors
- ALT
alanine transaminase
- APAP
acetaminophen
- ASC
apoptosis associated speck like protein
- ASK1
Apoptosis signal–regulating kinase 1
- AST
aspartate transaminase
- BDL
bile–duct ligated
- CAPS
cryopyrin-associated periodic syndromes
- CRIDs
cytokine release inhibitory drugs
- DAMPs
damage-associated molecular patterns
- EAE
experimental autoimmune encephalomyelitis
- ERKs
extracellular-signal-regulated kinases
- HFD
high-fat diet
- HMGB1
high-mobility group box protein 1
- Hsp90
heat shock protein 90
- IL
interleukin
- JNKs
c-Jun N-terminal kinases
- MAP
mitogen-activated protein
- MCD
methionine–choline-deficient
- MLKs
mixed lineage kinases
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- Nec-1
nectrostatin-1
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRs
nucleotide-binding domain and leucin-rich repeat receptor
- PAMPs
pathogen associated molecular patterns
- PRR
pattern-recognition receptors
- RIG
retinoic acid-inducible gene receptor
- RIP
receptor-interacting protein
- RLH
RIG-like helicase receptor
- ROS
reactive oxygen species
- TLRs
toll-like receptors
- TNF-R1
tumor necrosis factor-receptor 1
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
References
- 1.Levene AP, Goldin RD. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology. 2012;61(2):141–152. doi: 10.1111/j.1365-2559.2011.04145.x. [DOI] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34(3):274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 3.Brunt EM, Neuschwander-Tetri BA, Oliver D, Wehmeier KR, Bacon BR. Nonalcoholic steatohepatitis: histologic features and clinical correlations with 30 blinded biopsy specimens. Hum Pathol. 2004;35(9):1070–1082. doi: 10.1016/j.humpath.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 6.Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389–97.e10. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(3):547–555. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 9.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56(4):952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Wree A, Kahraman A, Gerken G, Canbay A. Obesity affects the liver - the link between adipocytes and hepatocytes. Digestion. 2011;83(1–2):124–133. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 11.Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis. 2010;30(4):391–401. doi: 10.1055/s-0030-1267539. [DOI] [PubMed] [Google Scholar]
- 12.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10(11):627–636. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 13.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 14.Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015;1(1):17–27. doi: 10.1016/j.jcmgh.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147(4):765–783.e4. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39(2):273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 17.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83(5):655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 18.Kroemer G, Galluzzi L, Vandenabeele P, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komatsu M. Liver autophagy: physiology and pathology. J Biochem. 2012;152(1):5–15. doi: 10.1093/jb/mvs059. [DOI] [PubMed] [Google Scholar]
- 20.Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5(11):897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 22.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90(3):1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12(5):815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 24.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gores GJ, Herman B, Lemasters JJ. Plasma membrane bleb formation and rupture: a common feature of hepatocellular injury. Hepatology. 1990;11(4):690–698. doi: 10.1002/hep.1840110425. [DOI] [PubMed] [Google Scholar]
- 26.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 27.Galluzzi L, Kepp O, Kroemer G. RIP kinases initiate programmed necrosis. J Mol Cell Biol. 2009;1(1):8–10. doi: 10.1093/jmcb/mjp007. [DOI] [PubMed] [Google Scholar]
- 28.Eguchi A, Wree A, Feldstein AE. Biomarkers of liver cell death. J Hepatol. 2014;60(5):1063–1074. doi: 10.1016/j.jhep.2013.12.026. [DOI] [PubMed] [Google Scholar]
- 29.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 30.Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther. 2004;308(3):1191–1196. doi: 10.1124/jpet.103.060129. [DOI] [PubMed] [Google Scholar]
- 31.Holsinger LJ, Coakley DF, Dener JM, et al. Efficacy of a reversible cathepsin B inhibitor in a rodent model of liver fibrosis and human pharmacokinetic profile. Hepatology. 2010;52:1128A. [Google Scholar]
- 32.Wu X, Zhang L, Gurley E, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47(6):1905–1915. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 33.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99(12):2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 34.Satapathy SK, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22(5):634–638. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- 35.Gogate P, Ambardekar P, Kulkarni S, Deshpande R, Joshi S, Deshpande M. Comparison of endothelial cell loss after cataract surgery: phacoemulsification versus manual small-incision cataract surgery: six-week results of a randomized control trial. J Cataract Refract Surg. 2010;36(2):247–253. doi: 10.1016/j.jcrs.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 36.Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135(6):1953–1960. doi: 10.1053/j.gastro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou C, Ma J, Wang X, et al. Lack of Fas antagonism by Met in human fatty liver disease. Nat Med. 2007;13(9):1078–1085. doi: 10.1038/nm1625. [DOI] [PubMed] [Google Scholar]
- 38.Valentino KL, Gutierrez M, Sanchez R, Winship MJ, Shapiro DA. First clinical trial of a novel caspase inhibitor: anti-apoptotic caspase inhibitor, IDN-6556, improves liver enzymes. Int J Clin Pharmacol Ther. 2003;41(10):441–449. doi: 10.5414/cpp41441. [DOI] [PubMed] [Google Scholar]
- 39.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 2012;92(5):713–723. doi: 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50(5):1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 41.Anstee QM, Concas D, Kudo H, et al. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol. 2010;53(3):542–550. doi: 10.1016/j.jhep.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 42.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46(2):324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 43.Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G. Clinical trial: the efficacy and safety of oral PF-03491390, a pancaspase inhibitor - a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther. 2010;31(9):969–978. doi: 10.1111/j.1365-2036.2010.04264.x. [DOI] [PubMed] [Google Scholar]
- 44.Barreyro FJ, Holod S, Finocchietto PV, et al. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015;35(3):953–966. doi: 10.1111/liv.12570. [DOI] [PubMed] [Google Scholar]
- 45.Ratziu V, Sheikh MY, Sanyal AJ, et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology. 2012;55(2):419–428. doi: 10.1002/hep.24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elbekai RH, Paranjpe MG, Contreras PC, Spada A. Carcinogenicity assessment of the pan-caspase inhibitor, Emricasan, in Tg.rasH2 mice. Regul Toxicol Pharmacol. 2015;72(2):169–178. doi: 10.1016/j.yrtph.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281(22):15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohl T, Gehrke N, Schad A, et al. Diabetic liver injury from streptozotocin is regulated through the caspase-8 homolog cFLIP involving activation of JNK2 and intrahepatic immunocompetent cells. Cell Death Dis. 2013;4:e712. doi: 10.1038/cddis.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J Hepatol. 2014;61(6):1376–1384. doi: 10.1016/j.jhep.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 51.Kim KY, Kim BC, Xu Z, Kim SJ. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-beta-induced apoptosis in hepatoma cells. J Biol Chem. 2004;279(28):29478–29484. doi: 10.1074/jbc.M313947200. [DOI] [PubMed] [Google Scholar]
- 52.Sharma M, Urano F, Jaeschke A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. J Hepatol. 2012;56(1):192–198. doi: 10.1016/j.jhep.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ibrahim SH, Gores GJ, Hirsova P, et al. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver Int. 2014;34(3):427–437. doi: 10.1111/liv.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang JX, Török NJ. MLK3 as a regulator of disease progression in non-alcoholic steatohepatitis. Liver Int. 2014;34(8):1131–1132. doi: 10.1111/liv.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 56.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780(11):1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto E, Dong YF, Kataoka K, et al. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension. 2008;52(3):573–580. doi: 10.1161/HYPERTENSIONAHA.108.112292. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa H, Maeda S, Hikiba Y, et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology. 2008;135(4):1311–1321. doi: 10.1053/j.gastro.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 59.Lim PL, Liu J, Go ML, Boelsterli UA. The mitochondrial superoxide/thioredoxin-2/Ask1 signaling pathway is critically involved in troglitazone-induced cell injury to human hepatocytes. Toxicol Sci. 2008;101(2):341–349. doi: 10.1093/toxsci/kfm273. [DOI] [PubMed] [Google Scholar]
- 60.Lin JH, Zhang JJ, Lin SL, Chertow GM. Design of a phase 2 clinical trial of an ASK1 inhibitor, GS-4997, in patients with diabetic kidney disease. Nephron. 2015;129(1):29–33. doi: 10.1159/000369152. [DOI] [PubMed] [Google Scholar]
- 61.Gilead S. ClinicalTrials.gov ID # NCT02234141. Bethesda, MD: National Institutes of Health; A phase 2, dose-ranging, randomized, double-blind, placebo-controlled study of GS-4997 in subjects with pulmonary arterial hypertension. [Google Scholar]
- 62.Gilead S. ClinicalTrials.gov ID # NCT02177786. Bethesda, MD: National Institutes of Health; A phase 2 double-blind, placebo-controlled, dose-ranging study evaluating the efficacy, safety, and tolerability of GS-4997 in subjects with diabetic kidney disease. [Google Scholar]
- 63.Gilead S. ClinicalTrials.gov ID # NCT02466516. Bethesda, MD: National Institutes of Health; A phase 2, randomized, open label study evaluating the safety, tolerability, and efficacy of GS-4997 alone or in combination with simtuzumab (sim) in subjects with nonalcoholic steatohepatitis (NASH) and fibrosis stages F2–F3. [Google Scholar]
- 64.Zhang D, Lin Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7(4):243–249. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang YF, He W, Zhang C, et al. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014;225(3):445–453. doi: 10.1016/j.toxlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 66.Takemoto K, Hatano E, Iwaisako K, et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li JX, Feng JM, Wang Y, et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014;5:e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gautheron J, Vucur M, Reisinger F, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6(8):1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mandal P, Berger SB, Pillay S, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56(4):481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deutsch M, Graffeo CS, Rokosh R, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Martinez I, Mehal WZ. DNA: adding injury to insult. Hepatology. 2015;61(1):35–36. doi: 10.1002/hep.27398. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Martinez I, Shaker ME, Mehal WZ. Therapeutic opportunities in damage-associated molecular pattern-driven metabolic diseases. Antioxid Redox Signal. 2015 doi: 10.1089/ars.2015.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mehal WZ. Cells on fire. Sci Am. 2015;312(6):44–49. doi: 10.1038/scientificamerican0615-44. [DOI] [PubMed] [Google Scholar]
- 74.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 75.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274(28):19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 76.Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26):11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323(5922):1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Venereau E, Casalgrandi M, Schiraldi M, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H. Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology. 2014;60(4):1336–1345. doi: 10.1002/hep.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marques PE, Oliveira AG, Pereira RV, et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology. 2015;61(1):348–360. doi: 10.1002/hep.27216. [DOI] [PubMed] [Google Scholar]
- 82.Bala S, Petrasek J, Mundkur S, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012;56(5):1946–1957. doi: 10.1002/hep.25873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kay E, Scotland RS, Whiteford JR. Toll-like receptors: Role in inflammation and therapeutic potential. Biofactors. 2014;40(3):284–294. doi: 10.1002/biof.1156. [DOI] [PubMed] [Google Scholar]
- 84.Di Virgilio F. P2X receptors and inflammation. Curr Med Chem. 2015;22(7):866–877. doi: 10.2174/0929867322666141210155311. [DOI] [PubMed] [Google Scholar]
- 85.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139(1):323–34.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4(4):659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mcdonald KA, Huang H, Tohme S, et al. Toll-like receptor 4 (TLR4) antagonist eritoran tetrasodium attenuates liver ischemia and reperfusion injury through inhibition of high-mobility group box protein B1 (HMGB1) signaling. Mol Med. 2014;20:639–648. doi: 10.2119/molmed.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37(12):3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 89.Garcia-Martinez I, Ouyang X, Santoro N, et al. Elevated plasma DNA in patients with NASH and reduced liver injury in mice with absence of TLR9 on Kupffer cells. Hepatology. 2014;60:517A. [Google Scholar]
- 90.Rice JW, Veal JM, Fadden RP, et al. Small molecule inhibitors of Hsp90 potently affect inflammatory disease pathways and exhibit activity in models of rheumatoid arthritis. Arthritis Rheum. 2008;58(12):3765–3775. doi: 10.1002/art.24047. [DOI] [PubMed] [Google Scholar]
- 91.Tago K, Tsukahara F, Naruse M, Yoshioka T, Takano K. Hsp90 inhibitors attenuate effect of dexamethasone on activated NF-kappaB and AP-1. Life Sci. 2004;74(16):1981–1992. doi: 10.1016/j.lfs.2003.07.056. [DOI] [PubMed] [Google Scholar]
- 92.Wang X, Wang S, Liu Y, et al. Comparative effects of SNX-7081 and SNX-2112 on cell cycle, apoptosis and Hsp90 client proteins in human cancer cells. Oncol Rep. 2015;33(1):230–238. doi: 10.3892/or.2014.3552. [DOI] [PubMed] [Google Scholar]
- 93.López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs. 2012;21(7):995–1007. doi: 10.1517/13543784.2012.690032. [DOI] [PubMed] [Google Scholar]
- 94.Hoque R, Vodovotz Y, Mehal W. Therapeutic strategies in inflammasome mediated diseases of the liver. J Hepatol. 2013;58(5):1047–1052. doi: 10.1016/j.jhep.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57(3):642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 96.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243(1):136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 97.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 98.Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wree A, McGeough MD, Peña CA, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 2014;92(10):1069–1082. doi: 10.1007/s00109-014-1170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Han YP, Zhou L, Wang J, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279(6):4820–4828. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moltó A, Olivé A. Anti-IL-1 molecules: new comers and new indications. Joint Bone Spine. 2010;77(2):102–107. doi: 10.1016/j.jbspin.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 102.Petrasek J, Bala S, Csak T, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122(10):3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mack M. ClinicalTrials.gov ID # NCT01809132. Bethesda, MD: National Institutes of Health; A phase 2, double-blind randomized controlled trial of anakinra, pentoxifylline, and zinc compared to methylprednisolone in severe acute alcoholic hepatitis. [Google Scholar]
- 104.Coll RC, Robertson A, Butler M, Cooper M, O’Neill LA. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS ONE. 2011;6(12):e29539. doi: 10.1371/journal.pone.0029539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mridha AWA, Robertson A, Teoh NC, et al. Blocking the NLRP3 inflammasome prevents inflammatory recruitment and fibrotic progression in experimental NASH. Paper presented at: the AASLD Liver Meeting; November 13–17, 2015; San Francisco, CA. [Google Scholar]