

Abstract
Mouse embryonic development comprises highly dynamic and coordinated events that drive key cell lineage specification and morphogenetic events. These processes involve cellular behaviors including proliferation, migration, apoptosis, and differentiation, each of which is regulated both spatially and temporally. Live imaging of developing embryos provides an essential tool to investigate these coordinated processes in three-dimensional space over time. For this purpose, the development and application of genetically encoded fluorescent protein (FP) reporters has accelerated over the past decade allowing for the high-resolution visualization of developmental progression. Ongoing efforts are aimed at generating improved reporters, where spectrally distinct as well as novel FPs whose optical properties can be photomodulated, are exploited for live imaging of mouse embryos. Moreover, subcellular tags in combination with using FPs allow for the visualization of multiple subcellular characteristics, such as cell position and cell morphology, in living embryos. Here, we review recent advances in the application of FPs for live imaging in the early mouse embryo, as well as some of the methods used for ex utero embryo development that facilitate on-stage time-lapse specimen visualization.
1. Introduction
During their gestation, embryos undergo coordinated complex morphogenetic changes as they develop from single fertilized eggs into neonates having all major organ systems in place for supporting adult life (Fig. 18.1). Embryonic development comprises of a dynamic three-dimensional orchestration of cellular interactions, including proliferation, apoptosis, movement, and differentiation, which occur in a temporally and spatially regulated manner (Arnold and Robertson, 2009; Nowotschin and Hadjantonakis, 2010; Rossant and Tam, 2009). Defining these processes requires time-lapse visualization of the embryo as development progresses. Thus, live imaging provides an essential tool for acquiring spatio-temporal information of the dynamic molecular mechanisms and cellular behaviors driving embryonic development in vivo.
Figure 18.1.

Development of (A) preimplantation and (B) postimplantation embryos. Requirements for dissection and culture media as well as gas content are shown for each embryonic stage. NCS, newborn calf serum; RS, rat serum; E, embryonic day (adapted from Nowotschin et al., 2010; with permission from Academic Press/Elsevier).
The mouse is the premier mammalian model organism used in embryological studies because of its genetic and physiological similarities to humans, as well as the ease with which its genome can be manipulated and analyzed. Key morphogenetic events in mouse and hence mammalian development have been studied for a few decades now, however recent advances in live imaging technologies have brought these studies to another level where these events can be visualized and examined at higher resolution and in utero. Today, protocols have been established and optimized that allow live imaging of mouse embryos ex utero, thus providing a way to study mouse embryonic development live and at high resolution (Garcia et al., 2011a,b,c, d; Nowotschin et al., 2010; Udan and Dickinson, 2010).
Fluorescence emitted from excited fluorophores has been exploited for cellular imaging and for the observation of single or groups of cells in developing embryos. Injection of vital dyes, such as the lipophilic tracers, DiI, or DiO (Serbedzija et al., 1992), inorganic semiconductor nanocrystals, known as quantum dots (Dubertret et al., 2002), and genetically encoded fluorescent proteins (FPs) (Nowotschin et al., 2009b) have been used as fluorophores to label cells in embryos. Of these, FPs have been most prominent for live cell imaging because of their high signal-to-noise ratio, minimal toxicity, non-reliance on the availability of antibodies for the protein of interest, and ease of use. FPs, such as the green fluorescent protein (GFP) and its variants, as well as the orange and red FPs are ideal reporters for live imaging since they can be expressed under the control of promoter/ cis-regulatory regions driving sufficient levels of expression of the FP for visualization. Moreover, FPs can be fused to any protein of interest to investigate both protein dynamics in vivo as well as to provide subcellular segmentation (Nowotschin et al., 2009b). The use of FPs for visualizing mouse embryonic development in live ex utero cultured mouse embryos (Hadjantonakis et al., 2003) has led to novel insights of the intrinsic cell behaviors underlying tissue morphogenesis and cell lineage specification in mouse embryos (Kwon et al., 2008; Plusa et al., 2008).
In this chapter, we provide an overview of some of the most commonly used FPs and their application for live imaging in mouse embryos. We also discuss the necessary tools for live imaging, such as standard confocal microscopy techniques available at most institutions. Finally, we provide a brief description of the methods and conditions that are routinely used in our and other laboratories for ex utero culturing and time-lapse imaging of mouse embryos.
2. Genetically Encoded FPs for Live Imaging Morphogenetic Events in the Early Mouse Embryo
2.1. Multispectral FPs
Currently, many FPs have been isolated and are readily available to use for live imaging applications (Table 18.1). Among them, the GFP and its variants are the most commonly used FPs to study the complex cell behaviors and organization of the embryo and the adult animal (Nowotschin et al., 2009b) due to their nontoxicity in eukaryotic cells. GFP and its variants have been widely applied in live cell imaging regimes either expressed in their native form throughout the cellular cytoplasm or, as will be discussed in the next section, fused in frame to a protein of interest so as to function as tags allowing for the visualization of specific protein localization, cell division, cell tracking, cell death, and cell morphology (Hadjantonakis and Papaioannou, 2004; Rhee et al., 2006). Moreover, cell-type-specific resolution within the tissues of the embryo can be achieved when these FPs are placed under the control of cis-regulatory elements (Ferrer-Vaquer et al., 2010; Kwon and Hadjantonakis, 2007; Kwon et al., 2006, 2008; Monteiro et al., 2008). Additional temporal control can be achieved by expressing the FP in a genetically inducible regime, as in genetically inducible fate mapping approaches (Joyner and Zervas, 2006). Further, the use of multiple spectrally distinct FPs enables the simultaneous study of several cell characteristics (Livet et al., 2007).
Table 18.1.
Characteristics of commonly used fluorescent proteins
| Characteristics
|
Used for live imaging in mouse embryos
|
||||||
|---|---|---|---|---|---|---|---|
| FPs | Absorbance (nm) | Emission (nm) | Conformation status | Cytoplasmic | Tagged | Referencesa | |
| GFP variant | ECFP | 433 | 475 | Monomer | – | – | |
|
| |||||||
| Cerulean | 433 | 475 | Monomer | – | ✓ | Stewart et al. (2009) | |
|
| |||||||
| EGFP | 488 | 507 | Monomer | ✓ | ✓ | Hadjantonakis and Papaioannou (2004), Kwon et al. (2006) | |
|
| |||||||
| EYFP | 512 | 528 | Monomer | ✓ | ✓ | Fraser et al. (2005) | |
|
| |||||||
| mVenus | 512 | 528 | Monomer | ✓ | ✓ | Rhee et al. (2006) | |
|
| |||||||
| Orange, red, and far-red | tdTomato 554 | 581 | Dimer | – | ✓ | Trichas et al. (2008) | |
|
| |||||||
| Katushka | 574 | 602 | Dimer | ✓ | – | Dieguez-Hurtado et al. (2011) | |
|
| |||||||
| mRFP1 | 584 | 607 | Monomer | ✓ | ✓ | Long et al. (2005), Hayashi-Takanaka et al. (2009) | |
|
| |||||||
| mCherry | 587 | 610 | Monomer | ✓ | ✓ | Viotti et al. (2011), Nowotschin et al. (2009a) | |
Examples of characteristic references describing the usage of corresponding FPs in live or fixed mouse embryos.
GFP emits green light when excited with ultraviolet (UV) or blue light. The engineering of GFP resulted in several spectral variants, the blue and cyan FPs (e.g., BFP, CFP, and Cerulean), the yellow FPs (e.g., YFP and Venus) which emit blue and yellow light, respectively, as well as in improved versions of the originally isolated wild-type GFP protein with brighter fluorescence and greater photostability (Cubitt et al., 1995; Heim et al., 1995). The most popular version for use in mammalian systems is the enhanced GFP (EGFP). Apart from GFP and its variants, spectrally distinct FPs that emit orange, red, and farred light are sought after for imaging in tissue specimens (Table 18.1). FPs with emission in the long-wavelength spectrum are advantageous in respect of their reduced cell toxicity, less background autofluorescence, deeper tissue penetration, which is important for larger specimens such as mouse embryos or whole animal imaging, as well as for their ease of covisualization with FPs possessing short-wavelength emission spectra. Though the list of RFPs is fairly short, some RFPs have already been used successfully in transgenic reporter mice (Kwon and Hadjantonakis, 2009; Viotti et al., 2011) and live imaging of mouse embryos. When expressed under the control of cis-regulatory elements, RFPs like mRFP and mCherry (‘m’ stands for monomeric) have been shown to provide cell-type-specific resolution during development of the early mouse embryo (Kwon and Hadjantonakis, 2009; Poche et al., 2009; Viotti et al., 2011). Transgenic mice ubiquitously expressing mRFP1 and mCherry in the cytoplasm (Fink et al., 2010; Long et al., 2005) have shown that the cytoplasmic expression of these RFPs is developmentally neutral. However, though mRFP1 is a monomeric variant of the tetramer DsRed, ubiquitous expression of histone (H2B) or myristoylated (myr) fusions of mRFP1 or mCherry are in some instances not developmentally neutral and thus incompatible with normal development in mice (Nowotschin et al., 2009a). Nevertheless, live imaging short-term lineage tracing experiments by injecting mRNA for H2B-RFP and myr-RFP in 8-cell stage embryos have been performed (Yamanaka et al., 2010). Another variant of mRFP1, the tandem dimer (td) (td)Tomato, characterized by its high brightness and photostability as well as its amenability to fusion tags has been used for live imaging in mice (Muzumdar et al., 2007; Trichas et al., 2008). Fusions to mCherry, such as histone H2B as a nuclear tag, have been shown to display a satisfactorily bright signal (Abe et al., 2011; Egli et al., 2007). Though, similar to mRFP1, constitutive widespread expression in mice of H2B-mCherry has proven to be toxic, its expression under specific cis regulatory elements has been shown to be non-teratogenic (Nowotschin et al., 2009a).
As already discussed, FPs with excitation and emission spectra in the far-red region of the spectrum can be advantageous when combined with GFP and its variants as spectral separation as well as deeper tissue penetration is crucial (Shcherbo et al., 2007). The dimer Katushka is currently the brightest FP among those with emission maxima above 620 nm to have been used in the mouse embryo. In fact, the first transgenic mouse line conditionally expressing Katushka was recently reported (Dieguez-Hurtado et al., 2011). Tissue-specific expression of Katushka was ubiquitous and strong without displaying any toxic effects in this transgenic line; nevertheless, usage of Katushka fused to protein tags has not been reported yet. New dimeric variants of Katushka with further red-shift emission as well as reduced cytotoxicity and brighter signal were recently reported. They were tested in tissue culture cells and Xenopus embryos, opening exciting possibilities for their use in mouse embryos (Shcherbo et al., 2010). However, the dimeric nature of Katushka and its variants has limited its potential for incorporation in protein fusions for subcellular localization.
Monomeric versions of far-red FPs have been developed, such as mKate, lacking the brightness of Katushka. However, the recently developed successor of mKate, mKate2, and a pseudo-monomeric Katushka have both been reported to work well with fusion tags while exhibiting bright fluorescence when imaged live in cells and transgenic Xenopus embryos (Shcherbo et al., 2009). Moreover, the development of nuclear-localized variants of mKate were recently reported that allowed observing tumor cells in living mice (Piatkevich et al., 2010). The development of novel monomeric red and far-red FPs are eagerly anticipated and should allow for their fusion with subcellular tags (Piatkevich and Verkhusha, 2010).
2.2. Fusion tags to FPs for labeling and tracking
The use of FP fusion proteins has provided a powerful tool to investigate mouse embryonic development in three dimensions through live imaging (Table 18.2) (Kwon and Hadjantonakis, 2007; Hadjantonakis et al., 2003). For example, fusions of FPs to the human histone H2B are bound to active chromatin and localize to the nucleus thus enabling tissue segmentation, and the visualization of cellular characteristics, such as division, movement, or apoptosis, as well as the tracking of daughter cells during developmental progression (Hadjantonakis and Papaioannou, 2004; Kanda et al., 1998). Histone fusions do not only facilitate tracking of cells, but also the subsequent segmentation of the spatially separated nuclei in cell lines and within the complex 3D structure of the pre- or postimplantation embryo during image data analysis (Fig. 18.2). Cell-type-specific combined with nuclear-specific resolution has been achieved by placing H2B-FP reporters under defined promoter and other cis-regulatory elements. For example, H2B-GFP driven by regulatory elements of the Pdgfra locus has been useful for lineage tracking in the preimplantation embryo (Artus et al., 2010; Plusa et al., 2008). In another study, H2B-GFP driven by TCF/Lef bound cis-regulatory elements have provided a nuclear-localized, single-cell reporter that can be used to live image the behavior and fate of cells responsive to Wnt signaling (Ferrer-Vaquer et al., 2010). Moreover, a recent report using H2B-GFP fusion reporter under the regulation of the epsilon-globin cis-regulatory elements allowed visualization of the first committed hematopoietic progenitors within the developing mouse embryo (Isern et al., 2011).
Table 18.2.
Fusion tags to fluorescent proteins that have been used for live imaging in mouse embryos
| Localization | Tags | Characteristics | References |
|---|---|---|---|
| Nucleus | H2B | Human histone | Hadjantonakis and Papaioannou (2004) |
| Plasma membrane | GPI | Glycosylphosphatidylinositol anchor | Rhee et al. (2006) |
| myr | Myristoyl anchor | Rhee et al. (2006), Abe et al. (2011) | |
| Lyn | Tyrosine kinase | Abe et al. (2011), Nowotschin and Hadjantonakis (2010) | |
| RAS | GTPase | Abe et al. (2011) | |
| GAP43 | Axonal membrane protein | Abe et al. (2011) | |
| pDisplay | Membrane targeting vector (Invitrogen) | Abe et al. (2011) | |
| Mitochondria | Mito | Cytochrome C oxidase subunit VIII A | Abe et al. (2011) |
| Golgi | Golgi | β-1,4-Galactosyltransferase 1 | Abe et al. (2011) |
| Microtubules | Tuba | α-Tubulin 2 | Abe et al. (2011) |
| hEMTB | Human microtubule associate protein 7 | Abe et al. (2011) | |
| EB1 | APC binding protein | Abe et al. (2011) | |
| Cytoskeletal proteins | Actin | β-Actin | Abe et al. (2011) |
| Moesin | ERM family protein (linker between plasma membrane and actin cytoskeleton) | Abe et al. (2011) | |
| Focal adhesions | Paxillin | Focal adhesion-associated adaptor protein | Abe et al. (2011) |
Examples of characteristic references describing the usage of corresponding FPs in live or fixed mouse embryos.
Figure 18.2.
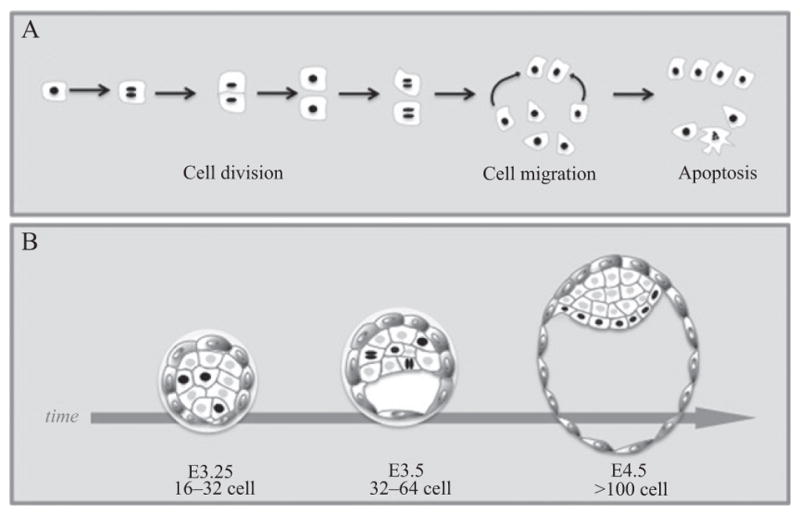
Histone fusions to FPs provide nuclear labeling. (A) Schematic representation of cells expressing a histone (H2B) fusion to fluorescent protein. The H2B-FP remains bound to chromatin, thus providing nuclear labeling. Cells expressing this fusion reporter can be tracked as they divide, migrate, or undergo apoptosis. (B) Cartoon for time-lapse imaging of preimplantation mouse embryo expressing H2B-FP under the control of the Pdgfra-α locus. This reporter labels the progenitors of the primitive endoderm lineage that lies adjacent to the blastocoel cavity in the E4.5 stage preimplantation embryo. Dynamic cell processes, such as division, migration, and apoptosis can be tracked by using this reporter (Plusa et al., 2008).
Fusions of FPs to plasma membrane proteins such as glycosylphosphati-dylinositol (GPI) or myristoylation (myr) and farnesylation (CAAX) tagging allows the visualization of cell morphology in vivo (Table 18.2) (Muzumdar et al., 2007; Rhee et al., 2006). Additional tags targeting FPs to the plasma membrane were recently reported (Table 18.2) (Abe et al., 2011). Importantly, these plasma membrane localized fusions can be used in combination with H2B fusions enabling the visualization of multiple cellular characteristics, such as nuclear position as well as cell morphology (Fig. 18.3); these combinations have been used in embryonic stem cell cultures and in mouse embryos (Abe et al., 2011; Nowotschin et al., 2009a; Stewart et al., 2009; Trichas et al., 2008). Recently, a mouse reporter line was developed that allows conditional expression of nuclear localized mCherry expression combined with plasma membrane GFP expression; this dual reporter was shown to be nontoxic for live imaging embryonic development and provides a powerful tool to mark and track distinct populations of cells in vivo (Shioi et al., 2011).
Figure 18.3.

Live imaging of multi-tagged mouse ES cell colonies. 2D images of an embryonic stem (ES) cell colony comprising two distinct transgenic cell populations expressing H2B-GFP ; myr-RFP or H2B-mCherry ; GPI-GFP. 1: GPI-GFP; 2: H2B-GFP; 3: H2B-mCherry; 4: myr-RFP. Bright field (Bf) image (A), green channel (B), red channel (C), and merge of green and red channel (D).
In addition, tags have recently been used to target FPs to subcellular locations, such as mitochondria, Golgi apparatus, microtubules, and actin filaments as well as focal adhesion points (Abe et al., 2011). This study also reported dual labeling of nucleus and Golgi apparatus, as well as nucleus and plasma membrane (Abe et al., 2011).
2.3. Photomodulatable FPs
Traditionally, labeling and tracking cells in live mouse embryos were performed using invasive techniques such as dye injections and tissue grafts, as well as binary transgenic strategies (Joyner and Zervas, 2006; Kinder et al., 1999, 2001; Nagy, 2000). Photomodulatable FPs can now be used to non-invasively label and track cells or proteins within the complex 3D structure of the mouse embryo (Table 18.3) (Nowotschin and Hadjantonakis, 2009a,b). This can be achieved at a specific embryonic region of interest and in a defined spatio-temporal resolution. There are two categories of photomo-dulatable FPs that change properties after excitation with UV or blue light: the photoactivatable FPs (PA-FPs) that change from a non-fluorescent to a fluorescent state and the photoconvertible FPs (PC-FPs) that change fluorescence absorbance and emission spectra and thus, convert from one color to another (Table 18.3). To date, photomodulatable FPs have been used successfully in live chick, Drosophila, zebrafish, and Xenopus embryos (Hatta et al., 2006; Murray and Saint, 2007; Stark and Kulesa, 2005; Wacker et al., 2007). However, their usage in mouse embryos has only recently started to be applicable (discussed below).
Table 18.3.
Characteristics of commonly used photomodulatable fluorescent proteins
| Characteristics
|
Used for live imaging in mouse embryos
|
|||||
|---|---|---|---|---|---|---|
| FPs | Change of fluorescence | Conformation status | Cytoplasmic | Tagged | Referencesa | |
| Photoactivatable | PAGFP | No fluorescence → green | Monomer | ✓ | – | Plachta et al. (2011) |
| KFP | No fluorescence ↔ red | Tetramer | – | – | ||
| Dronpa | No fluorescence ↔ green | Monomer | – | – | ||
| Photoconvertible | PS-CFP2 | Cyan → green | Monomer | – | – | |
| Kaede | Green → red | Tetramer | ✓ | – | Tomura et al. (2008) | |
| KikGR | Green → red | Tetramer | ✓ | ✓ | Nowotschin and Hadjantonakis (2009b), Abe et al. (2011) | |
Examples of characteristic references describing the usage of corresponding FPs in live or fixed mouse embryos.
Photoactivatable GFP was developed as a GFP variant with a single residue substitution that remains in a non-fluorescent state. However, PA-GFP yields a 100-fold increased green light fluorescence upon exposure to short-wavelength light as it irreversibly converts to a fluorescent state (Fig. 18.4). PA-GFP has been of special interest because of its monomeric nature and its potential to have no toxicity in live embryos. An example of PA-GFP application in mouse embryos has been to examine postnatal neo-cortex development (Gray et al., 2006). Moreover, PA-GFP was recently used to investigate lineage commitment in live preimplantation mouse embryos (Plachta et al., 2011). Use of other PA-FPs, such as PA-mCherry or the kindling FP (KFP) and Dronpa proteins, has not yet been reported for imaging applications in live mouse embryos (Table 18.3).
Figure 18.4.

Schematic representation of photoactivation in an embryo constitutively expressing a photoactivatable fluorescent protein fused to a histone. Initially, all cells express the fluorescent protein, which remains however in a non-fluorescent state. Upon excitation of certain cells (within a region of interest, ROI) with short-wavelength high-power (UV or blue) laser, the fluorescent protein switches to a fluorescence emitting state. These photoactivated cells can then be tracked over time. Fusion of the fluorescent protein to a histone protein (e.g., H2B) provides single-cell resolution, and cell tracking of labeled nuclei.
Regarding PC-FPs, transgenic mice constitutively expressing the photo-convertible Kaede, that emits green light and converts to a red fluorescent state upon exposure to UV or violet light, have been generated and used to study cell movements from lymphoid organs to other tissues (Tomura et al., 2008). Moreover, an in utero photoconversion procedure was recently reported by using the Kaede FP (Imai et al., 2010). Another PC-FP that has been used in embryonic stem cells and mouse embryos is the Kikume Green-Red protein (KikGR). KikGR exhibits similar photoconversion properties as Kaede and can be used to label and track cells in embryonic stem cell cultures and in live embryos. KikGR was compared to PA-GFP and two other PC-FPs, Kaede, and PS-CFP2 (which converts from cyan to green fluorescence) for applicability in murine embryonic stem cell lines and live mouse embryos (Nowotschin and Hadjantonakis, 2009b). It was shown that KikGR is most suitable for cell labeling and lineage studies because it is developmentally neutral, bright, and undergoes rapid and complete photoconversion. A recent study reported the development of a transgenic mouse with broad expression of KikGR from the Ubiqui-tin C promoter (Griswold et al., 2011). Although KikGR does not work well when fused to nuclear tags (perhaps due to its tetrameric nature), a recently reported conditionally expressed KikGR reporter with plasma membrane localization was developed, however its level of expression was weak (Abe et al., 2011). The recent development of additional PC-FPs that can convert from red to green or from orange to far-red fluorescence has opened the way for more options to be used for live imaging in the mouse embryo in the near future (Kremers et al., 2009; Piatkevich and Verkhusha, 2010).
3. Tools for Live Cell Imaging
“Seeing is believing” and in order to visualize FP reporters, appropriate microscopy equipment is required. Several optical imaging modalities are widely available (Table 18.4). Advances in imaging techniques have accelerated over the past decade and each type of microscope system provides certain advantages as well as limitations regarding live cell imaging (Table 18.4) (Walter et al., 2010). First, conventional widefield fluorescence microscopy (Lichtman and Conchello, 2005) was considered a powerful tool to observe whole embryos since it provides fast acquisition and flexible excitation at low cost. However, physical destruction could occur as a consequence of widespread exposure and out-of-focus light could interfere during image acquisition because of the thickness of the specimen. Image processing and deconvolution techniques can partially resolve out-of-focus interference; however, these methods have been applied efficiently for smaller specimens, such as bacteria. By contrast, distortion originating from the multiple layers of cells within the thick structure of an embryo is not easy to categorize and formulate, thus precluding use of deconvolution formulae as the system of choice for visualizing details within live embryos using widefield fluorescence.
Table 18.4.
Types of confocal microscope used for live imaging mouse embryos
| System configuration | Advantages | Disadvantages |
|---|---|---|
| Point laser scanning confocal | Most commonly used, best out-of-focus exclusion, best resolution | Slow, decreased sensitivity, high risk of phototoxicity, expensive |
| Slit laser scanning confocal | Fastest confocal, low risk of phototoxicity, high sensitivity | Low resolution |
| Spinning disk confocal | Fast, most gentle confocal, low risk of phototoxicity | Low resolution, not ideal for very thick specimens, works best with only one fluorophore |
| Light sheet based fluorescent | Ideal for very thick specimens, low risk of phototoxicity | Need for rotating device, huge amount of data are generated |
| Two- and multiphoton | Low risk of phototoxicity | Parameters need to be checked before experiment, red and far-red FPs cannot be used, difficult to perform multiphoton experiments |
By contrast, confocal microscopy excludes light originating from outside the focal plane and thus, provides optical sectioning. This allows observation within a complex specimen such as an embryo at subcellular and spatio-temporal resolution (Conchello and Lichtman, 2005). The different optical sections (usually referred to as a z-stack) can then be reconstructed into a 3D projection with appropriate software. For this reason, confocal fluorescence microscopy has become the tool of choice when visualizing live embryos. A list of several types of confocal technologies suitable for live embryo imaging is discussed next.
Point laser scanning (comparable to Zeiss LSM700 or 710 and Leica SP5 systems)
This is the most popular modality. It passes a single point of excitation laser light through a narrow cylinder across a specimen. The laser light illuminates all points of the specimen within the cylinder perpendicular to a specific focal plane. After excitation, fluorophores emit light that then passes through a pinhole. The pinhole functions to exclude light gathered from above and below the focal plane and provides a physical device to out-of-focus light. Thus, point scanning is less prone to be inaccurate because the pinhole function is not based on mathematical formulae (in contrast to deconvolution). A point scanning confocal is usually the system of choice in most laboratories because it provides good resolution and out-of-focus suppression as well as the highest multispectral flexibility. Point scanning with multispectral flexibility allows imaging of multiple probes simultaneously and is the most commonly used approach for performing photoactivation and photoconversion experiments. However, the process of image acquisition is relatively slow because of the time needed for the laser beam to scan the entire sample, which raises concerns about phototoxicity and photobleaching. For imaging live mouse embryos, it is preferable to increase the scan frequency and set up bidirectional xy scanning, thus resulting in a lower risk of photo-toxicity and photobleaching of the specimen. It is worth noting that the slower acquisition obtained with a point scanning confocal could be problematic for imaging fast biological dynamics in live specimens because there will always be a temporal difference from the pixel in the top left corner of the image to the bottom right pixel. Another point that should be taken into consideration regarding point laser scanning microscopy is the detectors used. The typical detector used is a Photomultiplier Tube (PMT). A PMT is less sensitive detector than a CCD camera based system. These devices are preferred because they acquire light quickly and allow the scanning speeds to be much faster than if they were acquired with a CCD. Therefore, commonly used point scanning confocal devices provide the best resolution, when thick specimens are visualized. The trade off is sensitivity, meaning that weaker fluorescent signals and thinner specimens do not resolve as well in point scanning modalities. Another disadvantage of point scanning confocal is its high cost due to the need for several lasers, each specific for only a subset of fluorophores.
Slit scanning (comparable to Zeiss LSM7LIVE systems)
This modality allows for more rapid image acquisition because it passes a fine slit, rather than a point, of laser light across the specimen. A pinhole is again used for limiting the gathering of light originating only from the focal plane. Even though this type of system does not provide better resolution or suppression of out-of-focus light compared to point confocal, it is the system of choice when observing rapid processes (e.g. microtubule or cilia movement) and when samples are sensitive to phototoxicity or photobleach. Moreover, slit scanning (as well as spinning disk confocals, discussed next) use CCD detectors instead of PMTs for detection and do not exclude as much out-of-focus light as point scanning does, allowing for higher sensitivity, especially when weaker signals or thinner specimens are visualized.
Spinning disk-Nipkow type (comparable to Perkin-Elmer UltraView RS5 and Leica SD6000 systems)
These systems are used in our laboratory for imaging pre- and early postimplantation embryos, as well as embryonic stem cells. This modality uses a pair of rotating disks each containing thousands of pinholes. Laser light is passed through these holes and is then projected to the specimen. The emitted light returns through the same holes providing a high-quality confocal image of the entire field of view. In this way the spinning disk confocal collects multiple points simultaneously rather than scanning a single point at a time, allowing for faster image acquisition. Spinning disk confocals, although much faster than point scanning confocals, are not considered faster than slit scanning but they do provide the least invasive imaging of the sample compared to all modalities discussed so far. This gentle acquisition is attributed to the fact that the whole field is excited by laser illumination and detected on a CCD chip. The illumination is not focused at high power on any one spot on the sample. Many different CCD options allow for extremely low laser power imaging for long-term time lapses, hence providing a powerful tool for reduced phototoxicity in live cell imaging. However, image resolution is decreased compared to the slower point laser scanning systems. Moreover, spinning disk confocal acquisition is well suited for imaging single fluorophores, but it does not exhibit great multispectral flexibility. Additionally, a spinning disk confocal may not be the most ideal system for performing photoactivation and photoconversion experiments. There are two additional considerations regarding the use of a spinning disk: the multiple pinholes allow light from one point in the sample to appear in multiple pinholes when imaging deeper into a sample. Therefore, the optical sectioning is only discrete to approximately 20–40 μm of penetration depth. After this depth, the images are closer to standard epifluorescence than confocal sections. It should also be noted that the disk contains multiple pinholes that are not adjustable to different apertures for different magnification objectives. That means the spinning disk (at least the Yokogawa versions) is only taking ideal optical sections with a 100× objective. At 63× the optical section is equivalent to 2 Airy Units (a pinhole of 1 Airy Unit gives the best signal-to-noise ratio), at 40× to 4 Airy Units and the lower the magnification selected, the closer the performance of the spinning disk to that of a standard fluorescence. Some spinning disk systems offer multiple disks with different size apertures depending on the magnification desired.
Light sheet-based fluorescence microscopes (LSFM, SPIM, DSLM) (Hell, 2003; Reynaud et al., 2008; Santi, 2011)
These modalities were further developed from spinning disk confocal systems and allow optical sectioning without illumination of the entire specimen (in contrast to point scanning confocal where the entire specimen is illuminated even though only a single focal plane is observed). As a consequence, phototoxic damage and photo-bleaching of the specimen is considerably reduced. The principle is to illuminate an xy plane at varying angles to the objective imaging the sample. This is achieved by moving the sample so that the optical sheet penetrates the sample at different angles. This modality becomes especially attractive when very thick specimens need to be visualized and other fluorescence techniques cannot be applied. The challenge is that the specimen has to be mounted in a device that can be rotated through 360°. A disadvantage for this sort of imaging is that it acquires a large amount of data which a computer needs to be able to render. Nevertheless, these methodologies are very attractive for imaging live embryos; to date these modalities have been used for zebrafish and Drosophila embryonic development and it is only a matter of time until they are used for mice (Huisken et al., 2006; Keller et al., 2008, 2010).
Super resolution fluorescence microscopy
These modalities encompass recent advances in light microscopy providing increased spatial resolution and allowing the visualization of unobserved details of biological functions and processes. Two of these systems, photoactivated localization microscopy (PALM) (Betzig et al., 2006) and stochastic optical reconstruction microscopy (STORM) (Rust et al., 2006) are not currently suited for imaging samples such as embryos. Both techniques rely on photomanipu-lation of a molecule within the specimen to activate fluorescence and then switch it off again to allow the concentration or localization of the original point of fluorescence. The challenge lies in the fact that the photoactiva-tion needs to take place at discrete x, y, and z coordinates. In a thick specimen, it would be impossible to identify the z plane from which the signal arises. However, the STED (stimulated emission depletion) technique could, in principle, be used for embryo imaging (Hell, 2003). The principle behind STED is to use a laser at one wavelength defocused slightly to compress or shrink fluorescent emission at a second wavelength thus providing higher resolution than a typical confocal image. The challenge is that two lasers of exact wavelengths must be used, and to date STED has only been demonstrated with certain fluorophores. It is also worth noting that to date only two color imaging is possible, and the cost of these systems is relatively high.
Two- and multiphoton microscopy (Helmchen and Denk, 2005; So et al., 2000)
Similarly to confocal systems multiphoton microscopes gather data originating from the focal plane within the specimen. Unlike confocals these microscopes only illuminate the focal plane rather than the entire specimen and they do not require pinholes. Therefore, multiphoton modalities are not considered part of confocal optical technologies. The basic principle for these systems is that the fluorophore is excited by two long-wavelength photons that have to strike it simultaneously. The fluorophore will then emit light of shorter wavelength than that of the excitation light, which is a major difference compared to conventional fluorescence microscopy. Based on this principle, fluorophores from outside the focal plane should not be excited; that makes two- and multiphoton systems powerful tools for live imaging with reduced instances of specimen phototoxicity and photobleaching these microscopes are more costly and not as easy to use and imaging parameters have to be arranged prior to the start of experiment. Also, many red and far-red FPs cannot be used with these types of systems because of their long-wavelength excitation spectra. Moreover, it is more difficult to perform multi-fluorophore experiments with multiphoton systems because of the broad and overlapping excitation spectra. Nevertheless, a recent report showed application of two-photon microscopy for live imaging of preimplantation mouse embryos, providing a framework for application of this powerful technology for imaging mouse embryos at different stages and with various reporters (McDole et al., 2011).
For live cell imaging, systems should be preferably fitted on an inverted microscope base. The maintenance of an environment with a stable temperature, humidity, and gas concentration, which is absolutely essential for proper embryo culture, is difficult to achieve on an upright microscope configuration.
The conditions of stable temperature, humidity, and gas concentration are achieved with use of an environmental chamber that encloses the microscope (Fig. 18.5). These are commercially available but because of their high cost, in-house environmental chambers can also be manufactured.
Figure 18.5.

Microscope set up for static on-stage live imaging of mouse embryos. (A) Inverted point laser scanning confocal microscope and (B) inverted spinning disk confocal microscope. Both systems include an environmental chamber that encloses the stage and optics carrier and provides a heated, humidified, and gassed environment for on-stage embryo culture. Both microscopes are positioned on an anti-vibration air table (adapted from Nowotschin et al., 2010; with permission from Academic Press/ Elsevier).
Confocal-based microscopes should be equipped with appropriate objectives, usually dry 5×, 10×, and 20× (with the latter also can be used multi-immersion), multi-immersion 40× and oil 63×. The 5× objective is used to scan the field of view and locate the embryos. The 10× objective can be used for low-magnification 3D time-lapse imaging. The 20× and 40× objectives are the most suitable for high-magnification 3D time-lapse image acquisition. The 63× objective is rarely used because it is too high a magnification to be suitable for whole embryo imaging. It is also worth considering different types of objectives and which might be most appropriate for live imaging. For example, Carl Zeiss provides two types of objectives: the Neofluar (Neo) lenses and the Plan Apochromat (Plan Apo) objectives. The Neo and Apo objectives provide different degrees of optical corrections (fluorite and apochromatic aberration correction respectively); the Apo objectives enjoy the highest correction for spherical and chromatic aberrations. Therefore, Neo objectives acquire a slightly thicker optical slice than the Apo series, meaning that when imaging through different z planes, each individual image is not as crisp but it is often brighter. However, the Apo lenses acquire a finer optical slice and each individual image is crisper. Specifically, the Apo lenses for the 63× objective have been developed for live cell imaging, providing optimal focus stability for time-lapse experiments.
Finally, a computer workstation(s) running appropriate software are needed for image acquisition and analysis. Software packages used for image acquisition often come with microscope systems and are available from Zeiss, Perkin-Elmer (Volocity), Molecular Devices (Metamorph). Software packages for image analysis are available from Bitplane (Imaris), Perkin-Elmer (Volocity), and Molecular Devices (Metamorph) websites to name a few. Image analysis can also be performed using open source applications such as ImageJ (http://rsbweb.nih.gov/ij/).
4. Methodology
4.1. Microscope setup
For live imaging of ex utero cultured embryos, inverted microscope systems are preferable. The microscope stage must be enclosed within an appropriate environmental chamber that will provide conditions closely resembling in utero development (Fig. 18.5). The chamber allows for embryo culturing with constant temperature at 37 °C as well as with humidity and stable gas content. The precise gaseous mixture comprising CO2/O2/N used for embryo culture depends on the embryonic stage (Fig. 18.1). Usually 5% CO2 is provided for all stages. Regarding oxygen concentrations, 5% O2 is used for preimplantation embryos and early postimplantation embryos (E5.5–E8 stages), and 20% O2 for later postimplantation embryos (E8.5–E10.5; Fig. 18.1).
The incubator should be turned on and allowed to come to temperature at least 1 h prior to starting an imaging experiment. Embryos within cultured media should be placed on glass coverslip chambers or culture dishes containing cover glass bottoms. Efficient imaging can only be acquired when the glass thickness does not exceed 1.5 mm (MatTek dishes or Lab-Tek coverslip chambers). The culture dish should be covered with embryo-quality mineral oil to prevent evaporation during on-stage culture.
In general, UV and laser light originating from other excitation spectra could be harmful to living embryos. Therefore, parameters for image acquisition will need to be optimized to minimize toxicity to embryos. Setting up these imaging adjustments depends on the confocal system being used. For example, when using slower point scanning confocal systems, laser power, and exposure time should be reduced whereas the size of the optical sections and the scan frequency can be increased to prevent photo-toxicity and photobleaching. Additionally, these adjustments greatly depend on the fluorophore that is used. Finally, imaging parameters also depend on the tissue or developmental stage being visualized.
4.2. Collecting and culturing early mouse embryos
Protocols have been developed for ex utero culture of pre- and postimplan-tation mouse embryos. We briefly overview the steps that need to be followed for collecting and preparing on-stage (static) cultures for live imaging preimplantation as well as postimplantation embryos. We also describe how to prepare roller cultures as a way to achieve the most optimal ex utero conditions for the development of postimplantation embryos.
Collection and on-stage culture of preimplantation embryos
After fertilization, mouse embryos float within the upper reproductive tract, making the process of sample recovery much simpler as compared to later postimplan-tation stages of development, which requires dissection. Culture conditions for preimplantation embryos are well established and require appropriate media, stable temperature, and gas content to resemble in utero conditions (Nagy et al., 2003). Briefly, preimplantation embryos are recovered in M2 media and cultured in KSOM media (both are commercially available by Millipore, Specialty Media). Both these media can also be made in-house (Nowotschin et al., 2010). In more detail:
Before starting the dissection of embryos, M2 and KSOM media are prewarmed at 37 °C. The glass bottom of a MatTek dish is covered with 2% Bacto agar (BD Medical) supplemented with 0.9% NaCl in order to prevent embryos sticking to the glass. Around 300–500 microliters of KSOM are then placed on the agar surface and covered with mineral oil to prevent evaporation. The dish should be placed for at least 30 min in a humidified incubator with a stable temperature of 37°C, gassed with 5% CO2.
The dish should not remain outside the incubator for long because changes in temperature and pH and exposure of the embryo in KSOM to atmospheric conditions are deleterious for culture.
After sacrificing a pregnant female (Nagy et al., 2003), the oviduct (when recovering E0.5–E2.5 embryos) or the uterus (when recovering E3.5–E4.5 embryos) is dissected and placed in prewarmed M2 media.
The dish is then placed under a stereomicroscope. The oviduct/uterus are flushed with prewarmed M2 media. A blunt 30-gauge needle is inserted into the oviduct infundibulum for flushing the oviduct. A 1 mL syringe with a 30- or 26-gauge needle is used for flushing the uterus.
Embryos are collected using a mouth pipette attached to a Pasteur pipette that has been pulled over a bunsen burner (Nagy et al., 2003). They are then transferred through several microdrops of KSOM (to wash off residual M2 media) and finally placed in equilibrated KSOM in the MatTek dish. It is essential that the zona pellucida of preimplan-tation embryos recovered after stage E2.5 has to be removed before they are placed on the MatTek dish; this can be performed by transferring the embryos through several microdrops of Acid-Tyrodes (Millipore, Specialty Media). A higher density of embryos enhances their development, thus embryos should be preferably transferred together.
Embryos placed within the equilibrated KSOM microdrop can then be visualized (Fig. 18.6). The steps followed for live imaging are discussed in the next section. Under these on-stage culture conditions, development occurs ex utero from zygote to blastocyst stages (Nagy et al., 2003).
Figure 18.6.
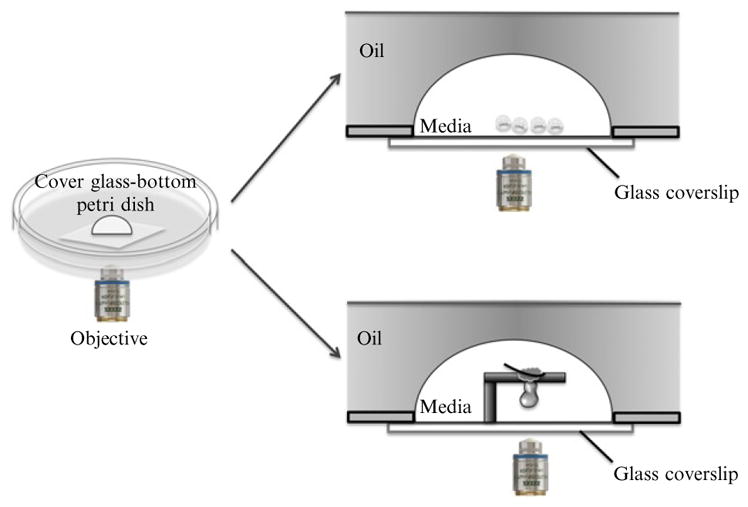
On-stage culture set-up and live imaging of pre- and postimplantation mouse embryos. After dissection, embryos are placed in culture media on glass-bottom dishes covered with mineral oil. The dishes are then positioned on the heated, humidified, and gassed microscope stage. Upper lateral section: preimplantation embryos are placed together in a drop of culture media covered with mineral oil. Bottom lateral section: postimplantation embryos are immobilized for on-stage culture using chamber gaskets. A region (for example the ectoplacental cone, see Nagy et al., 2003) is pierced with an eyelash, so that the embryo is immobilized and remains suspended in culture media in the hole of the gasket.
Collection and on-stage culture of postimplantation embryos
Around embryonic stage E4.0, embryos begin to implant into the maternal uterus and continue their development with a continuous physical connection with the uterine wall. Briefly, postimplantation embryos are dissected free of the uterine tissues in culture media and then cultured in either roller culture conditions (for optimum ex utero culture) or on-stage (static) conditions. The latter being necessary for live imaging and acquisition of time-lapse data.
Dissecting media for collecting postimplantation embryos are 95% DMEM/F12 (1:1) (Invitrogen) supplemented with 5% newborn calf serum (Lonza). After sacrificing the pregnant female, the uterus is removed and placed in a dish containing prewarmed dissecting media.
The uterus is placed under a stereomicroscope where embryos are dissected. Dissection details are provided in Nagy et al., 2003. Imperative for their ex utero development is that embryos are rapidly dissected and remain undamaged during dissection.
Ex utero embryo culture media are DMEM/F12 (1:1) (Invitrogen) supplemented with GLUTAMAX (Invitrogen), 1% penicillin/streptomycin (Invitrogen/Ginco), and rat serum (Harlan Bioproducts or made in-house; Nowotschin et al., 2010). The percentage of rat serum varies depending on the embryonic stage (compare with Fig. 18.1). Prior to dissection, culture media are placed in a culture dish, covered with embryo-tested mineral oil and incubated for at least 1 h in a humidified incubator providing a stable temperature of 37 °C and 5% CO2.
Embryos should be transferred with a transfer pipette into a dish with prewarmed culture media; and only the smallest amount of dissection media should be transferred along with the embryos into this dish. The dish should be incubated in an incubator providing 37 °C temperature and 5% CO2 for 15–20 min for embryos to equilibrate in culture conditions, before they are set up for on-stage culture.
Prior to setting up an on-stage culture, the chamber enclosing the microscope stage should be switched on and prewarmed at 37 °C for at least 1 h. The dish prepared in the previous step is then transferred to the microscope stage and CO2 is immediately provided. Embryos are then ready for imaging, details of which are discussed in the next section (Fig. 18.6).
Note: For some experiments, the embryo needs to be immobilized, so that specific regions are in close proximity to the objective and can be visualized (Fig. 18.6). To do this, embryos are suspended in culture using either suction holding pipette or in modified culture dishes (e.g., with a CoverWell chamber gasket). An eyelash can be inserted through a region not being imaged to position the embryo within the hole of the gasket (Fig. 18.6). After covering the media with mineral oil and transferring the dish to the microscope stage incubator (set at 37 °C and providing 5% CO2), immobilized embryos are ready to be visualized.
Roller culture of postimplantation embryos
Roller culture conditions, even though not suitable for time-lapse imaging, provide the best conditions for ex utero development of postimplantation mouse embryos. These conditions provide stable temperature and continuous gassing while the embryos move constantly within media.
The roller culture incubator is prewarmed at 37 °C prior to setting up the culture. Also, culture media have to be appropriately mixed based on the embryonic stage (Fig. 18.1) and transferred into the roller culture bottles within the incubator to be equilibrated with the appropriate gas composition for at least 1 h prior to starting the culture.
After collecting postimplantation embryos in dissecting media, embryos are transferred to roller culture bottles containing culture media, with approximately 1 mL of media per one embryo. Only the smallest amount of dissecting media should be transferred into the culture along with the embryos.
If using a close system, culture tubes have then to be regassed and sealed and be placed on the rotator that is located within the incubator (details provided in Nowotschin et al., 2010).
Culture media have to be regassed every 6 h (unless a roller culture system with a constant gassing is used) and may need to be replaced with freshly equilibrated media every 24 h.
Note: Live imaging of early postimplantation mouse embryos (E5.5–E6.0). Alternative media compositions have recently been reported for improved culture of very early postimplantation stage embryos (E5.5–E6.0) (Srinivas, 2010). Briefly, embryos are collected in prewarmed M2 media and stored for up to 1–2 h at 37 °C. The culture media used consist of a 1:1 mix of heat-inactivated mouse serum and CMRL medium (Invitrogen) supplemented with L-glutamine. DMEM can be used instead of CMRL, but CMRL appears to give more consistent results (Srinivas, 2010). After dissection, E5.5–E6.0 embryos are transferred in an equilibrated drop of culture media placed on cover glass-bottomed dish and covered with mineral oil. Embryos can then be visualized in static culture (on-stage) as described previously for pre- and postimplantation embryos.
4.3. Imaging early mouse embryos
For live imaging mouse embryos, laser power and exposure time should be decreased as much as possible to reduce the risk of photodamage. To prevent phototoxicity and ensure proper development, decreasing the frequency of scans and perhaps increasing the size of optical sections and scan speed also helps retaining embryo viability. For example, when using a point laser scanning confocal, bidirectional scan in “single track” mode, where all channels are acquired at once, can be performed to achieve faster acquisition. It is preferable to test these imaging parameters when visualizing the same fluorescent reporters in wild-type embryos rather than directly experimenting on embryos of a hard to come by mutant. We usually prefer to image every 15–20 min optical sectioning of 2–4 μm, acquiring z-stacks of up to 150 μm. However, imaging adjustments are determined empirically and depend on the microscope system used (e.g., point scanning vs. spinning disk), developmental stage of the embryo (e.g., the curvature of the embryo at certain postimplantation stages can increase the optical sections needed), and the brightness of the FP reporter. Bright fluorescent reporters are preferable for live imaging because the risk of phototoxicity and photobleaching of the fluorophore is reduced. It is also preferable to know of the exact excitation and emission maxima values of the FP reporter used. The wavelength of excitation should be close to the excitation maximum and the emission filters should capture as much of the emission spectrum as possible (ensuring a high signal-to-noise ratio).
Embryonic drift or movement during growth occurs and is problematic during time-lapse imaging acquisition. For example, preimplantation embryos float freely in the drop of culture media, whereas postimplantation embryos are buoyant, which makes them susceptible to small currents in the culture media and their heavier ectoplacental cone sinks toward the bottom of the dish often reorienting the embryo. To accommodate drifting, it is a good idea to set up a larger z-stack by extending the optical sectioning by several micrometers above and beneath the sample, in order to continuously visualize the embryo even if it slightly moves or grows. Specifically for imaging preimplantation embryos, the amount of media in the drop (too much or too little) can also affect embryo drift; it is worth trying to place several embryos together as it helps to immobilize them. For postimplantation embryos, only suction-holding pipette offers significant control over the orientation of the embryo. The choice of objectives is important; imaging with low magnification (e.g., 5×) could be advantageous when there is embryonic drift because the image field view may not be dramatically affected. Using such a low magnification does not permit high-resolution acquisition for single-cell resolution and thus, image analysis (e.g., cell tracking) can be problematic. Imaging with a higher magnification (e.g., 20× and 40 ×) is preferred for image analysis, but it may require monitoring the embryonic drift and adjusting the focus every few hours throughout the experiment. Alternatively, after time-lapse imaging is complete, drift can sometimes be corrected using software for image analysis.
It is imperative to check for fluctuations in gas flow and temperature during an experiment, as they may have deleterious effects on development of the embryo or time-lapse image acquisition. Also, morphologic characteristics (such as the heart beating, etc.) of the embryo should be monitored during the imaging process. Finally, when a time-lapse imaging experiment is complete the final morphological status of the embryo should be evaluated to ensure that development occurred normally, and that any defects occurred resulting from phototoxicity and/or photodamage.
5. Conclusions
Live imaging FPs in vivo has provided an exciting and rapidly advancing method to study the dynamic processes occurring in the early mouse embryo. However, even though the most recently developed genetically encoded and photomodulatable FPs have been tested successfully in embryos of other model organisms, such as Xenopus, Drosophila, and zebra-fish, their use for live imaging in mice has been limited. This could be attributed to the time-consuming process for the generation of mouse transgenesis, as well as the higher levels of fluorescence needed for proper imaging and the increased risk of phototoxicity. Therefore, it is preferable to use the most recently developed and brightest FPs for generating reporter expressing mice. Bright FPs will allow for the usage of low laser power and faster image acquisition and thus low risk of phototoxicity. It is also imperative for a live fluorescent reporter to be developmentally neutral. Many FPs with oligomeric conformation cannot be fused to subcellular tags because they result in toxicity for the embryo. Therefore, development of monomeric variants for these FPs would be very desirable. Nevertheless, even the generation of monomeric FPs fused to tags can be problematic.
Despite the difficulties that can be encountered when generating FP reporter-expressing, strains of mice, the exciting studies that can be performed using FPs are definitely worth the risk. The field is rapidly moving forward; for example, the multicolored approach exploited in BrainBow mice for labeling and tracking individual cells within a population is attractive for live imaging and tracking individual cells or clones of cells in vivo, providing an alternative to fusion tags for certain applications (Livet et al., 2007; Snippert et al., 2010). Moreover, the generation of “fluorescent timers” that have been tested in live imaging applications in Caenorhabditis elegans and Xenopus embryos hold great potential for use in mice (Chen et al., 2010; Terskikh et al., 2000). These specific FPs can be used to monitor both activation and downregulation of target promoters and thus, trace time-dependent expression (Chudakov et al., 2010; Piatkevich and Verkhusha, 2010). This type of approach would be powerful when investigating fluctuations in protein levels over time in vivo.
We have provided a general overview of the state of the field of live imaging FPs in mouse embryos. The field is rapidly moving forward, with new and improved reporters being continually generated. We thus apologize to the many investigators, whose work has not been discussed due to space constraints.
Acknowledgments
We thank Paul Carman (Carl Zeiss Microimaging) for advice on microscope systems. We thank Marilena D. Papaioannou, Silvia Munoz-Descalzo and Minjung Kang for comments on the manuscript. Work in our laboratory is supported by the Human Frontiers Research Program (HFSP), National Institutes of Health (NIH), and New York State Stem Cell Science (NYSTEM). S.N. is supported by a development grant from the Muscular Dystrophy Association (MDA).
References
- Abe T, Kiyonari H, Shioi G, Inoue KI, Nakao K, Aizawa S, Fujimori T. Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis. 2011;49:579–590. doi: 10.1002/dvg.20753. [DOI] [PubMed] [Google Scholar]
- Arnold SJ, Robertson EJ. Making a commitment: Cell lineage allocation and axis patterning in the early mouse embryo. Nat Rev Mol Cell Biol. 2009;10:91–103. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- Artus J, Panthier JJ, Hadjantonakis AK. A role for PDGF signaling in expansion of the extra-embryonic endoderm lineage of the mouse blastocyst. Development. 2010;137:3361–3372. doi: 10.1242/dev.050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Chen MR, Yang S, Niu WP, Li ZY, Meng LF, Wu ZX. A novel fluorescent timer based on bicistronic expression strategy in Caenorhabditis elegans. Biochem Biophys Res Commun. 2010;395:82–86. doi: 10.1016/j.bbrc.2010.03.143. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Conchello JA, Lichtman JW. Optical sectioning microscopy. Nat Methods. 2005;2:920–931. doi: 10.1038/nmeth815. [DOI] [PubMed] [Google Scholar]
- Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins. Trends Biochem Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- Dieguez-Hurtado R, Martin J, Martinez-Corral I, Martinez MD, Megias D, Olmeda D, Ortega S. A Cre-reporter transgenic mouse expressing the far-red fluorescent protein Katushka. Genesis. 2011;49:36–45. doi: 10.1002/dvg.20685. [DOI] [PubMed] [Google Scholar]
- Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science. 2002;298:1759–1762. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- Egli D, Rosains J, Birkhoff G, Eggan K. Developmental reprogramming after chromosome transfer into mitotic mouse zygotes. Nature. 2007;447:679–685. doi: 10.1038/nature05879. [DOI] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev Biol. 2010;10:121. doi: 10.1186/1471-213X-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink D, Wohrer S, Pfeffer M, Tombe T, Ong CJ, Sorensen PH. Ubiquitous expression of the monomeric red fluorescent protein mCherry in transgenic mice. Genesis. 2010;48:723–729. doi: 10.1002/dvg.20677. [DOI] [PubMed] [Google Scholar]
- Fraser ST, Hadjantonakis AK, Sahr KE, Willey S, Kelly OG, Jones EA, Dickinson ME, Baron MH. Using a histone yellow fluorescent protein fusion for tagging and tracking endothelial cells in ES cells and mice. Genesis. 2005;42:162–171. doi: 10.1002/gene.20139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MD, Udan RS, Hadjantonakis AK, Dickinson ME. Live imaging of mouse embryos. Cold Spring Harb Protoc. 2011a doi: 10.1101/pdb.top10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MD, Udan RS, Hadjantonakis AK, Dickinson ME. Preparation of postimplantation mouse embryos for imaging. Cold Spring Harb Protoc. 2011b;(4) doi: 10.1101/pdb.prot5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MD, Udan RS, Hadjantonakis AK, Dickinson ME. Preparation of rat serum for culturing mouse embryos. Cold Spring Harb Protoc. 2011c doi: 10.1101/pdb.prot5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MD, Udan RS, Hadjantonakis AK, Dickinson ME. Time-lapse imaging of postimplantation mouse embryos. Cold Spring Harb Protoc. 2011d doi: 10.1101/pdb.prot5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:2065–2075. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold SL, Sajja KC, Jang CW, Behringer RR. Generation and characterization of iUBC-KikGR photoconvertible transgenic mice for live time-lapse imaging during development. Genesis. 2011;49:591–598. doi: 10.1002/dvg.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis AK, Dickinson ME, Fraser SE, Papaioannou VE. Technicolour transgenics: Imaging tools for functional genomics in the mouse. Nat Rev Genet. 2003;4:613–625. doi: 10.1038/nrg1126. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, Papaioannou VE. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol. 2004;4:33. doi: 10.1186/1472-6750-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta K, Tsujii H, Omura T. Cell tracking using a photoconvertible fluorescent protein. Nat Protoc. 2006;1:960–967. doi: 10.1038/nprot.2006.96. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y, Yamagata K, Nozaki N, Kimura H. Visualizing histone modifications in living cells: Spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol. 2009;187:781–790. doi: 10.1083/jcb.200904137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hell SW. Toward fluorescence nanoscopy. Nat Biotechnol. 2003;21:1347–1355. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Imai JH, Wang X, Shi SH. Kaede-centrin1 labeling of mother and daughter centrosomes in mammalian neocortical neural progenitors. Curr Protoc Stem Cell Biol. 2010 doi: 10.1002/9780470151808.sc05a05s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isern J, He Z, Fraser ST, Nowotschin S, Ferrer-Vaquer A, Moore R, Hadjantonakis AK, Schulz V, Tuck D, Gallagher PG, Baron MH. Single-lineage transcriptome analysis reveals key regulatory pathways in primitive erythroid progenitors in the mouse embryo. Blood. 2011;117:4924–4934. doi: 10.1182/blood-2010-10-313676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner AL, Zervas M. Genetic inducible fate mapping in mouse: Establishing genetic lineages and defining genetic neuroanatomy in the nervous system. Dev Dyn. 2006;235:2376–2385. doi: 10.1002/dvdy.20884. [DOI] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Santella A, Khairy K, Bao Z, Wittbrodt J, Stelzer EH. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat Methods. 2010;7:637–642. doi: 10.1038/nmeth.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Kinder SJ, Tsang TE, Quinlan GA, Hadjantonakis AK, Nagy A, Tam PP. The orderly allocation of mesodermal cells to the extraembryonic structures and the anteroposterior axis during gastrulation of the mouse embryo. Development. 1999;126:4691–4701. doi: 10.1242/dev.126.21.4691. [DOI] [PubMed] [Google Scholar]
- Kinder SJ, Tsang TE, Wakamiya M, Sasaki H, Behringer RR, Nagy A, Tam PP. The organizer of the mouse gastrula is composed of a dynamic population of progenitor cells for the axial mesoderm. Development. 2001;128:3623–3634. doi: 10.1242/dev.128.18.3623. [DOI] [PubMed] [Google Scholar]
- Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. Photoconversion in orange and red fluorescent proteins. Nat Methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Fraser ST, Eakin GS, Mangano M, Isern J, Sahr KE, Hadjantonakis AK, Baron MH. Tg(Afp-GFP) expression marks primitive and definitive endoderm lineages during mouse development. Dev Dyn. 2006;235:2549–2558. doi: 10.1002/dvdy.20843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Hadjantonakis AK. Eomes::GFP—A tool for live imaging cells of the trophoblast, primitive streak, and telencephalon in the mouse embryo. Genesis. 2007;45:208–217. doi: 10.1002/dvg.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Hadjantonakis AK. Transthyretin mouse transgenes direct RFP expression or Cre-mediated recombination throughout the visceral endoderm. Genesis. 2009;47:447–455. doi: 10.1002/dvg.20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Viotti M, Hadjantonakis AK. The endoderm of the mouse embryo arises by dynamic widespread intercalation of embryonic and extraembryonic lineages. Dev Cell. 2008;15:509–520. doi: 10.1016/j.devcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Conchello JA. Fluorescence microscopy. Nat Methods. 2005;2:910–919. doi: 10.1038/nmeth817. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Long JZ, Lackan CS, Hadjantonakis AK. Genetic and spectrally distinct in vivo imaging: Embryonic stem cells and mice with widespread expression of a monomeric red fluorescent protein. BMC Biotechnol. 2005;5:20. doi: 10.1186/1472-6750-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDole K, Xiong Y, Iglesias PA, Zheng Y. Lineage mapping the pre-implantation mouse embryo by two-photon microscopy, new insights into the segregation of cell fates. Dev Biol. 2011;355:239–249. doi: 10.1016/j.ydbio.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro RM, de Sousa Lopes SM, Bialecka M, de Boer S, Zwijsen A, Mummery CL. Real time monitoring of BMP Smads transcriptional activity during mouse development. Genesis. 2008;46:335–346. doi: 10.1002/dvg.20402. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Saint R. Photoactivatable GFP resolves Drosophila mesoderm migration behaviour. Development. 2007;134:3975–3983. doi: 10.1242/dev.005389. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: The universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. Manipulating the Mouse Embryo. [Google Scholar]
- Nowotschin S, Eakin GS, Hadjantonakis AK. Dual transgene strategy for live visualization of chromatin and plasma membrane dynamics in murine embryonic stem cells and embryonic tissues. Genesis. 2009a;47:330–336. doi: 10.1002/dvg.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Eakin GS, Hadjantonakis AK. Live-imaging fluorescent proteins in mouse embryos: Multi-dimensional, multi-spectral perspectives. Trends Biotechnol. 2009b;27:266–276. doi: 10.1016/j.tibtech.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Ferrer-Vaquer A, Hadjantonakis AK. Imaging mouse development with confocal time-lapse microscopy. Methods Enzymol. 2010;476:351–377. doi: 10.1016/S0076-6879(10)76020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Hadjantonakis AK. Photomodulatable fluorescent proteins for imaging cell dynamics and cell fate. Organogenesis. 2009a;5:135–144. doi: 10.4161/org.5.4.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Hadjantonakis AK. Use of KikGR a photoconvertible green-to-red fluorescent protein for cell labeling and lineage analysis in ES cells and mouse embryos. BMC Dev Biol. 2009b;9:49. doi: 10.1186/1471-213X-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Hadjantonakis AK. Cellular dynamics in the early mouse embryo: From axis formation to gastrulation. Curr Opin Genet Dev. 2010;20:420–427. doi: 10.1016/j.gde.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Hulit J, Subach OM, Wu B, Abdulla A, Segall JE, Verkhusha VV. Monomeric red fluorescent proteins with a large Stokes shift. Proc Natl Acad Sci USA. 2010;107:5369–5374. doi: 10.1073/pnas.0914365107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Verkhusha VV. Advances in engineering of fluorescent proteins and photoactivatable proteins with red emission. Curr Opin Chem Biol. 2010;14:23–29. doi: 10.1016/j.cbpa.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachta N, Bollenbach T, Pease S, Fraser SE, Pantazis P. Oct4 kinetics predict cell lineage patterning in the early mammalian embryo. Nat Cell Biol. 2011;13:117–123. doi: 10.1038/ncb2154. [DOI] [PubMed] [Google Scholar]
- Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis AK. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development. 2008;135:3081–3091. doi: 10.1242/dev.021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poche RA, Larina IV, Scott ML, Saik JE, West JL, Dickinson ME. The Flk1-myr::mCherry mouse as a useful reporter to characterize multiple aspects of ocular blood vessel development and disease. Dev Dyn. 2009;238:2318–2326. doi: 10.1002/dvdy.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud EG, Krzic U, Greger K, Stelzer EH. Light sheet-based fluorescence microscopy: More dimensions, more photons, and less photodamage. Hfsp J. 2008;2:266–275. doi: 10.2976/1.2974980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JM, Pirity MK, Lackan CS, Long JZ, Kondoh G, Takeda J, Hadjantonakis AK. In vivo imaging and differential localization of lipid-modified GFP-variant fusions in embryonic stem cells and mice. Genesis. 2006;44:202–218. doi: 10.1002/dvg.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J, Tam PP. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development. 2009;136:701–713. doi: 10.1242/dev.017178. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi PA. Light sheet fluorescence microscopy: A review. J Histochem Cytochem. 2011;59:129–138. doi: 10.1369/0022155410394857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbedzija GN, Bronner-Fraser M, Fraser SE. Vital dye analysis of cranial neural crest cell migration in the mouse embryo. Development. 1992;116:297–307. doi: 10.1242/dev.116.2.297. [DOI] [PubMed] [Google Scholar]
- Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, Verkhusha VV, Pletnev VZ, Hazelwood KL, Roche PM, Lukyanov S, Zaraisky AG, et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem J. 2009;418:567–574. doi: 10.1042/BJ20081949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo D, Shemiakina II, Ryabova AV, Luker KE, Schmidt BT, Souslova EA, Gorodnicheva TV, Strukova L, Shidlovskiy KM, Britanova OV, Zaraisky AG, Lukyanov KA, et al. Near-infrared fluorescent proteins. Nat Methods. 2010;7:827–829. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi G, Kiyonari H, Abe T, Nakao K, Fujimori T, Jang CW, Huang CC, Akiyama H, Behringer RR, Aizawa S. A mouse reporter line to conditionally mark nuclei and cell membranes for in vivo live-imaging. Genesis. 2011;49:570–578. doi: 10.1002/dvg.20758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- So PT, Dong CY, Masters BR, Berland KM. Two-photon excitation fluorescence microscopy. Annu Rev Biomed Eng. 2000;2:399–429. doi: 10.1146/annurev.bioeng.2.1.399. [DOI] [PubMed] [Google Scholar]
- Srinivas S. Imaging cell movements in egg-cylinder stage mouse embryos. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5539. [DOI] [PubMed] [Google Scholar]
- Stark DA, Kulesa PM. Photoactivatable green fluorescent protein as a single-cell marker in living embryos. Dev Dyn. 2005;233:983–992. doi: 10.1002/dvdy.20385. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Jang CW, Hong NW, Austin AP, Behringer RR. Dual fluorescent protein reporters for studying cell behaviors in vivo. Genesis. 2009;47:708–717. doi: 10.1002/dvg.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P. “Fluorescent timer”: Protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, Kanagawa O. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA. 2008;105:10871–10876. doi: 10.1073/pnas.0802278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trichas G, Begbie J, Srinivas S. Use of the viral 2A peptide for bicistronic expression in transgenic mice. BMC Biol. 2008;6:40. doi: 10.1186/1741-7007-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan RS, Dickinson ME. Imaging mouse embryonic development. Methods Enzymol. 2010;476:329–349. doi: 10.1016/S0076-6879(10)76019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viotti M, Nowotschin S, Hadjantonakis AK. Afp::mCherry, a red fluorescent transgenic reporter of the mouse visceral endoderm. Genesis. 2011;49:124–133. doi: 10.1002/dvg.20695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker SA, Oswald F, Wiedenmann J, Knochel W. A green to red photoconvertible protein as an analyzing tool for early vertebrate development. Dev Dyn. 2007;236:473–480. doi: 10.1002/dvdy.20955. [DOI] [PubMed] [Google Scholar]
- Walter T, Shattuck DW, Baldock R, Bastin ME, Carpenter AE, Duce S, Ellenberg J, Fraser A, Hamilton N, Pieper S, Ragan MA, Schneider JE, et al. Visualization of image data from cells to organisms. Nat Methods. 2010;7:S26–S41. doi: 10.1038/nmeth.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka Y, Lanner F, Rossant J. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development. 2010;137:715–724. doi: 10.1242/dev.043471. [DOI] [PubMed] [Google Scholar]