

Abstract
Langerhans cells (LC) are antigen presenting cells responsible for initiating an immune response against human papillomaviruses (HPV) entering the epithelial layer in vivo as they are the first immune cell that HPV comes into contact with. LC become activated in response to foreign antigens, which causes internal signaling resulting in the increased expression of co-stimulatory molecules and the secretion of inflammatory cytokines. Functionally activated LC are then capable of migrating to the lymph nodes where they interact with antigen specific T cells and initiate an adaptive T cell response in vivo. However, HPV has evolved in a manner that suppresses LC function, and thus the induction of antigen specific T cells is hindered. While many methods exist to monitor the activity of LC in vitro, the migration and induction of cytotoxic T-cells is ultimately indicative of a functional immune response. Here, methods in analyzing functional migration and induction of antigen specific T cells after stimulation of LC with HPV virus-like particles in vitro are described.
Keywords: HPV16, Langerhans cells, ELISpot, Antigen-specific T cells, Migration, In Vitro Immunization
1. Introduction
High-risk human papillomaviruses (HPV) are sexually transmitted viruses that cause several cancers including cervical cancer [1]. Of the different cancer-causing HPV genotypes, HPV type 16 (HPV16) is by far the most common, leading to more than 50% of all cervical cancers [2]. During its natural life cycle, HPV16 infects the basal cells of the epithelium in vivo where it interacts with Langerhans cells (LC), the resident antigen presenting cells (APC) of the epithelium [3]. Due to their location, LC are responsible for initiating immune an adaptive immune response against pathogens entering the epithelial layer [4].
The expression of HPV16 viral genes and the production of new infectious virions is dependent on the differentiation of basal epithelial cells into mature keratinocytes [5]. This has led much of the papillomavirus research field to utilize virus-like particles (VLP) to study specific aspects of viral internalization and HPV-induced immune responses. There are 360 copies of the major capsid protein L1, which self-assemble into L1-only VLP when expressed alone, and possess an icosahedral structure composed of 72 L1 pentamers [6]. If L1 is expressed with the minor capsid protein L2, there are between 12 and 72 L2 proteins incorporated per capsid [6,7].
Initial HPV-LC immune responses are mediated through the interaction between HPV capsid proteins and LC surface receptors making VLP a valuable tool for studying HPV capsid protein-mediated immune responses in vitro. Although L1 is sufficient to form a VLP, L1L2 VLP more closely resemble wild type infectious virions. Furthermore, we have recently demonstrated that LC exposed to the L2 protein in HPV16 L1L2 VLP do not become activated. It has also been shown that epithelial LC respond differently to HPV16 VLP when compared to dermal dendritic cells (DC) [11] which suggests that HPV16 has evolved in a specific mechanism to manipulate this unique epithelial APC. Interestingly, L1 VLP lacking the L2 protein were shown to activate LC, and those activated LC were functional in initiating an adaptive immune response including the induction of HPV specific cytotoxic T-cells [8,9].
Proper antigenic stimulation leads to a unique signaling cascade within LC shortly after initial contact. Specifically, activation of the PI3K/AKT signaling cascade has been observed in activated LC, and the HPV16 L2 minor capsid protein manipulates this pathway [8,10-12]. The PI3K/AKT signaling cascade is activated within minutes in response to certain stimuli. The downstream consequences of activation-associated signal cascades within LC include the upregulation of genes associated with T cell stimulation such as CD80 and CD86, the release of inflammatory cytokines such as TNFα and IL-12, and the translocation of MHC class II to the extracellular surface [8,13]. Functionally activated LC then migrate to the lymph nodes where they induce antigen specific cytotoxic T-cells with a milieu of proper co-stimulatory molecules and cytokines in an adaptive immune response [14-16].
Here, two different modalities for analyzing the functional activation of LC exposed to HPV16 VLP in vitro are described in detail. Each assay uses HPV VLP to assess different aspects of functional LC activation from the migration of LC towards a chemokine gradient and the induction of HPV-specific CD8+ cytotoxic T cells. Likewise, the initial steps for the activation of LC are constant for each experiment, and each assay will differ in the methods necessary for detection and analysis. For instance, LC capable of migrating through polycarbonate filters are counted, which would be indicative of a functional LC expressing a chemokine receptor and capable of migrating to a lymph node. Then, an Enzyme-linked Immunosorbant Spot Assay will be described for detection of LC-induced HPV-specific T cells from an In Vitro Immunization.
2. Materials
1. Langerhans Cells
LC can be directly purified from epithelial tissue via separation techniques or can be derived in vitro from CD34+ progenitor cells or peripheral blood monocytes as previously described [8, 13]. However, the activation assays described herein have been consistently performed on LC derived from monocytes. (Section 3.1). Peripheral blood mononuclear cells (PBMC) from healthy donors are obtained by leukapheresis. Leukocytes are purified by Ficoll gradient centrifugation (Nycomed, Oslo, Norway) and stored in liquid nitrogen prior to differentiation (∼150 × 106 PBMC/cryovial; see Note 1).
2. Virus-Like Particles
HPV16L1 VLP, HPV16L1L2 VLP and HPV16 L1L2-E7 chimeric-VLP (cVLP) can be produced using a recombinant baculovirus expression system in insect cells as previously described [6]. Western blot analyses confirms the presence of L1 and L2 while an ELISA and transmission electron microscopy can be used to confirm the presence of intact particles. An E-toxate kit (Sigma-Aldrich) is used to semi-quantitate endotoxin. Baculovirus DNA used in VLP production procedure has been shown not to activate LC, however endotoxin levels in preparations need to be shown to not activate LC; levels of less than 0.06 in our own preparations do not activate LC. VLP are stored at −80°C until needed (see Note 2).
3. Solutions
LC/DC complete medium: RPMI 1640 containing 10 mM sodium pyruvate, 10 mM non-essential amino acids (NEAA; Life Technologies), 100 μg/ml penicillin-streptomycin, 55 μM 2-mercaptoethanol and 10% heat-inactivated FBS
Phosphate buffered saline (PBS)
PBS+0.5% Tween-20 filter sterilized
PBS+0.05% Tween-20 for plate washing
PBS+0.5% BSA
0.05 M Sodium Acetate buffer pH 5.0
0.2 M Sodium Acetate
0.2 M glacial acetic acid
30% H2O2
Dimethyl Formamide (DMF; Sigma)
Sterile MACS Buffer (PBS+2mM EDTA+0.5%BSA)
PBS+1%BSA filter sterilized
13. Cytokines and Chemokines
GM-CSF: granulocyte macrophage colony stimulating factor (Genzyme) reconstituted in complete medium at 1 × 105 units/mL (18 ng/μL).
rhIL-4: recombinant human interleukin 4 (Invitrogen) reconstituted in PBS at 40 μg/mL.
TGF-β: Transforming growth factor beta (BioSource International) reconstituted in PBS at 5 μg/mL. After reconstituted, cytokines were and stored following the manufacturer's instructions.
Recombinant human CCL21 (rCCL21; R&D, 25 μg/mL stock (100x)).
rhIL-7: recombinant human interleukin 7 (Peprotech) reconstituted in PBS/1%BSA at 5 μg/mL.
rhIL-10: recombinant human interleukin 10 (Peprotech) reconstituted in PBS/1%BSA at 5 μg/mL.
β2-microglobulin: (Sigma M4890) Dissolve 250 μg of β2-microglobulin in 830 μL of sterile water for 300 μg/mL stock. Spin filter through 0.22 μm membrane.
8. Antibodies and Peptides
IFNγ capture antibody (Mabtech mouse anti-human IFNγ, Clone 1-D1K, 1 mg/mL)
IFNγ detection antibody (Mabtech mouse anti-human IFNγ-biotin, Clone 7-B6-1, 1 mg/mL)
Streptavidin-Horse Radish Peroxidase conjugate, 1 mg/ml (Sigma)
Lipopolysaccharide (LPS; Sigma). LPS was reconstituted in PBS at 1 mg/mL and stored following manufacturer's instructions.
Peptides: Weigh out a few mg of necessary peptide for IVI and ELIspot. Dissolve in DMSO for a concentration of 10 mg/mL. Add sterile PBS to dilute to a concentration of 2 mg/mL.
6. Dry Chemical
3-amino-9-ethyl-carbazole (AEC), 20 mg tablets (Sigma)
2. Dry Material
Millipore Multiscreen HTS IP 96 well plates
Disposable culture tubes - glass (VWR)
Costar 5.0 μm pore size 6.5mm diameter insert Transwell 24-well plates (Corning)
Miltenyi MACS Human CD8 T cell Isolation Kit (Cat. 130-094-156)
Ziess Elispot Reader
Beckman Coulter Z1 Particle Counter (optional)
Upright or inverted microscope capable of counting cells
8. Methods
3.1 Generation of Langerhans Cells
As previously mentioned, primary LC can be isolated from tissue or derived from precursor cells. The following steps are used to derive LC from peripheral blood monocytes.
Day 1 (or Day -9 for an In Vitro Immunization Assay: Section 3.4)
Thaw frozen PBMC and wash once with LC complete medium. To do this, add thawed cells to ≥20 mL warm LC medium, centrifuge at 500 RCF for 5 min, and decant supernatant to wash away dimethyl sulfoxide (DMSO) used for liquid nitrogen storage. Repeat wash step. Resuspend cell pellet in 10 mL complete medium.
Plate ∼150 × 106 cells (PBMC should have been counted before liquid nitrogen storage) in a 175-cm2 tissue culture flask for 2 h at 37°C to select for plastic adherent cells. Do not disturb culture flasks during this time period.
Pour off medium and gently wash away excess non-adherent cells away with warm PBS.
Culture remaining adherent cells for 7 days in 30 mL fresh LC complete medium, and add 1000 U/mL GM-CSF, 1000 U/mL IL-4, and 10 ng/ml TGF-β; thaw date is day 1.
Replenish 50% GM-CSF and IL-4 on day 3 and 100% on day 5; replenish 100% TGFB on days 3 and 5 (see Note 3 for feeding details).
At the end of 7 days, optional phenotyping can be performed to test for proper differentiation (see Note 4) and an LC in-vitro migration assay may be performed.
1. Quantification of VLP Prep
Thaw and quantify VLP in mg/mL. Though quantification can be done before cold storage, it is recommended to re-quantify after thawing. The quantification of VLP is preferably done with a coomassie blue stain of an electrophoresed reducing gel where the protein content of the L1 band is quantified according to a predetermined quantified standard (see Note 5 for tips on coomassie quantification).
If using different VLP preps (i.e. HPV16 L1 VLP, HPV16 L1L2 VLP, HPV16 L1L2-E7 cVLP or VLP from different HPV genotypes), bring all VLP to the same concentration of 20 μg/100 μL PBS.
3.3 LC In-Vitro Migration Assay
The migration of LC to local lymphoid organs during an infection indicates the functional capacity of LC to perform their role of presenting antigen to responder cells in T-cell rich areas of lymph nodes and spleen.
Day 7
Collect LC for the assay and count cells (2 ×106 LC are necessary per treatment).
Incubate LC with VLP at 37°C 5% CO2 in PBS for 1 hour.
Transfer cells to a 12-well plate in a volume of 2 mL complete RPMI with 500 U/mL of GM-CSF. Incubate at 37°C 5% CO2 for 4-6 hours.
Add LPS (1-5 μg/mL) to wells or add appropriate activators to the wells. Incubate plates for an additional 24-48 hours.
Pre-wet transwells with 600 μl of complete RPMI by adding media to the lower chamber of each well. Incubate overnight at 37°C.
Day 9
-
6
Collect LC from 12-well plates. Resuspend cells in 600 μl of complete RPMI. Count the number of LC and adjust concentration to 2 ×106 cells/mL.
-
7
Move the transwell inserts to an empty well using forceps. Remove media from the lower transwell chambers.
-
8
Add 600 μL media containing 250 ng/mL CCL21 to each well in row B of each plate. Add 600 μl media each well in row C of each plate. Replace the transwell inserts. Each sample is tested in triplicate wells, with and without CCL21. Each plate contains 12 inserts; 2 treatment conditions can be tested using one plate.
-
9
Add 100 μL of LC to transwell inserts corresponding to a treatment condition. Each condition requires 6 wells. Incubate plates for 4 hours at 37°C 5% CO2.
-
10
After incubation, remove the transwell inserts with forceps. Transfer cells in the entire volume into coulter counting vials (see Note 6). Keep each well separate.
-
11
Add 9.4 mL of counting sheath fluid or PBS to each vial. Count cells using Coulter Counter. Change output to number of cells counted in the metered volume (0.5 mL).
-
12
To calculate the number of cells that migrated into the lower chamber, multiply value by 20 (dilution factor).
-
13
Calculate the migration index as number of cells migration to CCL21/number cells migrating spontaneously.
1. In-Vitro Immunization Assay (IVI)
In Vitro Immunization assay assesses a donor's CD8+ T cell capability in responding to various HPV16 VLP exposed LC in a 6 week experiment. An ELISpot assay (see Section 3.5) follows to assess the number of E7-specific CD8+ IFNγ releasing T cells.
Table 1. Sample Schedule. Start date of IVI (Day -9) may be moved to different days of the week.
Table 1. Sample In Vitro Immunization Assay Schedule.
| Sunday | Monday | Tuesday | Wednesday | Thursday | Friday | Saturday | |
|---|---|---|---|---|---|---|---|
| Week 1 | Day -9: Start LC (A), Feed LC (A) Full1 | Day -7: Feed LC (A) half | Day -5: Feed LC (A) Full | ||||
| Week 2 | Day -2: Activate LC (A) and Start LC (B), Feed LC (B) Full | Day 0: Initial start of co-culture of CD8+ cells with LC (A), IL-7 to co-cultures, Feed LC (B) half | Day 1: IL-10 to co-cultures | Day 2: Feed LC (B) Full | |||
| Week 3 | Day 5: Activate LC (B) and Start LC (C), Feed LC (C) full | Day 7: Restimulation with LC (B), Feed LC (C) half | Day 8: IL-10 to co-cultures | Day 9: Feed LC (C) Full, IL-2 to co-cultures | |||
| Week 4 | Day 11: IL-2 to co-cultures | Day 12: Activate LC (C) and Start LC (D), Feed LC (D) full | Day 14: Restimulation with LC (C), Feed LC (D) half | Day 15: IL-10 to co-cultures | Day 16: Feed LC (D) Full, IL-2 to co-cultures | ||
| Week 5 | Day 18: IL-2 to co-cultures | Day 19: Activate LC (D) | Day 21: Restimulation with LC (D) | Day 22: IL10 to co-cultures | Day 23: IL-2 to co-cultures | ||
| Week 6 | Day 25: IL-2 to co-cultures | Day 28: ELISpot2 |
see Note 3
refer to Section 3.5 for full protocol on ELISpot assay
Day -9 of IVI: Start LC
Follow protocol from Section 3.1.
Day -2, 5, 12, and 19 of IVI: Activate LC
-
2
Activate LC with the necessary treatment for the experiment. Incubate LC with 20 μg of HPV16 L1L2-E7 cVLP per 106 LC in 500 μl of PBS for 1 hour at 37°C (see Note 7).
-
3
Transfer cells to a T-25ul flask (1-2×106 LC in 8 mL) or T-75 (>2×106 LC in 20 mL) flask for 3 hours prior to adding activators (LPS, TLR agonist, or nothing).
-
4
Incubate flask for 48 hours at 37°C 5%CO2.
-
5
Follow protocol from section 3.1 to start a new batch of LC from the same donor (do not start new donor on Day 19).
Day 0 of IVI: Set up and start IVI assay
-
6
Thaw ∼250×106 non-adherent PBL (from same donor) for every 4 LC treatments planned. Need 25×106 CD8+ T cells/plate (1 plate = 4 treatment groups) (see Note 8).
-
7
Wash cells twice with complete medium and resuspend cells in 20 mL of complete medium.
-
8
Incubate cells in a 50 mL conical tube for 1 hour at 37°C 5% CO2 with loosen lid to allow air exchange.
Peptide Pulsing LC
-
9
Collect LC that need peptide pulsing from Day -2 of IVI in a 15 mL conical tube.
-
10
Top off with PBS/1%BSA and spin down at 500 RCF for 5 minutes.
-
11
Wash LC again in 10 mL PBS/1%BSA and spin down at 500 RCF for 5 minutes.
-
12
Resuspend cells in supplemented RMPI (serum free) media/1%BSA.
-
13
Pulse with HLA-A2-binding peptide or peptide pool with 10ul of β2-microglobulin (final concentration of 3 μg/ml).
-
14
Add 10-15 ml of peptide (2 mg/ml stocks) to the tube for a final concentration of 25 μM.
-
15
Incubate treated cells for 4 hours at 37°C. Resuspend cells every hour.
Magnetic Separation of CD8+ Cells (MACS Protocol)
-
16
Centrifuge PBL that were thawed from step 8 (after 1 hour incubation) at 500 RCF for 5 minutes.
-
17
Resuspend cell pellet in 50 mL of cold sterile MACS buffer. Count cells with cell counter.
-
18
Spin down at 500 RCF for 5 minutes.
-
19
Follow instructions on MACS CD8+ Isolation kit.
-
20
Count the number of CD8+ cells isolated.
Irradiating LC
-
21
After peptide pulsing LC (following step 15), collect peptide pulsed LC and LC that did not need peptide pulsing in their individual tubes.
-
22
Wash cells once with medium and resuspend in 10 mL for irradiation.
-
23
Irradiate all LC for 30 Gray (3000 Rad).
-
24
Spin down at 500 RCF for 5 minutes and wash twice with complete medium.
-
25
Resuspend cells in 1 mL and count LC in each treatment group, adjust concentration to 1.25×105 LC/mL.
Plating out CD8+ cells and LC (see Figure 1)
Figure 1.
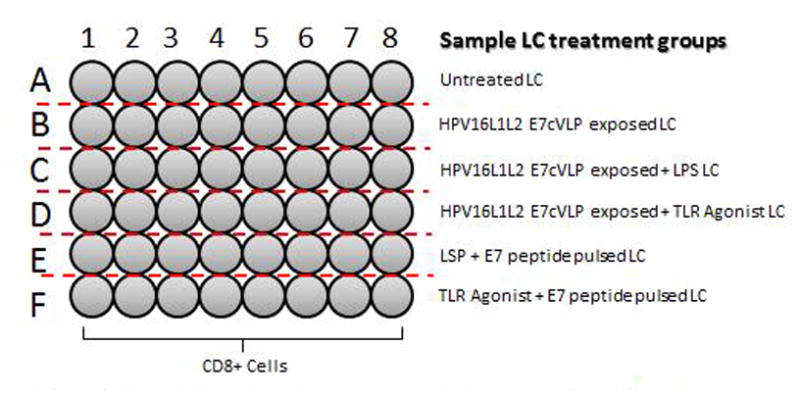
Example plate set up for an in vitro immunization. All wells contain 5×105 cells per well. Each row contains a different LC exposed treatment group (2×104 LC/well).
-
26
Adjust concentration of CD8+ cells to 2.5×106 cells/mL (from step 20). Add 200 μL (5×105 CD8+ cells) to each well in a 48 well plate.
-
27
Add 200 μL of appropriate irradiated LC (2×104 LC) to 8 wells according to their treatment groups.
-
28
Add 100 μl of IL-7 to each well (final concentration of 10 ng/mL).
-
29
Incubate plates at 37°C 5% CO2 for 24 hours.
Day 1 of IVI: Cytokines
-
30
Add 50ul of IL-10 to each well (final concentration of 10 ng/mL).
-
31
Incubate for 6 days. Do not shake plates.
Day 7 of IVI: Restimulation of cultures
-
32
Follow steps 9-15 and 21-14 from Day 0 protocol for peptide pulsing.
-
33
Adjust concentration of LC to 8.5×104 LC/mL.
-
34
Remove 300 μL of medium from each well.
-
35
Add 300 μL of appropriate irradiated LC to each treatment row (2.5×104 cells).
-
36
Incubate at 37°C 5% CO2 for 7 days.
Day 8 of IVI: Cytokines
-
37
Add 50 μL of IL-10 to each well (final concentration of 10 ng/mL)
Day 9 and 11 of IVI: Cytokines
-
38
Add 50 μL of 500 U/mL IL-2 (final concentration of 50 U/mL).
Day 14 of IVI: Restimulation of Cultures
-
39
Follow Day 7 protocol steps 32-36.
Day 15 of IVI: Cytokines
-
40
Add 50 μL of IL-10 to each well (final concentration of 10 ng/mL)
Day 16 and 18 of IVI: Cytokines
-
41
Add 50 μL of 500 U/mL IL-2 (final concentration of 50 U/mL).
Day 21 of IVI: Restimulation of Cultures
-
42
Follow Day 7 protocol steps 32-36.
Day 22 of IVI: Cytokines
-
43
Add 50 μL of IL-10 to each well (final concentration of 10 ng/mL)
Day 23 and 25 of IVI: Cytokines
-
44
Add 50 μL of 500 U/mL IL-2 (final concentration of 50 U/mL).
Day 28 of IVI: ELISpot
-
45
Follow instructions from Section 3.5. (Day 1 of ELISpot assay should be performed on Day 27 of IVI).
3.5 Enzyme-linked Immunosorbant Spot Assay (ELISpot)
To assess whether LC were capable of inducing CD8+ T cell specificity towards a certain antigen, an ELIspot assay may determine IFNγ CD8+ releasing T cells. CD8+ cells may be isolated from murine organs, in-vitro immunization, and other samples.
Day 1 of ELISpot (all reagents need to remain sterile, perform in Biosafety Cabinet)
Dilute capture IFNγ antibody to 10 μg/mL with PBS, 10mL of working solution is necessary for 1 plate.
Prewet PVDF HTC filter plates with 20 μL of 70% sterile ethanol for a maximum of 1 minute. As soon as the filter turns from white to gray, flick to remove ethanol. Wash plate 3 times with 200 μL of PBS. Blot on paper towels between washes (see Note 9).
Add 100 μL coating antibody per well.
Wrap in plastic wrap and incubate overnight at 4°C (see Note 10).
Day 2 of ELISpot (all reagents need to remain sterile, perform in Biosafety Cabinet)
-
5
Flick out antibody solution from PVDF HTC filter plates from Day 1 of ELISpot.
-
6
Wash wells once with 200 μL of 0.5% sterile PBST. Wash twice with 200 μL of sterile PBS. Blot on paper towels between washes.
-
7
Block wells with 200 μL of complete RPMI. Incubate at 37°C 5% CO2 for a minimum of 2 hours.
-
8
Collect CD8+ cells in a 50 mL tube, spin down at 500 RCF for 5 minutes and resuspend in 30 mL of complete RMPI. Let T cells rest in a polypropylene tube with loosened cap for 2 hours in 37°C 5% CO2 incubator (see Note 11).
-
9
Prepare a working concentration of peptide appropriate for T-cell specificity such that CD8+ cells will be in a final concentration of 2-10 μg/mL in each well. Store peptides at 4°C until ready.
-
10
Prepare PHA positive control. Dilute stock concentration of PHA to a working solution of 30 μg/mL. 50 μL will be added per well for a final concentration of 10 μg/mL. Store PHA at 4°C until ready to use.
-
11
Collect resting CD8+ T cells, spin down at 500 RCF for 5 minutes and resuspend cells in 15 mL of complete media. Count and adjust cell concentration to 1 ×106 T cells/mL. A minimum of 4 ×106 cells are necessary per group.
-
12
Flick out blocking medium from HTC PVDF filter plates, blot on paper towel prior to adding any solutions/cells.
-
13
Add 50 μL of medium (background count), working peptide solution to appropriate wells, or PHA to necessary wells prior to adding cells. Add an extra 50 μL of medium to rows C+D and G+H.
-
14
For each treatment, add 1 ×105 (100 μL) to row A+B (or row E+F). Add 5 ×104 cells (50 μL) to row C+D (or row G+H). Add cells to middle of wells slowly to avoid pushing cells to outer edge of well.
-
15
Incubate plates for 16-18 hours in 37°C incubator that is infrequently used. Do not disturb plates during incubation (e.g. moving or vibrations from opening door).
Day 3 of ELISpot (maybe performed outside of the Biosafety Cabinet)
-
16
Flick plates empty and wash plates 6 times with 0.05% PBST, soaking 3 minutes between every other wash. Leave 0.05% PBST in the last wash.
-
17
Prepare biotin-IFNγ antibody to 1 μg/ml in PBS/0.5% BSA (10 mL antibody solution is necessary for 1 plate). Filter sterilize solution through a 0.22 μm Millex LGV low-protein-binding syringe filter.
-
18
Discard 0.05% PBST wash buffer from plate. Blot plate on paper towel and apply 100ul antibody working solution per well. Incubate at room temperature for 2 hours.
-
19
Discard antibody and wash plates 6 times with 0.05% PBST. Leave 0.05% PBST in the last wash.
-
20
Dilute Strepavidin-Horse Radish Peroxidase conjugate 1/4000 in PBS/0.5% BSA solution.
-
21
Discard 0.05% PBST wash buffer from plate. Blot plate on paper towel and apply 100 μL of SA-HRP solution per well. Incubate at room temperature for 1 hour.
AEC Substrate
-
22
Prepare AEC solution 10 minutes prior to finishing the 1 hour incubation (from step 21 Section 3.4). (Performed in fume hood). Dissolve 20 mg tablet of AEC in 2.5 mL DMF in a disposable glass test tube. Wait until completely dissolved (5 minutes).
-
23
Prepare fresh 0.05 M acetate buffer pH 5.0 (see Note 12).
-
24
To 47.5 mL of 0.5 M acetate buffer, add dissolved 2.5 mL of AEC tablet/DMF mixture to tube and mix well. Add 25 μL of 30% H2O2 to the 50 mL solution. Filter through a 0.45 μm millex syringe filter. Use immediately (step 25 Section 3.4) and discard remaining solution (see Note 13 for waste procedure).
Developing plate
-
25
After plates are ready (1 hour incubation with SA-HRP). Wash plates 3 times with 0.05% PBST and then wash 3 times with PBS. Leave PBS in wells during the last wash.
-
26
Discard PBS wash. Blot on paper towel to dry. With a multichannel pipette, apply 100 μL AEC substrate per well. Start a timer for 5 minutes when AEC substrate has been added to the last row of well.
-
27
Stop reaction by washing extensively with deionized water (see Note 14).
-
28
Remove underdrain to rinse the underside of wells. Shake out water from wells and blot on paper towels to dry. Gently blot the underside of plates and allow the plates to air dry upside down in the dark.
Reading plates and Analysis
-
29
3-5 days subsequently to development, transfer filter wells to a punch sheet (see Note 15). Follow factory instructions from the Zeiss ELISpot reader.
-
30
Normalize the number of counts to 1×106 cells per well (Multiply wells that received 1×105 cells by 10 and wells that received 5×104 cells by 20).
-
31
Normalized the number of spots to the background count.
32. Notes
PBMC should be stored in liquid nitrogen with freezing medium containing 50% LC/DC complete medium, 40% FBS, and 10% DMSO. Experiments call for ∼200 × 106 cells to be cultured per culture flask so ideally freeze this number of PBMC/1mL freezing medium per cryovial.
When possible, avoid multiple freeze-thaw cycles of VLP preparations.
-
For cytokines reconstituted at concentrations suggested in Section 2.4, add cytokines as follows:
Day 1: 300 μL GM-CSF, 200 μL IL-4, 60 μL TGF-β
Day 3: 150 μL GM-CSF, 100 μL IL-4, 60 μL TGF-β
Day 5: 300 μL GM-CSF, 200 μL IL-4, 60 μL TGF-β
Properly derived LC express certain surface markers that can be detected with extracellular immuno-staining and flow cytometry. For example, LC express CD207 (Langerin), E-cadherin, and CD1a.
Many protein quantification methods (ex. Bradford and coomassie) depend on a reagent binding to available primary amine groups on the protein of interest, which causes a change in absorption of the reagent at a pre-determined wavelength. VLP have many of these available amine groups hidden within the stable capsid structure, therefore a method that breaks apart the capsid and linearizes the individual capsid proteins such as separation with SDS-PAGE in reducing conditions followed by a coomassie blue stain has been most consistent in our own experiences, though other methods may also work. For a coomassie stain of VLP, first prepare VLP running samples for reducing SDS-PAGE (VLP sample with loading buffer and reducing agent). Heat running samples to 95°C for ten minutes to melt VLP into individual components (i.e monomeric L1 and L2). Load samples and predetermined protein standards (i.e. BSA in increasing concentrations) into a 10% BT gel along with a protein ladder. Electrophorese gel at 160 V for 1 hr. Pre-fix gel with 20 mL fixing solution (43% acetic acid and 7% methanol added to deionized H20) for 15 min. Wash with approx. 100 mL deionized H20 3 × 5 m. Add 20 mL GelCode Blue Stain Reagent (Thermo) until protein bands appear (approx. 10-30 m). Wash gel with 100 mL deionized H20 3 × 5 min. Optimization and modifications can be made per manufacturer's instructions. Scan with infrared scanner and quantify L1 bands (L1 appears as a strong band at ∼55 kDa with 700 nm excitation) according to protein standards.
Cells may be counted manually using a microscope and hemacytometer or via other cell-counter apparatus, please follow instructions of other devices for total cell count.
cVLP must be used in activating LC for an IVI. HPV16 L1L2-E7 cVLP contains E7 peptide linked to the L2 monomer. Treatment groups may include the following: untreated LC, HPV16L1L2-E7 cVLP exposed LC, HPV16L1L2-E7 cVLP + LPS exposed LC, LPS + E7 peptide pulsed LC, etc.
Assume 10-15% CD8+ cells in PBL population.
Plates should be flicked into a biohazard waste container. Never let the membrane of the plates dry out.
Plates maybe coated on Day 2 of ELISpot if forgotten on Day 1 of ELISpot. Execute Day 1 of ELISpot and incubate plates at 37°C for 2 hours (replacing step 4 of Section 3.4).
This step quenches remaining cytokines that CD8+ may be releasing that will bias IFNγ results.
To prepare fresh 0.05M acetate buffer pH 5.0, to 54.7 mL MilliQ water, add 5.5 mL 0.2M sodium acetate and 2.3 mL 0.2 M glacial acetic acid.
Discard any unused AEC substrate in special waste container in fume hood, label correctly and contact biosafety department for disposal procedure.
Plates may be submerged into a bucket of deionized water to quench reaction. Do not overdevelop plate or background will be high.
To remove reddish/pink background on filter plate, allow plates to sit in a lit-room until background fades to a lighter yellow/white hue. Do not overexpose plates to light.
Figure 2.
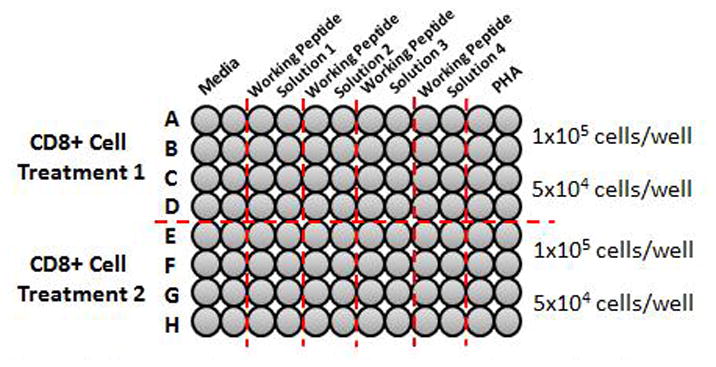
Example plate set up. CD8+ cells may be tested against more than 1 antigen of choice (Working peptide solution x). Two CD8+ treatments may be tested on 1 plate.
References
- 1.Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. The Journal of Pathology. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Bosch FMM, Munoz N, Sherman M, Jansen A, Peto J, Schiffman M, Moreno V, Kurman R, Shah K. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on servical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 3.Stanley MAP MR, Coleman N. HPV: from infection to cancer. Biochemical Society Transactions. 2007;35:1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- 4.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nature reviews Immunology. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 5.Cumming SC-I T, Milligan S, Graham SV. Human Papillomavirus type 16 late gene expression is regulated by cellular RNA processing factors in response to epithelial differentiation. Biochemical Society Transactions. 2008;36:522–524. doi: 10.1042/BST0360522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, et al. Arrangement of L2 within the papillomavirus capsid. Journal of virology. 2008;82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahey LM, Raff AB, Da Silva DM, Kast WM. A major role for the minor capsid protein of human papillomavirus type 16 in immune escape. Journal of immunology. 2009;183:6151–6156. doi: 10.4049/jimmunol.0902145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, et al. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology. 2004;324:297–310. doi: 10.1016/j.virol.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 10.Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: a possible immune escape mechanism used by human papillomaviruses. Journal of immunology. 2002;169:3242–3249. doi: 10.4049/jimmunol.169.6.3242. [DOI] [PubMed] [Google Scholar]
- 11.Fausch SCDSD, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Research. 2003;63:3478–3482. [PubMed] [Google Scholar]
- 12.Fausch SC, Fahey LM, Da Silva DM, Kast WM. Human papillomavirus can escape immune recognition through Langerhans cell phosphoinositide 3-kinase activation. Journal of immunology. 2005;174:7172–7178. doi: 10.4049/jimmunol.174.11.7172. [DOI] [PubMed] [Google Scholar]
- 13.Peiser M, Koeck J, Kirschning CJ, Wittig B, Wanner R. Human Langerhans cells selectively activated via Toll-like receptor 2 agonists acquire migratory and CD4+T cell stimulatory capacity. Journal of leukocyte biology. 2008;83:1118–1127. doi: 10.1189/jlb.0807567. [DOI] [PubMed] [Google Scholar]
- 14.Larsen CP, Steinman RM, Witmer-Pack M, Hankins DF, Morris PJ, et al. Migration and maturation of Langerhans cells in skin transplants and explants. The Journal of experimental medicine. 1990;172:1483–1493. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 16.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]