

Abstract
Background
On trigeminal ganglion neurons, pain-sensing P2X3 receptors are constitutively inhibited by brain natriuretic peptide via its natriuretic peptide receptor-A. This inhibition is associated with increased P2X3 serine phosphorylation and receptor redistribution to non-lipid raft membrane compartments. The natriuretic peptide receptor-A antagonist anantin reverses these effects. We studied whether P2X3 inhibition is dysfunctional in a genetic familial hemiplegic migraine type-1 model produced by introduction of the human pathogenic R192Q missense mutation into the mouse CACNA1A gene (knock-in phenotype). This model faithfully replicates several properties of familial hemiplegic migraine type-1, with gain-of-function of CaV2.1 Ca2+ channels, raised levels of the algogenic peptide calcitonin gene-related peptide, and enhanced activity of P2X3 receptors in trigeminal ganglia.
Results
In knock-in neurons, anantin did not affect P2X3 receptor activity, membrane distribution, or serine phosphorylation level, implying ineffective inhibition by the constitutive brain natriuretic peptide/natriuretic peptide receptor-A pathway. However, expression and functional properties of this pathway remained intact together with its ability to downregulate TRPV1 channels. Reversing the familial hemiplegic migraine type-1 phenotype with the CaV2.1-specific antagonist, ω-agatoxin IVA restored P2X3 activity to wild-type level and enabled the potentiating effects of anantin again. After blocking calcitonin gene-related peptide receptors, P2X3 receptors exhibited wild-type properties and were again potentiated by anantin.
Conclusions
P2X3 receptors on mouse trigeminal ganglion neurons are subjected to contrasting modulation by inhibitory brain natriuretic peptide and facilitatory calcitonin gene-related peptide that both operate via complex intracellular signaling. In the familial hemiplegic migraine type-1 migraine model, the action of calcitonin gene-related peptide appears to prevail over brain natriuretic peptide, thus suggesting that peripheral inhibition of P2X3 receptors becomes insufficient and contributes to trigeminal pain sensitization.
Keywords: Trigeminal ganglia, adenosine triphosphate (ATP), nociception, purinergic receptor, lipid raft, calcitonin gene-related peptide, familial hemiplegic migraine type-1
Background
Migraine is a common, disabling, and undertreated episodic brain disorder with incompletely understood pathophysiology.1 Transgenic mouse models with human monogenic migraine gene mutations are useful to identify and unravel the pathophysiological mechanisms potentially involved in migraine.2,3 Familial hemiplegic migraine type-1 (FHM1) is a rare subtype of migraine4 caused by missense mutations in the CACNA1A gene.5,6 This gene encodes the pore-forming α1A subunit of neuronal voltage-gated calcium channel type 2.1 (CaV2.1, P/Q-type) that triggers neurotransmitter release.7,8 Transgenic knock-in (KI) mice with the FHM1 R192Q mutation (FHM1 R192Q KI) in the orthologous mouse CACNA1A gene show gain-of-function of CaV2.1 channels,8–11 increased neurotransmission, cortical (glutamatergic) hyperexcitability that might explain the increased susceptibility to cortical spreading depression,8,9,12 and signs of migraine-like pain behavior.13,14 Previous studies show that the R192Q mutation also affects the trigeminovascular system that plays an integral role in migraine pain transduction.2 In fact, the gain-of-function of CaV2.1 channels in trigeminal ganglion (TG) of KI mice leads to increased calcitonin gene-related peptide (CGRP) release15,16 and neuronal hyperexcitability.17 Furthermore, in trigeminal sensory neurons, the R192Q mutation causes sensitization of ATP-gated P2X3 receptors,18 which are considered to play a major role in transducing peripheral nociception, including migraine pain.19,20 In KI trigeminal neurons, the constitutively higher amplitude of P2X3 currents is supported by larger ambient CGRP levels, and it is associated with decreased serine P2X3 phosphorylation and preferential receptor localization to lipid raft membrane compartments.21,22
In our search for endogenous mechanisms controlling the function of P2X3 receptors, we have recently observed that in the TG of wild-type (WT) mice, these receptors are constitutively downregulated by endogenous brain natriuretic peptide (BNP) via activation of its receptor natriuretic peptide receptor-A (NPR-A).23,24 This phenomenon is readily observed by suppressing BNP synthesis or inhibiting its receptors implying that even rather small concentrations of this peptide are sufficient for NPR-A receptor-mediated inhibition of P2X3 receptors.24 Interestingly, sustained inactivation of the BNP/NPR-A pathway in WT TG neurons enhances membrane currents mediated by P2X3 receptors, changes their membrane distribution and decreases their serine phosphorylation, making the WT phenotype of TG neurons close to the KI one.24 One attractive hypothesis is that a deficit in the BNP/NPR-A system might account for the sensitization of P2X3 receptors in KI TG. The implication of this hypothesis is that for at least a genetic type of migraine, pain might originate from lack of intrinsic inhibition of P2X3 receptors, thus paving the way for potentiation by ambient CGRP.
The present study examined this hypothesis by investigating the expression and function of the BNP/NPR-A system in R192Q KI TG, as well as its ability to regulate P2X3 receptor membrane distribution and serine phosphorylation. To this end, we used the selective NPR-A blocker anantin to suppress NPR-A signaling23,24 and to test the consequences on P2X3 receptor function. For the purpose of comparison, we checked if BNP could affect TRPV1 channels that are a less sensitive target for this peptide modulation.23 Thereafter, we employed the CaV2.1 antagonist ω-agatoxin IVA or the CGRP receptor antagonist CGRP 8-37 to reverse the upregulated P2X3 receptor activity of KI neurons18,22,25 and to find out the role (if any) of the NPR-A system.
Methods
Animals and TG primary culture preparations
FHM1 R192Q KI and WT littermates were used for the experiments.9 All animal procedures were conducted in accordance with the guidelines of the Italian Animal Welfare Act. All treatment protocols were approved by the Scuola Internazionale Superiore di Studi Avanzati ethics committee and are in accordance with the European Union guidelines and in accordance with the 3R principles (using cultures and reducing the number of mice). Genotyping was performed by PCR, as previously reported.9 TG primary cultures were obtained from animals at the age of P12–14 as described previously following general anesthesia with slowly raising levels of CO2 and employed after 24 h from plating.26 Ganglion tissue samples and cultures were collected and processed in parallel for WT and R192Q KI mice.
Chemicals and treatments
All reagents were purchased from Sigma–Aldrich (Milan, Italy) except anantin, which was purchased from US Biologicals (Salem, MA, USA); ω-agatoxin IVA, which was purchased from Tocris Bioscience (Bristol, UK); and recombinant mouse BNP, which was purchased from Phoenix Pharmaceuticals (Burlingame, CA, USA). The specific NPR-A receptor antagonist anantin, the CGRP receptor antagonist CGRP 8-37, ω-agatoxin IVA, and BNP peptide were dissolved in sterile water and freshly diluted from the stock to the desired concentrations before the experiment. Anantin was applied for 30 min up to 24 h at the concentration of 500 nM to fully block NPR-A receptors.27 CGRP 8-37 was applied overnight at 1 μM concentration to effectively block CGRP receptors.28–30 BNP was applied at the dose of 100 ng/mL or 500 ng/mL, as previously reported for sensory ganglia.23,31 We used ω-agatoxin IVA (200 nM) to selectively inhibit P/Q-type (CaV2.1) Ca2+ channels.32
Membrane fractionation and Western blot
Total membrane protein extraction and fractionation into lipid raft and non-raft membrane fractions were performed as reported by Gnanasekaran et al.22 Immunoprecipitation of P2X3 receptors was described earlier.33 Equal amounts of proteins were separated in 10% sodium dodecyl sulfate-polyacrilamide gel and transferred to nitrocellulose membrane. Immunoblotting was done using the following primary antibodies validated in former studies21,23,24: anti-NPR-A (1:1000; Abcam, Cambridge, UK), anti-P2X3 (1:1000; Santa Cruz Biotechnology, Heidelberg, Germany), anti-β-actin (1:5000; A5441, Sigma), anti-β-tubulin III (1:2000; T5076, Sigma), anti-flotillin 1 (1:250; BD Biosciences, Milan, Italy), anti-phospho-Serine (1:500; Millipore, Milan, Italy). Signals were revealed after incubation with secondary antibodies conjugated with horseradish peroxidase by using the ECL detection system (Amersham Biosciences, Piscataway, NJ, USA) and recorded by the Alliance 4.7 (UVITEC, Cambridge, UK) digital imaging.
Immunocytochemistry
Immunocytochemistry was performed as previously described23 on primary TG cultures prepared from P12-P14 R192Q KI and WT mice. Primary antibodies anti-NPR-A (1:1000, Abcam), anti-BNP (1:100, Phoenix Pharmaceuticals), and β-tubulin III (1:2000, Sigma) were used. The specificity of the antibodies used in this study has been previously validated.23,31 Secondary antibodies conjugated with Alexa Fluor-488 or Alexa Fluor-594 were purchased from Invitrogen (1:500; Milan, Italy). Nuclei were counterstained with DAPI (Sigma). Cells were mounted with Vectashield (Vector laboratories) and analyzed at confocal microscope (Leica TCS SP2, Wetzlar, Germany).
Enzyme-linked immunosorbent assay
Basal and stimulated intracellular cGMP concentrations were evaluated in primary TG cultures after 24 h in vitro. A commercial enzyme-linked immunosorbent assay (ELISA) kit for cGMP (MBL International Corporation, Woburn, MA, USA) was used, following the manufacturer’s instructions as previously described.23 Protein concentrations in cell lysates were determined by the bicinchoninic acid method (Sigma). The cGMP concentrations in pmol/µg of protein were extrapolated from a best-fit line calculated from serial dilutions of a cGMP sample standard.
To determine BNP concentration in medium samples, a commercial ELISA kit for mouse BNP was used (Abnova, Hidelberg, Germany) following the instructions of the manufacturer, as previously reported.23 After 24 h from plating cells, culture medium was collected. All samples were centrifuged at 1200 r/min for 5 min, and the supernatants immediately processed for BNP measurement. Protein concentrations in cell lysates were determined by the bicinchoninic acid method (Sigma). All samples were run in triplicate and values averaged.
Electrophysiology
Electrophysiological experiments were performed according to the previously reported protocols.26,28 After 24 h incubation, trigeminal cultures were continuously superfused during the experiment (3 mL/min) with physiological solution containing (in mM): 152 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH). Neurons were patch-clamped in the whole-cell configuration using patch pipettes with 3–4 MΩ resistance and filled with a solution containing (in mM): 140 KCl, 0.5 CaCl2, 2 MgCl2, 2 Mg2ATP3, 2 GTP, 10 HEPES, and 10 EGTA (pH adjusted to 7.2 with KOH). Recordings were obtained from small and medium size trigeminal neurons,28 with holding potential equal to −65 mV after correction for liquid junction potential. A total of 1 kHz filtering was used during recording; currents were acquired by means of a DigiData 1200 interface and pClamp8.2 software (Molecular Devices, Sunnyvale, CA, USA). To activate P2X3 receptors, the stable synthetic agonist α,β-methylene-adenosine-5’-triphosphate (α,β-meATP; Sigma)34 was applied for 2 s at the concentration of 10 µM23,34,35 to produce large current responses that we showed to be strongly enhanced by anantin.23 Our previous study has demonstrated that anantin application caused a leftward shift in the α,β-meATP concentration response plot without significant change in the maximal response.23 α,β-meATP was administered using a fast superfusion system (Rapid Solution Changer RSC-200; BioLogic Science Instruments, Claix, France). Being a non-hydrolysable analog of the natural P2X3 ligand adenosine triphosphate (ATP), α,β-meATP is chemically very stable and, in addition, possesses almost identical efficacy and potency compared to ATP itself.34,36
Capsaicin (1 µM, 3 s) was used to activate TRPV 1 receptors.18,28 For statistical analysis, data from at least four individual experiments were collected, with number of cells equal or greater than 25. Capsaicin- and α,β-meATP-induced responses were measured as peak current amplitudes, and current density values (pA/pF) were then calculated to normalize current size to the cell capacity and to eliminate differences originated from cell size variation.
Statistical analysis
Data are expressed as mean ± standard deviation (for molecular biology experiments) or mean ± standard error of the mean (for patch clamp experiments), where n indicates the number of independent experiments or the number of investigated cells, as indicated in figure legends. Statistical analysis was performed using non-parametric Mann–Whitney rank sum test, or the Student’s t test, after the software directed choice of non-parametric or parametric data, respectively (Matlab; Sigma Plot and Sigma Stat Software, Chicago, IL, USA), or Kruskal–Wallis test for multiple comparison. A p value of <0.05 was accepted as indicative of a statistically significant difference.
Results
Untethered P2X3 receptor activity from BNP/NPR-A inhibition in KI neurons
In line with our previous reports on WT cells,23,24 sustained (3–24 h) block of NPR-A receptors with anantin (500 nM)25,37 demonstrated significant enhancement of P2X3 receptor currents evoked by their selective agonist α,β-meATP (10 μM) (Figure 1(a)). This facilitation indicates that anantin had reversed the P2X3 downregulation by endogenous BNP.23,24 The effect by anantin on WT cells took P2X3 currents up to the value usually observed in KI neurons (Figure 1(a)18). When applied to KI neurons, anantin failed to further potentiate P2X3 receptor currents after 3 or 24 h treatment (Figure 1(a)), suggesting loss of modulation by the endogenous BNP/NPR-A pathway.
Figure 1.

Anantin effects on WT or KI trigeminal neurons. (a) Representative traces obtained with α,β-meATP (10 µM, 2 s) application show P2X3 current amplitudes in control and after 3 h anantin treatment (500 nM) in WT and KI trigeminal neurons. Histograms show average P2X3 current density values from WT or KI neurons in control or after 3 h or 24 h pretreatment with anantin (n = 25, 25, and 26 for WT; 42, 40, 34 for KI cells, respectively; *p < 0.05, Mann–Whitney rank sum test). Note that P2X3 currents in KI control are already potentiated compared to WT and cannot be further enhanced by anantin application. (b) Western immunoblotting shows the amount of P2X3 pSer and total amount of P2X3 receptors in control and after 24 h anantin (500 nM) application for WT and KI trigeminal cultures. β-tubulin was used as loading control. Histograms on the right show statistically lower P2X3 pSer (relative optical density value) in WT cultures after anantin application. In KI samples, pSer level is already low and is not altered by anantin (n = 4; *p < 0.05, Mann–Whitney rank sum test). (c) Top panel is a representative example of Western immunoblotting showing the amount of P2X3 receptors in raft (R) and non-raft (NR) membrane fractions in control or after treatment with anantin (500 nM, 24 h). Flotillin bands indicate lipid raft membrane fractions. Lower panel shows total amount of P2X3 receptors for each experimental condition; β-tubulin was used as loading control. Histograms on the right quantify mean P2X3 relative optical density values in lipid raft and non-raft membrane fractions (n = 4; *p < 0.05, Kruskal–Wallis test).
In TG neurons, the BNP/NPR-A pathway increases P2X3 serine phosphorylation (pSer) level and favors P2X3 receptor distribution to non-lipid raft membrane compartments.24 These two processes are critical regulators of P2X3 receptor function.21,22 Figure 1(b) compares pSer levels of immunopurified membrane P2X3 receptors from WT or KI cultures in control or anantin-treated condition. In the absence of anantin, P2X3 pSer was low in KI cells compared to that in WT cells and was not changed by anantin application (Figure 1(b)). As expected,24 treatment with anantin resulted in a decreased pSer in WT samples (Figure 1(b)).
Figure 1(c) shows P2X3 receptor distribution between lipid raft (flotillin-labeled; see methods) and non-raft membrane compartments in control or anantin-treated cultures. Under control conditions, more P2X3 receptors were distributed to the lipid raft compartment in KI TG neurons compared with WT, as previously reported by Gnanasekaran et al.22 It is noteworthy that, after anantin application to KI cultures, the predominant P2X3 receptor distribution to lipid rafts remained intact (Figure 1(c)) unlike the previously reported redistribution to the lipid raft domain in WT samples.24 The data in Figure 1 globally suggested that the upregulated function of P2X3 receptors in KI neurons was not constitutively inhibited by the NPR-A system. We, therefore, searched for any deficit in the expression of BNP and NPR-A receptors in KI cells.
BNP and NPR-A expression and function in KI cells
In primary TG cultures from WT mice, NPR-A receptor immunoreactivity was readily detected in 91.7 ± 1.3% of neurons (Figure 2(a), (b)) as shown by its colocalization with β-tubulin III in accordance with previous data.23 In primary TG cultures from KI mice, the percentage of β-tubulin III-positive cells expressing NPR-A was even higher (98.7 ± 1.3%; p < 0.05) than in WT, indicating that almost all KI trigeminal neurons expressed this receptor (Figure 2(b)). Western blot analysis did not reveal such difference at the NPR-A protein level between the two genotypes, either in TG primary cultures from young (P12) mice (Figure 2(c), left) or in TG tissue in situ from adult (P30) animals (Figure 2(c), right).
Figure 2.

BNP and NPR-A in WT and KI TGs. (a) Representative immunocytochemical examples of endogenous NPR-A expression in WT (left panel) and KI (right panel) mouse TG cultures. Nuclei are visualized with DAPI (blue); scale bar = 50 µm. Note extensive costaining of NPR-A (red) and β-tubulin (green). (b) The histograms quantify percentage of NPR-A positive cells in WT and KI cultures (n = 4; *p < 0.05, Kruskal–Wallis test). (c) Western blot showing protein expression of NPR-A in TG from P30 mice (right) and TG cultures from P12 mice (left). β-Actin was used as a loading control. Histograms (bottom) quantify NPR-A relative optical density values for each examined group. (d) ELISA-based quantification of intracellular cGMP in TG cultures from WT and KI mice. Cultures were treated with BNP (500 ng/ml for 5, 30, or 60 min) and/or anantin (500 nM, 30 min) or vehicle (indicated as 0). Note similar dynamics of cGMP increase after BNP application and similar decrease after treatment with anantin alone (500 nM) throughout. Data were normalized to the total protein content in each sample and are presented as mean value ± standard deviation (n = 3 in which samples were run in duplicate; *p < 0.05, Kruskal–Wallis test). (e) Immunocytochemical analysis of BNP (red) in WT and KI mouse TG culture. Nuclei are visualized with DAPI (blue). Scale bar, 15 µm. (f) Histograms show percent value of BNP-positive cells in WT and KI populations (DAPI-labeled cells).
Next, we investigated whether the NPR-A receptor system was functional in KI TG. To this end, since activation of NPR-A by BNP has been shown to increase intracellular cGMP production,38 we investigated if application of BNP to WT and KI cultures could increase intracellular cGMP to comparable levels. Figure 2(d) shows that basal cGMP levels were similar in WT and KI samples. Application of 500 ng/mL BNP (vs. vehicle control) caused a similar rise in cGMP in WT and KI samples in a time-related fashion. These effects were prevented by 500 nM anantin. It is noteworthy that in WT and KI TG cultures, anantin per se significantly decreased the basal level of cGMP (Figure 1(d)), suggesting constitutive activity of NPR-A receptors.
Consistent with previous findings that proposed a mainly paracrine BNP action,23 immunocytochemistry revealed scant presence of BNP-positive cells, in which peptide staining appeared as cytoplasmic granules (Figure 1(e)). In untreated TG cultures from WT animals, 2% ± 0.2 DAPI-stained cells were identified as BNP-positive, while in KI ganglion culture, a slight increase in immunopositive cells was observed (3.7 % ± 0.7; p < 0.05) (Figure 1(f)). We measured, with ELISA assay, the concentration of BNP peptide in the culture medium from WT or KI TG, which was found to be 3.8 ± 1.6 ng/mL and 4.1 ± 1.8 ng/mL, respectively, with no significant difference between WT and KI.
KI cultures, thus, possessed an intact (or even slightly enhanced) BNP/NPR-A system, indicating that in these cells, the downstream pathway linking NPR-A to P2X3 was perhaps severed. To check if BNP could retain its modulation of other pain-sensing receptors, we explored if inhibition of TRPV1 receptors by exogenous BNP23 persisted in KI neurons. Thus, pre-incubating KI cultures for 24 h with 100 ng/mL BNP significantly depressed currents evoked by the TRPV1 agonist capsaicin (Figure 3(a)), while it did not alter responses mediated by P2X3 receptors (Figure 3(b)). The inhibitory effect on TRPV1 receptors seen after BNP application was fully prevented by concomitant administration of 500 nM anantin (Figure 3(a)).
Figure 3.
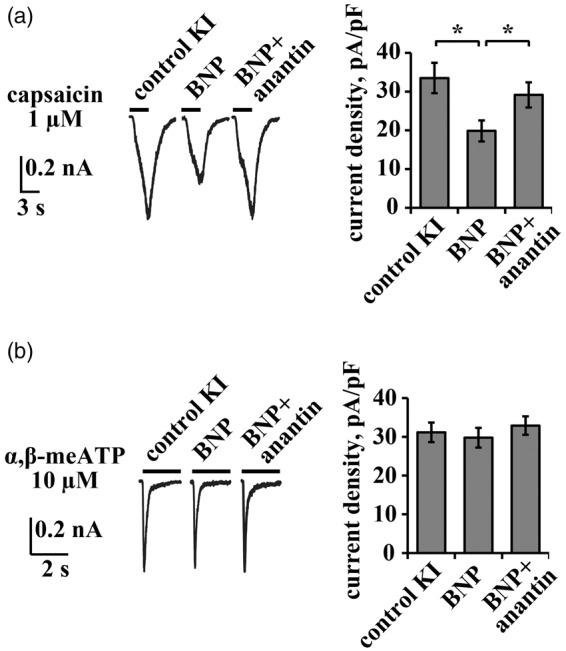
Effect of exogenous BNP on TRPV1- and P2X3-mediated responses in KI TG neurons. (a) Representative traces of capsaicin (1 µM, 3 s)-induced TRPV1 currents of KI trigeminal neurons in control and after application of BNP (100 ng/mL, 24 h) alone or in combination with anantin (500 ng/mL, 24 h). Histograms on the right summarize mean current density values of TRPV1 currents. Note that BNP suppresses TRPV1 responses and that the effect is fully blocked by anantin (n = 39, 42, 47, *p < 0.05, Mann–Whitney rank sum test). (b) Representative traces of α,β-meATP (10 µM, 2 s)-evoked P2X3 responses of KI trigeminal neurons in control and after application of BNP (100 ng/mL, 24 h) or BNP together with anantin (500 ng/mL, 24 h). Histograms on the right summarize mean current density values of P2X3 currents. Note that exogenous BNP does not affect P2X3 currents (n = 38, 51, 33, *p ≥ 0.05, Mann–Whitney rank sum test).
Overall, these data indicated rather small differences in the BNP/NPR-A system between the two genotypes, suggesting that loss of constitutive inhibition of P2X3 receptors could not be attributed to deficient agonist or receptor signal of the BNP/NPR-A system in KI TG.
Reversing the gain-of-function phenotype of KI cells restores NPR-A dependent inhibition of P2X3 receptor activity
The gain-of-function of CaV2.1 (P/Q-type) Ca2+ channels in KI TG neurons is selectively reversed by ω-agatoxin IVA (200 nM) that also restores P2X3 currents to WT level.18 Figure 4(a) indicates that overnight pre-treatment with ω-agatoxin IVA significantly decreased P2X3 currents in KI neurons18,22 and that anantin could then regain its facilitatory action on P2X3 receptors. In addition, ω-agatoxin IVA increased P2X3 pSer (Figure 4(b)) and shifted P2X3 receptors preferentially to the non-raft membrane compartments (Figure 4(c)). When anantin was coapplied with ω-agatoxin IVA, pSer P2X3 levels were significantly reduced (Figure 4(b)). Nonetheless, after ω-agatoxin IVA had decreased the amount of P2X3 receptors in the lipid raft compartment of KI samples, anantin could not change this effect (Figure 4(c)).
Figure 4.

ω-agatoxin IVA restores anantin effects on P2X3 receptors in KI TG neurons. (a) Representative traces of α,β-meATP (10 µM, 2 s)-evoked P2X3 responses in control and after application of ω-agatoxin IVA (200 nM, 24 h) alone or in a combination with anantin (500 ng/mL; coapplied during the last 3 h of ω-agatoxin IVA treatment). Histograms on the right quantify current density values of P2X3 currents for each condition. Note that ω-agatoxin IVA reduces P2X3 currents and that anantin can up-regulate them to the control level (n = 23, 24, 25, *p < 0.05, Mann–Whitney rank sum test). (b) Western immunoblotting shows the amount of P2X3 pSer and total amount of P2X3 receptors in control, after 24 h ω-agatoxin IVA or ω-agatoxin IVA plus anantin (500 nM, 3 h) treatment. β-tubulin III was used as loading control. Histograms on the right show statistically higher P2X3 pSer value after ω-agatoxin IVA application compared to control (n = 4; *p < 0.05, Mann–Whitney rank sum test) with reversal by anantin application. (c) Top panel is a representative example of Western immunoblotting showing the amount of P2X3 receptors in raft (R) and non-raft (NR) membrane fractions in control and after treatment with ω-agatoxin IVA (200 nM, 24 h) or a combination of ω-agatoxin IVA and anantin (500 ng/mL; 3 h). Flotillin bands indicate lipid raft membrane fractions. Lower panel shows total amount of P2X3 receptors for each experimental condition; β-tubulin was used as loading control. Histograms on the right quantify mean P2X3 values in lipid raft and non-raft membrane fractions (n = 4; *p < 0.05, Kruskal–Wallis test).
CGRP receptor antagonism facilitates the action of anantin in KI neurons
CGRP is a powerful positive regulator of P2X3 function.16,39–41 In TG neurons, CGRP stimulates P2X3 receptor neosynthesis and trafficking to the membrane.42 The basal potentiation of P2X3 receptor activity of KI neurons is largely due to higher release of endogenous CGRP from neuronal and non-neuronal sources.28 We investigated whether, in KI TG cultures, BNP-mediated inhibition of P2X3 receptors was swamped by the enhancing action of ambient CGRP. Consistent with previous data,28 treatment with the selective CGRP receptor antagonist peptide CGRP 8-37 (1 µM, overnight) decreased the mean current density of P2X3 responses in KI cells (Figure 5(a)). Application of anantin during the last 3 h of CGRP 8-37 administration restored α,β-meATP-elicited currents to a level close to that in the control situation (Figure 5(a), right). Nonetheless, neither CGRP 8-37 alone nor its combination with anantin changed pSer P2X3 levels of KI TG (Figure 5(b)). The distribution of P2X3 receptors between raft and non-raft membrane compartments of KI TG is illustrated in Figure 5(c). Antagonizing CGRP receptor function reduced P2X3 protein levels both in the lipid raft and in the non-raft compartments. Coapplication of CGRP 8-37 with anantin increased the fraction of P2X3 receptors in the raft compartment with a corresponding fall in the non-raft compartment (Figure 5(c)).
Figure 5.

CGRP receptor antagonist CGRP 8-37 restores anantin effects on P2X3 receptors of KI TG neurons. (a) Representative traces of P2X3 responses induced by application of α,β-meATP (10 µM, 2 s) in control and after application of CGRP 8-37 (1 µM, 24 h) alone or in a combination with anantin (500 ng/mL; coapplied for the last 3 h of CGRP 8-37 treatment). Histograms on the right quantify current density values of P2X3 currents for above mentioned conditions. Note that CGRP 8-37 reduces P2X3 currents and anantin can reverse them (n = 30, 33, 27, *p < 0.05, Mann–Whitney rank sum test). (b) Representative example of Western immunoblotting showing the amount of P2X3 pSer and total amount of P2X3 receptors in control, after CGRP 8-37 (1 µM, 24 h) or co-application of CGRP 8-37 and anantin (500 nM, 3 h). β-tubulin was used as loading control. Histograms on the right show P2X3 pSer values for each experimental condition (n = 4). (c) Top panel is a representative example of Western immunoblotting showing the amount of P2X3 receptors in raft (R) and non-raft (NR) membrane fractions in control and after treatment with CGRP 8-37 (1 µM, 24 h) or a combination of CGRP 8-37 and anantin (500 ng/mL; 3 h); flotillin bands indicate lipid raft membrane fractions. Lower panel shows total amount of P2X3 receptors for each experimental condition; β-tubulin was used as loading control. Histograms on the right quantify mean P2X3 values in lipid raft and non-raft membrane fractions (n = 3; *p < 0.05, Kruskal–Wallis test). Note a reduction in the amount of P2X3 receptors in R and NR samples after CGRP 8-37 application. Anantin relocates P2X3 receptors from non-raft to lipid raft membrane compartments when applied after CGRP 8-37 pretreatment.
Discussion
The primary finding of the study is the demonstration that the BNP/NPR-A system, which downregulates P2X3 receptors in WT trigeminal neurons, lost this ability in a transgenic mouse model of FHM1. This novel observation suggests that sensitization of P2X3 receptors may occur not only because of enhancing action of migraine mediators like CGRP19,43 but also because an endogenous inhibitor becomes poorly functional. The observation that overwhelming majority of TG neurons expresses NPR-A receptors is compatible with their proposed role in controlling activity of sensory neurons.
Intact BNP/NPR-A system in KI TG
Increased P2X3 activity of KI TG neurons is associated with preferential P2X3 receptor redistribution to lipid raft membrane compartments22 and reduced receptor pSer level.21 Notably, in WT cells, these properties of P2X3 receptors are controlled by the BNP/NPR-A system in the opposite direction via BNP itself rather than related peptides.23,24 The present study demonstrated that when NPR-A receptors of KI neurons were blocked by anantin, no increase in P2X3 function ensued, indicating ineffective constitutive inhibition. The KI phenotype was, therefore, not influenced by the BNP/NPR-A system despite normal expression level of BNP and NPR-A, and cGMP synthesis.
On the other hand, while TRPV1 receptor function in KI trigeminal cultures is unchanged,18,28 the BNP/NPR-A system could still negatively modulate TRPV1-mediated responses when BNP was exogenously applied. A multifarious role of endogenous BNP (depending on its extracellular concentration) may, therefore, be exerted on different pain-sensing modalities mediated by P2X3 or TRPV1 receptors. Experiments in vivo are necessary to clarify this suggestion particularly in view of the report that the plasma level of the BNP peptide precursor is elevated in migraine patients.44
NPR-A-dependent downregulation of P2X3 receptors is restored by reversing the KI phenotype
Next, we investigated what made P2X3 receptors of KI neurons apparently disjointed from BNP/NPR-A constitutive inhibition. Previous studies show that enhanced P2X3 receptor activity in KI sensory neurons could be normalized to the WT level upon prolong inactivation of CaV2.1 channels with the selective inhibitor ω-agatoxin IVA.18,22 It is noteworthy that this toxin does not change P2X3 receptors in WT neurons.18 When applied to KI cultures, P2X3-mediated responses went back to the WT level and were associated with raised pSer and membrane redistribution of P2X3 receptors. Over this scenario of reversed functional phenotype, application of anantin could then potentiate P2X3 receptors (thereby unmasking constitutive inhibition) and concomitantly decrease their pSer level. Nevertheless, anantin did not change P2X3 raft membrane distribution. This observation was somewhat unexpected in view of the P2X3 receptor redistribution that anantin causes in WT TG.23 These data, thus, suggest that low pSer and lipid raft compartmentalization were individual actors in the complex process of P2X3 upregulation, and that they are not mutually exclusive.
Contrasting role of CGRP and BNP on P2X3 receptors
Increased release of endogenous CGRP in the KI TG is an important mechanism regulating P2X3 membrane trafficking and function.15,16 Accordingly, the CGRP antagonist CGRP 8-37 reduces P2X3-mediated responses of KI neurons without affecting WT neurons28 (Figure 5). When CGRP activity was blocked in KI TG, constitutive inhibition by BNP/NPR-A could then be restored because anantin regained its delayed effect on P2X3 lipid raft membrane distribution and current magnitude. These observations suggest that, in the dynamic equilibrium between the opposite actions of CGRP and BNP on KI P2X3 receptors, the effect of CGRP was usually dominant. The functionally opposite control by ambient CGRP and BNP on P2X3 receptors did not include contrasting regulation of P2X3 pSer level that was independent from background CGRP concentration.
It should be noted that in KI TG in situ and in culture, there is a sterile neuroinflammatory environment35,45 with related increase in CGRP release from neuronal and non-neuronal cells.16 Thus, the present data suggest that there are multiple processes amplifying the function of trigeminal P2X3 receptors in KI ganglia, and that these mechanisms are potent enough to overcome constitutive inhibition by BNP, at least in this genetic model of migraine.
While the synthetic agonist α,β-meATP used in the present study is selective for P2X3 homomeric receptors, it can also activate heteromeric P2X2/3 receptors that mediate a slowly desensitizing current component.46–48 Nonetheless, colocalization of P2X2 and P2X3 subunits has been previously reported in a minority of mouse TG neurons.49 Furthermore, the contribution of the slow-desensitizing P2X2-mediated component to the total α,β-meATP-induced membrane current is relatively small and detectable in a few trigeminal neurons only.26 This condition precluded a systematic evaluation of BNP/NPR-A inhibition on heteromeric P2X2/3 receptors in trigeminal cultures.
Conclusions
Figure 6 summarizes our view on the dual regulation of P2X3 receptors by BNP and CGRP. In WT cultures, ambient CGRP is minimal and the NPR-A receptors, highly sensitive even to rather low BNP concentrations, generate via cGMP and cGMP-dependent protein kinase (PKG) activity an inhibitory control over P2X3 receptor function. This condition is associated with two phenomena: relatively high pSer and significant presence of P2X3 receptors in non-raft compartments.
Figure 6.

Scheme of BNP/NPR-A-induced modulation of P2X3 receptors expressed on the membrane of WT and KI trigeminal neurons. (a) In WT neurons, endogenous BNP acting through its receptor NPR-A constitutively restrains P2X3 receptor activity. This process is cGMP- and cGMP-dependent protein kinase-dependent and involves high pSer (indicated by circled P) and receptor redistribution from lipid rafts (red) to non-raft (white) membrane compartments. Low activity of CaV2.1 channels and virtually silent CGRP pathway do not generate enough P2X3 stimulation to overcome BNP-mediated inhibition. (b) In KI trigeminal neurons, elevated concentration of ambient CGRP and gain-of-function of CaV2.1 extensively enhance P2X3 receptor activity to the level when the inhibitory BNP/NPR-A pathway becomes ineffective. This is associated with lower level of pSer and receptor preferential redistribution to the lipid raft membrane fraction. CGRP-R denotes the CGRP receptor complex. GC = guanylyl cyclase.
In KI neurons with enhanced Ca2+ influx through CaV2.1 channels, the NPR-A and P2X3 receptors became functionally uncoupled for yet unknown reason, as, despite intact cGMP responses to endogenous BNP, the downstream signal could no longer impact the activity of P2X3 receptors. It seems likely that the process of P2X3 up-regulation underlying trigeminal sensitization to pain19,43 might be triggered because excessive levels of “migraine mediators” like CGRP swamp the negative BNP control that preserves a physiological pain threshold. This notion accords with the observed stronger excitability of KI TG neurons.17 While additional experiments are required to clarify the exact molecular mechanisms involved in this process, identifying strategies to enhance intrinsic inhibition, even at peripheral level, may be useful to control pain. In this sense, future in vivo experiments to study any physiological relevance of the described modulation will be necessary to further support the functional impact of the current results.
Authors' contributions
All authors read and approved the final manuscript. AM, SV, and NN contributed to the design of experiments and collection of data; AMJMVDM contributed to the supply of mouse genetic model; AN contributed to the project supervision; AM, SV, AN, and AMJMVDM contributed to MS writing. AM and SV contributed equally to the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the EU FP7 grant EuroHeadPain (#602633).
References
- 1.Ferrari MD, Klever RR, Terwindt GM, et al. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 2015; 14: 65–80. [DOI] [PubMed] [Google Scholar]
- 2.Gasparini CF, Sutherland HG, Griffiths LR. Studies on the pathophysiology and genetic basis of migraine. Curr Genomics 2013; 14: 300–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 2013; 75: 365–391. [DOI] [PubMed] [Google Scholar]
- 4.Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004; 24: 9–160. [DOI] [PubMed] [Google Scholar]
- 5.Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2 + channel gene CACNL1A4. Cell 1996; 87: 543–552. [DOI] [PubMed] [Google Scholar]
- 6.de Vries B, Frants RR, Ferrari MD, et al. Molecular genetics of migraine. Hum Genet 2009; 126: 115–132. [DOI] [PubMed] [Google Scholar]
- 7.Pietrobon D. Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr Opin Neurobiol 2005; 15: 257–265. [DOI] [PubMed] [Google Scholar]
- 8.Tottene A, Conti R, Fabbro A, et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in CaV2.1 knockin migraine mice. Neuron 2009; 61: 762–773. [DOI] [PubMed] [Google Scholar]
- 9.van den Maagdenberg AMJM, Pietrobon D, Pizzorusso T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004; 41: 701–710. [DOI] [PubMed] [Google Scholar]
- 10.Gao Z, Todorov B, Barrett CF, et al. Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2 + -dependent K + -channel activators in Cacna1aS218L mutant mice. J Neurosci 2012; 32: 15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchitel OD, Inchauspe CG, Urbano FJ, et al. CaV2.1 voltage activated calcium channels and synaptic transmission in familial hemiplegic migraine pathogenesis. J Physiol Paris 2012; 106: 12–22. [DOI] [PubMed] [Google Scholar]
- 12.Eikermann-Haerter K, Dileköz E, Kudo C, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest 2009; 119: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chanda ML, Tuttle AH, Baran I, et al. Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain 2013; 154: 1254–1262. [DOI] [PubMed] [Google Scholar]
- 14.Langford DJ, Bailey AL, Chanda ML, et al. Coding of facial expressions of pain in the laboratory mouse. Nat Methods 2010; 7: 447–449. [DOI] [PubMed] [Google Scholar]
- 15.Fioretti B, Catacuzzeno L, Sforna L, et al. Trigeminal ganglion neuron subtype-specific alterations of CaV2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J Physiol 2011; 589: 5879–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceruti S, Villa G, Fumagalli M, et al. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 knock-in mice: implications for basic mechanisms of migraine pain. J Neurosci 2011; 31: 3638–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hullugundi SK, Ansuini A, Ferrari MD, et al. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience 2014; 266: 244–254. [DOI] [PubMed] [Google Scholar]
- 18.Nair A, Simonetti M, Birsa N, et al. Familial hemiplegic migraine Ca(v)2.1 channel mutation R192Q enhances ATP-gated P2X3 receptor activity of mouse sensory ganglion neurons mediating trigeminal pain. Mol Pain 2010; 6: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabbretti E. ATP P2X3 receptors and neuronal sensitization. Front Cell Neurosci 2013; 7: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan J, Dussor G. Ion channels and migraine. Headache 2014; 54: 619–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair A, Simonetti M, Fabbretti E, et al. The Cdk5 kinase downregulates ATP-gated ionotropic P2X3 receptor function via serine phosphorylation. Cell Mol Neurobiol 2010; 30: 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gnanasekaran A, Sundukova M, van den Maagdenberg AMJM, et al. Lipid rafts control P2X3 receptor distribution and function in trigeminal sensory neurons of a transgenic migraine mouse model. Mol Pain 2011; 7: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vilotti S, Marchenkova A, Ntamati N, et al. B-type natriuretic peptide-induced delayed modulation of TRPV1 and P2X3 receptors of mouse trigeminal sensory neurons. PloS One 2013; 8: e81138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marchenkova A, Vilotti S, Fabbretti E, et al. Brain natriuretic peptide constitutively downregulates P2X3 receptors by controlling their phosphorylation state and membrane localization. Mol Pain 2015; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdelalim EM, Tooyama I. NPR-A regulates self-renewal and pluripotency of embryonic stem cells. Cell Death Dis 2011; 2: e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simonetti M, Fabbro A, D'Arco M, et al. Comparison of P2X and TRPV1 receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol Pain 2006; 2: 11. [DOI] [PMC free article] [PubMed]
- 27.Yu Y-C, Cao L-H, Yang X-L. Modulation by brain natriuretic peptide of GABA receptors on rat retinal ON-type bipolar cells. J Neurosci Off J Soc Neurosci 2006; 26: 696–707. [DOI] [PMC free article] [PubMed]
- 28.Hullugundi SK, Ferrari MD, van den Maagdenberg AMJM, et al. The mechanism of functional up-regulation of P2X3 receptors of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine type 1 (FHM-1). PloS One 2013; 8: e60677. [DOI] [PMC free article] [PubMed]
- 29.Giniatullin R, Di Angelantonio S, Marchetti C, et al. Calcitonin gene-related peptide rapidly downregulates nicotinic receptor function and slowly raises intracellular Ca2+ in rat chromaffin cells in vitro. J Neurosci Off J Soc Neurosci 1999; 19: 2945–53. [DOI] [PMC free article] [PubMed]
- 30.Chiba T, Yamaguchi A, Yamatani T, et al. Calcitonin gene-related peptide receptor antagonist human CGRP-(8-37). Am J Physiol 1989; 256: E331–5. [DOI] [PubMed]
- 31.Zhang F-X, Liu X-J, Gong L-Q, et al. Inhibition of Inflammatory Pain by Activating B-Type Natriuretic Peptide Signal Pathway in Nociceptive Sensory Neurons. J Neurosci 2010; 30: 10927–38. [DOI] [PMC free article] [PubMed]
- 32.Mintz IM, Venema VJ, Swiderek KM, et al. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature 1992; 355: 827–9. [DOI] [PubMed]
- 33.D'Arco M, Giniatullin R, Leone V, et al. The C-terminal Src inhibitory kinase (Csk)-mediated tyrosine phosphorylation is a novel molecular mechanism to limit P2X3 receptor function in mouse sensory neurons. J Biol Chem 2009; 284: 21393–401. [DOI] [PMC free article] [PubMed]
- 34.Sokolova E, Skorinkin A, Moiseev I, et al. Experimental and modeling studies of desensitization of P2X3 receptors. Mol Pharmacol 2006; 70: 373–82. [DOI] [PubMed]
- 35.Franceschini A, Nair A, Bele T, et al. Functional crosstalk in culture between macrophages and trigeminal sensory neurons of a mouse genetic model of migraine. BMC Neurosci 2012; 13: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokolova E, Skorinkin A, Fabbretti E, et al. Agonist-dependence of recovery from desensitization of P2X(3) receptors provides a novel and sensitive approach for their rapid up or downregulation. Br J Pharmacol 2004; 141: 1048–58. [DOI] [PMC free article] [PubMed]
- 37.El-Ayoubi R, Menaouar A, Gutkowska J, et al. Urinary responses to acute moxonidine are inhibited by natriuretic peptide receptor antagonist. Br J Pharmacol 2005; 145: 50–6. [DOI] [PMC free article] [PubMed]
- 38.Pandey KN. Biology of natriuretic peptides and their receptors. Peptides 2005; 26: 901–32. [DOI] [PubMed]
- 39.Cady RJ, Glenn JR, Smith KM, et al. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain 2011; 7: 94. [DOI] [PMC free article] [PubMed]
- 40.Yasuda M, Shinoda M, Kiyomoto M, et al. P2X3 receptor mediates ectopic mechanical allodynia with inflamed lower lip in mice. Neurosci Lett 2012; 528: 67–72. [DOI] [PubMed]
- 41.Simonetti M, Giniatullin R, Fabbretti E. Mechanisms mediating the enhanced gene transcription of P2X3 receptor by calcitonin gene-related peptide in trigeminal sensory neurons. J Biol Chem 2008; 283: 18743–52. [DOI] [PubMed]
- 42.Fabbretti E, D'Arco M, Fabbro A, et al. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci Off J Soc Neurosci 2006; 26: 6163–71. [DOI] [PMC free article] [PubMed]
- 43.Giniatullin R, Nistri A, Fabbretti E. Molecular mechanisms of sensitization of pain-transducing P2X3 receptors by the migraine mediators CGRP and NGF. Mol Neurobiol 2008; 37: 83–90. [DOI] [PubMed]
- 44.Uzar E, Evliyaoglu O, Yucel Y, et al. Serum cytokine and pro-brain natriuretic peptide (BNP) levels in patients with migraine. Eur Rev Med Pharmacol Sci 2011; 15: 1111–6. [PubMed]
- 45.Franceschini A, Vilotti S, Ferrari MD, et al. TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PloS One 2013; 8: e52394. [DOI] [PMC free article] [PubMed]
- 46.Tsuda M, Koizumi S, Kita A, et al. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J Neurosci Off J Soc Neurosci 2000; 20: RC90. [DOI] [PMC free article] [PubMed]
- 47.Kawashima E, Estoppey D, Virginio C, et al. A novel and efficient method for the stable expression of heteromeric ion channels in mammalian cells. Receptors Channels 1998; 5: 53–60. [PubMed]
- 48.Chen X, Gebhart GF. Differential purinergic signaling in bladder sensory neurons of naïve and bladder-inflamed mice. Pain 2010; 148: 462–72. [DOI] [PMC free article] [PubMed]
- 49.Staikopoulos V, Sessle BJ, Furness JB, et al. Localization of P2X2 and P2X3 receptors in rat trigeminal ganglion neurons. Neuroscience 2007; 144: 208–16. [DOI] [PMC free article] [PubMed]