

Abstract
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by abnormally long CAG-repeats in the huntingtin gene that encode an expanded polyglutamine (polyQ) domain near the N-terminus of the huntingtin (htt) protein. Expanded polyQ domains are directly correlated to disease-related htt aggregation. Htt is found highly associated with a variety of cellular and subcellular membranes that are predominantly comprised of lipids. Since cholesterol homeostasis is altered in HD, we investigated how varying cholesterol content modifies the interactions between htt and lipid membranes. A combination of Langmuir trough monolayer techniques, vesicle permeability and binding assays, and in situ atomic force microscopy were used to directly monitor the interaction of a model, synthetic htt peptide and a full-length htt-exon1 recombinant protein with model membranes comprised of total brain lipid extract (TBLE) and varying amounts of exogenously added cholesterol. As the cholesterol content of the membrane increased, the extent of htt insertion decreased. Vesicles containing extra cholesterol were resistant to htt-induced permeabilization. Morphological and mechanical changes in the bilayer associated with exposure to htt were also drastically altered by the presence of cholesterol. Disrupted regions of pure TBLE bilayers were grainy in appearance and associated with a large number of globular aggregates. In contrast, morphological changes induced by htt in bilayers enriched in cholesterol were plateau-like with a smooth appearance. Collectively, these observations suggest that the presence and amount of cholesterol in lipid membranes play a critical role in htt binding and aggregation on lipid membranes.
Graphical abstract
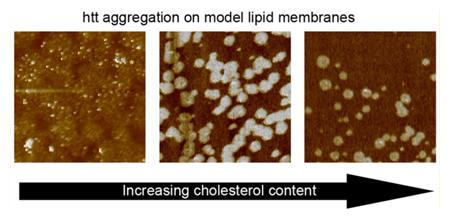
Huntington’s disease, a neurodegenerative genetic disorder, is caused by an expanded polyglutamine (polyQ) domain near the N-terminus of the huntingtin (htt) protein.1 PolyQ expansion promotes the self-assembly of htt into fibrils and other types of aggregates that accumulate in the hallmark inclusion bodies found in HD brain tissues.2 Furthermore, age of onset and disease severity are strongly correlated with the length of the polyQ domain, with a critical expansion length of at least ~35 glutamines required for disease.3 Full-length htt is over 3000 amino acids long, but it undergoes proteolysis, resulting in a variety of truncation products.4,5 Specifically, N-terminal htt fragments, such as the first exon of htt, have been shown to have a potentially important role in HD. For example, expression of htt exon1 with an expanded polyQ tract causes a progressive neurological phenotype in transgenic mice.2,6 Furthermore, N-terminal fragments similar to exon1 are detected in knock-in mouse models expressing full length htt,7 and fragments on the order of exon1 have been detected in HD patients.5
While htt appears to be a multifunctional protein,8–12 there is considerable published support that it intimately interacts with a variety of lipid membranes.13–17 Htt localizes with brain membrane fractions18 and is essential for the normal development of several perinuclear membrane organelles, including mitochondria and the ER.19–21 As a result of its normal function, htt localizes to specific subcellular compartments8 and has been implicated in the transport of lipid vesicles (endocytic, synaptic or lysosomal), especially along microtubules.11,22–24 Furthermore, the first 17 amino acids of htt (Nt17) that flank the polyQ domain facilitate the binding of N-terminal fragments to lipid membranes.25–27 Additionally, polyQ length correlates with magnitude of htt insertion and disruption of lipid membranes.13,28
Cholesterol is an important structural component of biological membranes and is involved in regulating the fluidity of lipid bilayers. The brain is the most cholesterol-rich organ in the mammalian body, with ~23% of total body cholesterol. ~70% of this cholesterol is contained within the myelin sheaths and is a crucial element for proper neuron and astrocyte function.29,30 Cholesterol affects the functional properties of membrane-resident proteins (ion channels and transmitter receptors) as well as plays a role in signal transduction, synaptogenesis, and neurotransmitter release.31–33 These functions are attributed to its regulatory role of the membrane physical properties.34,35 The presence of cholesterol is necessary for the formation of lipid rafts, used for cellular communication and signal transduction, via hydrogen bonding with the sphingosine hydroxyl group of sphingomyelin and alignment of the hydrophobic planar core causing condensation of the fatty acid domains.36,37 Cholesterol in the brain is synthesized endogenously, due to its inability to cross the blood-brain barrier.
Abnormalities in cholesterol metabolism and homeostasis have been observed in cellular and animal models of HD, as well as in HD patient tissues.38–40 However, the specifics of altered cholesterol content associated with HD remains controversial. Numerous studies suggest a decrease in cholesterol levels in HD models;41–44 however, other reports appear to demonstrate an accumulation or increase in cholesterol.45–47
The importance of htt aggregation in HD has long been appreciated, and lipid bilayers have been shown to heavily influence the aggregation of a variety of N-terminal htt fragments.25 However, the impact of specific lipid components, such as cholesterol, on modulating htt aggregation is poorly understood. Here, we characterize the interaction of N-terminal htt fragments with total brain lipid extract (TBLE) lipid membranes that contain varying amounts of exogenously added cholesterol.
EXPERIMENTAL PROCEDURES
Peptide Preparation
The synthetic, model htt peptide used in this study contains 35 glutamines (which is sufficient for aggregation48), a C-terminal polyproline (polyP) domain that is 10 residues in length, and the previously mentioned Nt17 on the N-terminus. This peptide is referred to as Nt17-Q35-P10-KK. The lysines were added to the C-terminus to aid in solubility. Nt17-Q35-P10-KK peptides were obtained via custom synthesis (Keck Biotechnology Resource Laboratory, New Haven, Connecticut). Disaggregation and solubilization of Nt17-Q35-P10-KK peptides were achieved using established protocols.49 Briefly, peptide was dissolved overnight in a 1:1 mixture of trifluoroacetic acid (TFA, Acros Organics) and hexafluoroisopropanol (HFIP, Acros Organics) at a concentration of 0.5 mg/mL. After vigorous vortexing, a stream of N2 was used to evaporate the solvent. To remove any residual solvent, samples were then placed in a Vacufuge concentrator for 3 h (Eppendorf, Hauppauge, NY), resulting in thin peptide films. These peptide films were resuspended in ultrapure water that had been adjusted to pH 3 with TFA to 200 μM, which was diluted in phosphate-buffered saline (PBS) (10 mM phosphate, 140 mM NaCl, 2.7 mM KCl) (Life Technologies) to a final concentration of 20 μM at a pH of 7.3.
Purification of GST-htt-exon1 Fusion Proteins
Glutathione S-transferase (GST)–htt exon1 fusion proteins were purified as previously described.50 For experiments presented here, htt exon1 with 51 repeat glutamines was used. In short, the GST-htt fusion proteins were expressed by induction in Escherichia coli with isopropyl β-d-thiogalactoside at 30 °C for 4 h. The cells were lysed with lysozyme (0.5 mg/mL). Fusion proteins were purified from lysate by liquid chromatography (LPLC, BioRad) using a GST affinity column. Relevant fractions to collect and purity were determined by gel electrophoresis. Cleavage of the GST moiety was achieved by Factor Xa (Promega, Madison, WI). Cleavage initiates aggregation. Prior to the addition of the cleaving agent, solutions of fusion proteins were centrifuged at 20000g for 30 min at 4 °C to remove preexisting aggregates. Experiments were carried out in buffer A (50 mM Tris–HCl, pH 7.0, 150 mM NaCl). To ensure efficient GST cleavage, the GST-htt exon1 fusion protein and respective cleavage agents were incubated for 1 h on ice prior to their addition to any assay.
Liebermann-Buchard Assay
To determine the cholesterol content of the stock TBLE, a Liebermann-Buchard assay was performed.51 To prepare 100 mL of Liebermann-Buchard reagent, 10 mL of concentrated sulfuric acid was added to to 60 mL of acetic anhydride with continuous stirring in an ice bath. Next, 30 mL of acetic acid and 0.6 g of anhydrous sodium sulfate were added. Standard cholesterol samples were used to calibrate. 0.5 mL of the TBLE stocks dissolved in chloroform were added to 5 mL of Liebermann-Buchard reagent, vortexed, and placed in a water bath at 35 °C for 10 min. Absorbance was measured at 620 nm using a GENESYS 20 Visible spectrophotometer (Thermo Scientific).
Langmuir Trough Configuration
All lipid monolayer isotherm and peptide insertion experiments were conducted in a Teflon Langmuir trough with symmetric automated Teflon barriers (large inverted microscopy model, NIMA Technologies, Coventry, England). Surface pressure at the air/buffer interface was measured with a stationary Wilmhelmy balance (NIMA Technologies, Coventry, England). All measurements were collected at 30 ± 0.5 °C, and the trough temperature was maintained with a circulating heated water bath (Isotemp 3016D water circulator, Thermo Fisher Scientific).
Lateral Compression Experiments
Lipid monolayer isotherms were conducted to characterize monolayer phase behavior as a function of lipid-packing density and subsequent altering effects of cholesterol. Stock TBLE (Avanti Polar Lipids) was obtained in chloroform and used without further purification. Cholesterol (Sigma-Aldrich) was received in powder form and was dissolved in chloroform (Sigma-Aldrich). TBLE and cholesterol were mixed in different ratios to a total spreading concentration of 0.4 mg/mL and stored in glass vials at −20 °C. The lipid monolayer was formed by dropwise addition of spreading solution on the PBS (pH = 7.3) surface; chloroform was allowed to evaporate for 15 min. Because of the lack of available quantitative characterization information on TBLE lipid components, the area per molecule at the interface was calculated assuming an average TBLE molecular weight of 850 g/mol. The barriers compressed the monolayer at 6 cm2/min and isotherm measurements in the form of surface pressure (mN/m) versus mean area per lipid molecule (Å2/molecule) were taken at 1 s intervals until monolayer collapse.
Peptide Insertion into a Lipid Monolayer
Insertion experiments were performed to measure the amount of Nt17-Q35-P10-KK peptide insertion into TBLE and TBLE/cholesterol monolayers. The Langmuir trough setup was identical to an isotherm experiment, but the lipid monolayer was compressed to a surface pressure of 30 mN/m to mimic the lateral density of a cellular bilayer52 and then the barriers were set to feedback control to maintain constant surface pressure. Lyophilized peptide was rehydrated in TFA/PBS as described above and immediately injected beneath the monolayer for a final peptide concentration of 900 nM. After 10 min, if no increase in average area per molecule to signify peptide insertion was observed, the surface pressure was reduced by 4 mN/m increments down to a minimum of 14 mN/m. Percent insertion was determined as a % increase in area ((Afinal − Ainit)/Ainit) × 100) at each surface pressure.
Preparation of Vesicles for Leakage Assay
Large unilamellar vesicles (LUVs) of TBLE and TBLE/cholesterol were prepared to determine leakage caused by htt peptide insertion. Lipid films, evaporated with a stream of N2 from a chloroform stock, were hydrated in 70 mM calcein (Sigma) via 5 min of vortexing and 5 min of sonication. The vesicles went through seven freeze–thaw cycles in dry ice/ethanol and warm water baths promoting unilamellar vesicles, followed by 11 extruder filtrations through a 100 nm pore polycarbonate filter (Avanti). Excess dye was removed with size exclusion chromatography using Sephadex G-50 beads (Sigma-Aldrich) and ultrapure water eluent. 120 nm diameter size uniformity was confirmed with dynamic light scattering (Zetasizer ZS90 [Malvern Worcestershire, UK]). Lipid concentration was determined by a phosphorus assay adapted from Anderson and Rouser.53,54 Briefly, a lipid fraction (500 μL) and 8.9 N H2SO4 (225 μL) were vortexed and heated for 20 min at 155 °C. After cooling, 6% H2O2 (75 μL) was added and heated for an additional 40 min at 155 °C. After cooling, 1.95 mL of ultrapure water, 5% molybdic acid (225 μL), and 10% ascorbic acid (225 μL) were added, and the solution was placed in a boiling water bath for 10 min. Absorbance at 820 nm was measured using a Jasco V-550 UV/visible spectrophotometer (Jasco, Easton, MD) and compared to a KH2PO4 standard reference plot to calculate final lipid concentration.
Vesicle Leakage Assay
TLBE LUVs with exogenous cholesterol ranging from 0 to 16 wt % were exposed to Nt17-Q35-P10-KK peptide resulting in a range from 1:1 to 64:1 lipid:peptide molar ratios at 25 °C. Lyophilized peptides were hydrated as outlined above and used promptly after hydration to minimize presence of non-monomeric species. Calcein fluorescence at 515 nm (495 nm excitation) was measured at room temperature prior to peptide exposure and 1 min after peptide administration using a RF-1501 spectrofluorometer (Shimadzu, Columbia, MD). Vesicles were then completely lysed using 1% Triton X-100 (Sigma) to measure a maximum fluorescence. Vesicle leakage caused by peptide was calculated by
| (1) |
where IF is fluorescent intensity after peptide addition, II is initial fluorescence of the LUVs, and IT is total fluorescence after complete lysis.
TBLE/PDA Lipid Vesicle Protein Binding Assay
TBLE/polydiacetylene (PDA) lipid binding assays were performed to measure the interaction between htt-exon1(51Q) and lipid vesicles. TBLE/PDA assays were performed using previously reported protocols.55,56 In short, diacetylene monomers 10,12-tricosadiynoic acid (GFS Chemicals, Columbus, OH), TBLE, and an appropriate amount of cholesterol were dissolved in a solution of 1:1 chloroform/ethanol. Total lipid (TBLE + cholesterol) and PDA was always mixed in a 2:3 molar ratio. The solution was evaporated off in a rotary evaporator, leaving a thin, dry film. 1× Tris-buffered saline (TBS) heated to 70 °C was added to the film and sonicated for 5 min at 100 W using a sonic dismembrator (FisherSci). To ensure self-assembly of the vesicles, the suspension was stored at 4 °C overnight. The diacetylene monomers were polymerized by irradiation at 254 nm with 7 lm for 10 min (room temperature with stirring), resulting in a brilliant blue color. As controls, each TBLE/PDA vesicle sample was exposed to 1× Buffer A (negative control) or NaOH (pH 12) (positive control). Polymerized TBLE/PDA vesicles were exposed to htt-exon1(51Q) at a final concentration of 20 μM. Experiments were performed in triplicate in a 96-well format, and the colorimetric response of each well was recorded over 16 h using an Infinite M1000 Pro plate reader (TECAN, Switzerland) at 25 °C measuring both the blue component (640 nm) and the red component (500 nm) of the spectrum. A quantitative value representative of blue-to-red color transition was obtained by determining the % colorimetric response (%CR), defined as55,56
| (2) |
where PB is a red/blue ratio of absorbance (A) defined as Ablue/(Ablue + Ared). Ablue is the absorbance at the “blue” component in the UV–vis spectrum (~640 nm); Ared is absorbance at the “red” component (~500 nm), PB0 is the red/blue ratio of the control sample (before induction of color change), and PBI is the value obtained for the vesicle solution after addition of peptides.
AFM Imaging Conditions
In situ AFM experiments were performed with a Nanoscope V MultiMode scanning probe microscope (Bruker, Santa Barbara, CA) equipped with a closed-loop “vertical engage” J-scanner and a sealable tapping fluid cell. Images were acquired with silicon cantilevers (VISTAprobes) with nominal spring constants of ~0.1 N/m. Scan rates were ~2 Hz with cantilever drive frequencies ranging from ~8 to 10 kHz. The free amplitude of the cantilever was ~25 nm, and the tapping amplitude was set at 75% of free amplitude. The Matlab image processing toolbox (Mathworks, Natick, MA) was used to analyze AFM images. Topographic and phase images were captured simultaneously. In phase imaging, the shift in the phase of the oscillating probe tip with respect to the oscillating drive is mapped, and these phase shifts highlight compositional contrast of the surface. In solution, phase-shifts are sensitive to the local elastic response of the sample and can be used to map local elasticity of samples like lipid membranes.57
Preparation of Supported TBLE Bilayers for AFM Experiments
TBLE (Avanti Polar Lipids) and cholesterol were dissolved in chloroform (Fisher Scientific) and mixed at appropriate ratios to have samples of pure TBLE and TBLE spiked with 10%, 20%, or 30% exogenously added cholesterol based on mass. The chloroform was evaporated in the fume hood in the air. The resulting lipid films were resuspended in Buffer A (pH 7.3) at a concentration of 0.5 mg/mL. Bilayers and multilayer lipid sheets were formed by at least five sequential freeze–thaw cycles. The lipid suspensions were then sonicated for 7 or 8 min to promote vesicle formation. 35 μL of the vesicle solution was injected directly into the AFM fluid cell and onto freshly cleaved mica. The formation of a continuous bilayer via vesicle fusion was directly observed by continual AFM imaging. AFM experiments were performed between 23 and 25 °C.
RESULTS
Addition of Cholesterol Reduces the Insertion of htt into Lipid Membranes
To understand how cholesterol affects interactions between htt and the cell membrane, monomeric peptide insertion into a TBLE monolayer with varying amounts of exogenous cholesterol was measured. The amount of cholesterol in the stock TBLE (no exogenous cholesterol) was 22 wt % as measured by a Liebermann-Buchard assay. This wt % of cholesterol with respect to total lipid content is similar to that found in the human brain.58 When peptide is introduced into an aqueous solution, the peptide molecules partition between the bulk and the air/buffer interface until a certain surface pressure, the equilibrium spreading pressure, is attained. It has been previously shown that Nt17-Q35-P10-KK has appreciable surface activity (equilibrium spreading pressure or ESP of 17.3 mN/m at 900 nM) due to the inclusion of the N-terminal lipid binding domain, and this, along with the polyP domain, promotes insertion into pure TBLE lipid monolayers.25 A TBLE lipid monolayer with either 0, 7, or 16 wt % exogenous cholesterol at the air/buffer interface, serving as a model for the outer leaflet of a cell membrane, was compressed to a physiologically relevant surface pressure of 30 mN/m,52 held at this pressure, and then freshly prepared peptide was injected into the subphase beneath the monolayer. Change in average area per molecule at the interface, which indicates peptide insertion, was monitored until the monolayer equilibrated. Then the pressure was stepped down by a 4 mN/m increment and the process was repeated. The addition of cholesterol to TBLE monolayers systematically decreased the amount of htt peptide insertion at each surface pressure (Figure 1A), but the effect was not dramatic with total insertion into TBLE of 18.2 ± 1.3%, TBLE with 7 wt % cholesterol of 16.1 ± 1.1%, and TBLE with 16 wt % cholesterol of 15.4 ± 1.0%. Of note in all cases, regardless of the amount of exogenous cholesterol, the peptide inserted into the lipid monolayer at a surface pressure higher than the ESP indicating a driving force for the peptide to interact with the membrane.
Figure 1.
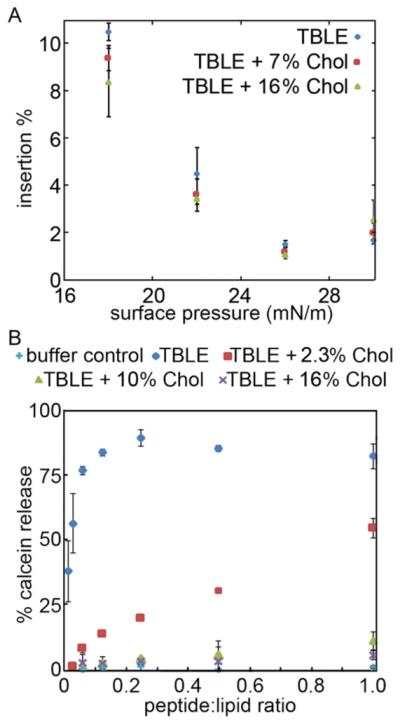
(A) Insertion of Nt17-Q35-P10-KK into lipid monolayers of varying TBLE:cholesterol ratios; expressed as % area increase at each surface pressure. (B) Calcein leakage, measured as relative fluorescence, from LUVs composed of different TBLE:cholesterol ratios exposed to Nt17-Q35-P10-KK (or buffer acting as a control) as a function of peptide:lipid ratio. Error bars indicate one standard deviation (n = 3).
To understand why the addition of cholesterol to TBLE membranes would affect htt peptide insertion, individual surface pressure versus mean molecular area isotherms were measured for a series of binary mixtures of TBLE and cholesterol and additivity plots constructed (Supplemental Figure 1).58 Comparison of the mean molecular area of the binary mixtures to that of a theoretical mean area consistent with ideal mixing of the two components provides evidence that exogenous cholesterol causes condensation or a stiffening of the monolayer (Supplemental Figure 1B) which may affect the ability of the htt peptide to insert into the layer.
Cholesterol Reduces the Susceptibility of Vesicles to htt-Induced Permeability
It has been previously shown that Nt17-Q35-P10-KK peptides can permeablize TBLE bilayer lipid membranes.25 To determine if additional cholesterol altered the susceptibility of membranes to permeabilization, calcein dye leakage assays with LUVs were performed. Disruption of the membrane by an external agent, via mechanisms ranging from pore formation to complete vesicle lysis, results in calcein release, dilution, and fluorescence emission from the unquenched dye; this allows permeabilization to be correlated with fluorescence increases. Solutions of predominately monomeric peptides were mixed with vesicles comprised of TBLE containing varying amounts of exogenously added cholesterol. Several lipid:peptide ratios ([peptide] = 7.34–470 nM) were used and fluorescence measured as a function of time, though a signal maximum was typically reached within several minutes. Nt17-Q35-P10-KK was able to induce significant calcein release from LUVs comprised of pure TBLE compared to control volumes of TFA/PBS buffer (Figure 1B). As the cholesterol content of the LUVs was increased, the extent of calcein dye leakage induced by exposure to Nt17-Q35-P10-KK drastically decreased, with minimal leakage at the highest cholesterol levels. The addition of just 2.3 wt % cholesterol to TBLE LUVs reduced permeabilization by more than half at all but the highest peptide concentrations. Reduced susceptibility of cholesterol containing LUVs to Nt17-Q35-P10-KK induced permeabilization was surprising as the peptide still had an appreciable affinity for monolayers containing up to 16% exogenous cholesterol as measured by Langmuir isotherms. This suggests that the peptides are still inserting into the membrane, but not causing a similar amount of disruption leading to content leakage.
Enriching TBLE with Cholesterol Alters the Extent of htt-exon1 Binding to Lipid Vesicles As Determined by a TBLE/PDA Binding Assay
To verify that altering the lipid content of TBLE would modulate the interaction of full-length htt-exon1 with lipids, we performed a TBLE/PDA vesicle lipid binding assay (Figure 2). This is a colorimetric assay in which mixed vesicles composed of phospholipids and polymerized PDA lipids exhibit rapid visible color changes from blue to red induced by interactions between peptides and the phospholipid moieties within the vesicles. The colorimetric response can be used to quantify peptide–membrane interactions and interfacial membrane processes. The TBLE/PDA vesicles were systematically enriched with 0% (pure TBLE), 10%, 20%, or 30% cholesterol by mass. These vesicles were exposed to 20 μM htt-exon1 (51Q) and %CR was measured for 16 h (Figure 2). Typically, there is an initial steady increase in %CR as TBLE/PDA vesicles are exposed to htt-exon1(51Q). With time, the % CR tends to level off to a quasi-steady state. As the exogenous cholesterol content was enriched from 0% to 30%, the binding of htt-exon1(51Q) to the vesicles was reduced. That is, the initial increase in %CR was less steep with increasing amounts of cholesterol. The quasi-steady state %CR is also significantly smaller in a cholesterol dose-dependent manner, indicating that cholesterol reduces htt-exon1(51Q) overall binding to and insertion into lipid membranes. Of note, there is still an appreciable interaction of htt-exon1(51Q) with the lipid vesicles at high exogenous cholesterol levels which parallels results seen in the htt peptide monolayer insertion measurements.
Figure 2.
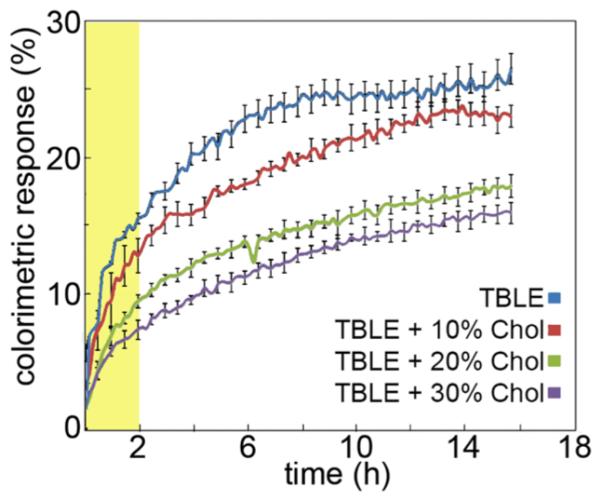
Percent colorimetric response (%CR) of TBLE/PDA vesicles containing various amounts of cholesterol upon exposure to htt-exon1(51Q) plotted as a function of time. Error bars indicate one standard deviation (n = 3). The highlighted area indicates the time frame accessible with AFM experiments described later.
Cholesterol Modulates the Morphological and Mechanical Changes in Lipid Bilayers Associated with Exposure to htt
To investigate the influence of cholesterol on the aggregation of htt on lipid membranes, supported TBLE bilayers containing various amounts of cholesterol were exposed to htt-exon1(51Q) and continuously imaged using in situ AFM, allowing for the direct tracking of protein aggregation on the bilayer. Supported lipid bilayers retain several properties, such as lateral fluidity, associated with free membranes,59 and have been used as model surfaces to study the interaction of several amyloid-forming proteins with lipid membranes.60–65
Bilayers comprised of pure TBLE, TBLE + 10% cholesterol, TBLE + 20% cholesterol, and TBLE + 30% cholesterol were used. First, it was confirmed that continuous supported bilayers of the different lipid mixtures would form via vesicle fusion and remain structurally stable, i.e., no observed morphological changes, when imaged for several hours (Supplemental Figure 2). Only continuous TBLE bilayers (40 × 40 μm in size), as assessed by AFM imaging, were exposed to htt-exon1(51Q), and observations were limited to these verified continuous bilayers. After formation of an appropriate supported bilayer, the fluid cell was washed with neat buffer to reduce the amount of lipid components not bound to the mica substrate. Pure TBLE bilayers and those with exogenously added cholesterol were all smooth in appearance (RMS roughness less than 0.2 nm) with no indication of distinct lipid domains in either AFM height or phase images (Supplemental Figure 2).
Having established that supported bilayers appropriate for this study could be made from the different lipid mixtures, these model bilayers were systematically exposed to freshly prepared aliquots of htt-exon1(51Q) at a final protein concentration of 20 μM in the fluid cell. When pure TBLE bilayers were exposed to 20 μM solutions of htt exon1(51Q), extensive areas of the bilayer displayed disrupted morphology characterized by increased surface roughness and the accumulation of discrete htt aggregates within 10 min. These regions increased in size and became steadily rougher as a function of time (Figure 3 and Supplemental Movie 1). The disrupted regions appeared to be mechanically altered from the unperturbed regions of the bilayer, as there is distinct contrast observed in AFM phase images. This is consistent with previous reports that correlated the morphological changes associated with exposure of TBLE bilayers to htt-exon1(51Q) with decreased compression modulus of the membrane.25,28 During the 120 min of observation, the phase contrast between the different regions remains consistent, suggesting that differences in elasticity between these distinct regions persist.
Figure 3.

(A) Sequential AFM height and phase images taken in solution of supported TBLE bilayers exposed to htt-exon1(51Q). Blue arrows indicate oligomer-like structures. (B) AFM height images taken as a function of time demonstrating the rough, grainy morphological changes induced in a pure TBLE bilayer by htt-exon1(51Q). Blue lines correspond to the height profile presented directly below the images.
Aggregates observed on the pure TBLE bilayer appeared globular in nature (examples indicated by blue arrows in Figure 3). These aggregates were often stable, appearing in subsequent images of the same region; however, they did increase in volume as a function of time. In contrast to incubation experiments that demonstrate that htt-exon1(51Q) forms fibrils in the absence of lipids within an hour and increase in number thereafter (Supplemental Figure 3),66 fibrils were not observed on lipid bilayers during the time course of the experiment. This suggests that the pure TBLE bilayer either promotes the formation of aggregates off pathway to fibrils or that aggregate intermediates are stabilized by interaction with the membrane.
When TBLE + 10% cholesterol bilayers were exposed to 20 μM htt-exon1(51Q), morphological changes in the bilayer were observed that evolved with time (Figure 4 and Supplemental Movie 2). As the development of these domains could be directly tracked by continuous AFM imaging, these morphological changes arose from direct interaction of htt-exon1(51Q) with the bilayer. Initially, small domains of slightly increased height were detected in the TBLE + 10% cholesterol bilayers, and these domains were associated with clear contrast in AFM phase images, suggesting that these regions differed in their elastic properties compared to the regions of unaffected bilayer. With time, these domains grew in size and increased in height. The increase in height was correlated with a loss of contrast in corresponding phase images, indicating that these thicker domains had similar elastic properties compared with unperturbed bilayer. While a few globular aggregates were observed on the TBLE + 10% cholesterol bilayer, they were relatively rare compared to those that formed on the pure TBLE bilayers
Figure 4.

(A) Sequential AFM height and phase images taken in solution of supported TBLE bilayers containing 10% exogenously added cholesterol exposed to htt-exon1(51Q). (B) AFM height images taken as a function of time demonstrating the smooth morphological changes induced in a TBLE + 10% cholesterol bilayer by htt-exon1(51Q). Blue lines correspond to the height profile presented directly below the images.
The domains of altered morphology that developed in TBLE + 10% cholesterol bilayers were distinct compared to changes associated with exposure of pure TBLE bilayers to htt-exon1(51Q) (Figures 3B and 4B). While the disrupted regions that appeared in the pure TBLE bilayers were grainy in appearance (Figure 3B), the domains that developed in the TBLE + 10% cholesterol bilayers were plateau-like with a smoother appearance (Figure 4B). Both types of altered morphologies increased in height as presumably more material accumulated in these regions; however, the height increase was smaller in pure TBLE bilayers except when there was a discrete, globular aggregate. The smoother domains that developed on the TBLE + 10% cholesterol gradually grew from being ~1 nm to ~4 nm above the regions of unperturbed bilayer. In the absence of htt, these domains in TBLE bilayers with additional cholesterol did not develop.
When the amount of exogenously added cholesterol in the TBLE bilayers was increased to +20% or +30%, changes in membrane morphology similar to those associated with the +10% cholesterol bilayer were observed (Supplemental Figure 4). Specifically, small domains of slightly altered height and clear phase contrast were detected at initial time points. With time, these domains grew and transitioned toward the smooth, plateau-like morphology. However, as the cholesterol content was increased, the time required for this transition in morphology was extended. Furthermore, surface area affected by exposure to htt-exon1(51Q) was reduced, consistent with the reduced insertion and binding of htt to lipid membranes via Langmuir and vesicle binding assays. The initial sharp phase contrast that dissipated with the development of these domains was also observed.
To quantify the interaction of htt-exon1(51Q) with the supported bilayers containing various amounts of cholesterol, the percentage of the surface that displayed any altered morphology was determined as a function of time via AFM image analysis (Figure 5). The extent of bilayer disruption consistently decreased as a function of increasing cholesterol content. This result is consistent with the TBLE/PDA lipid binding assay (compare with the highlighted region in Figure 2), as it also demonstrated a significant decrease in the interaction between htt-exon1(51Q) and the lipid vesicles within the first 2 h of the assay.
Figure 5.
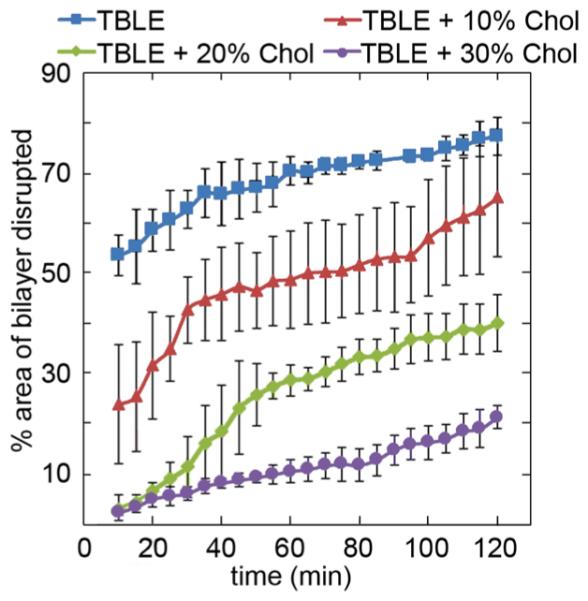
Percent area of disrupted bilayer morphology associated with exposure of lipid bilayers containing various amounts of cholesterol to htt-exon1(51Q) presented as a function of time. Error bars indicate one standard deviation (n = 3).
DISCUSSION
In this study, we investigated the role of cholesterol in modulating the binding of model htt peptides (both synthetic and recombinant) to lipid membranes and the associated consequences. Importantly, these two peptide systems have been shown to interact similarly with lipid bilayers.25 Here, we report several findings. We demonstrate that increased cholesterol content reduces the insertion and binding of N-terminal htt fragments to lipid membranes. While there is still appreciable binding of htt-exon1 to membranes with up to 30% exogenously added cholesterol, there is a stark decrease in the ability of Nt17-Q35-P10-KK peptides to permeabilize lipid vesicles at additions as small as 2.3 wt % cholesterol. Furthermore, the morphological change in TBLE bilayers associated with exposure to htt-exon1 are drastically altered upon the addition of cholesterol. Pure TBLE bilayers develop regions of grainy, disrupted morphology that contain numerous globular aggregates. These regions are associated with stark contrast in AFM phase images which is indicative of altered elastic properties. However, the morphological changes induced in bilayers by exposure to htt-exon1 are drastically altered with the addition of cholesterol. Under these circumstances, discrete, thicker domains develop on the surface, and relatively few globular htt-exon1 aggregates are observed. Furthermore, the extent of bilayer that exhibits altered morphology as a result of exposure to htt-exon1 is significantly reduced as cholesterol content increases.
While it is clear that cholesterol homeostasis is altered in HD, the exact impact on cholesterol levels in the brain in HD remains controversial, as conflicting reports are present in the literature. In clonal striatal-derived cells that overexpress htt with expanded polyQ domains, mRNA levels of important genes associated with cholesterol biosynthesis appear to be reduced.67 This has also been observed in brain HD mice and in post-mortem HD cerebral specimens.41 Reduced levels of cholesterol precursors were detected, via a variety of biochemical and mass spectrometry assays, during the initial stages of disease in brains from transgenic mouse and rat models of HD, and decreased levels of sterols/cholesterol were observed in these same models as disease progressed.41–43 Alterations in cholesterol homeostasis also appear in early stages of HD in humans.68,69 However, there are also conflicting reports that cholesterol accumulation occurs in mouse and human HD tissues and cell cultures.45–47 These reports suggest that htt with an expanded polyQ domain interacts with caveolin-1, resulting in the accumulation of intracellular cholesterol in primary neurons and in brains from YAC72 transgenic mice.45 The accumulation of cholesterol in HD cellular and animal models, as well is in HD human brains, contributed to NMDA-mediated excitotoxicity.47 It has been suggested that limitations on the assays used to quantify cholesterol levels in these different studies may be responsible for the diverging results.70 Despite these conflicting reports on the fate of cholesterol in HD, our results clearly demonstrate that the presence of cholesterol has pronounced effects on htt aggregation and its ability to bind and permeablize lipid membranes. Our observations either illustrate a mechanism through which reduced cholesterol levels in neurons contribute to the misfolding of mutant htt leading to subsequent membrane perturbation or alternatively show how increased cholesterol levels in HD brains could represent a means by which neurons are protected from disruption induced by misfolded htt. This further justifies the need to resolve the controversy regarding altered cholesterol homeostasis in HD.
An interesting observation in cells expressing mutant htt was an increased number of membrane rafts associated with accumulation of cholesterol.47 Here, we observed the development of large domains that were reminiscent of raft domains in bilayers that had been enriched with cholesterol and exposed to htt-exon1(51Q). These domains were, however, much thicker than is normally associated with lipid raft domains observed in pure lipid bilayers,71,72 but this may be partially due to the incorporation of the htt protein into these domains. Presumably, the formation of these domains was facilitated by the presence of htt-exon1(51Q) and the excess levels of cholesterol contained within the bilayer; therefore, it is reasonable to speculate that these domains are enriched in both htt and cholesterol. Lipid/htt interactions are also relevant in the htt traflcking within the cell,9,73,74 and cholesterol appears to mediate the affinity of htt for lipid surfaces.
There appears to be a subtle balance between the initial affinity of htt-exon1(51Q) to bind or insert into the lipid bilayer, which appears to be impeded by increasing cholesterol content, and the availability of cholesterol to aid in forming these domains once htt-exon1(51Q) has inserted into the bilayer. Both Nt17-Q35-P10-KK and htt-exon1(51Q) binding and insertion is impeded by the condensation or a stiffiening effect of adding cholesterol to the membrane. Furthermore, the htt-exon1 that does insert into cholesterol enriched membranes behaves very differently, in how it alters the bilayer morphology and aggregates. Importantly, the development of these domains does not appear to be associated with membrane leakage induced by exposure to htt. However, it should be noted that the leakage assays were performed with Nt17-Q35-P10-KK, and the AFM studies were performed with htt-exon1(51Q). Another potential scenario by which cholesterol reduces the affinity of htt for lipid membranes is by restricting the efficient transition of the Nt17 into an amphipathic α-helix, a structure which is associated with its ability to bind lipids.25–27,75 Hydrogen bonding with phospholipid headgroups appears to stabilize α-helix formation in Nt17, facilitating binding to lipid membranes.76 It is possible that additional cholesterol content may interfere with the formation of these important hydrogen bonds.
It has been hypothesized that the interaction of htt with membranous surfaces may mediate pathogenesis in a numerous ways, including stabilization or promotion of specific aggregate species, altered mechanical integrity of membranes leading to their dysfunction, and in traflcking of htt within the cell. The polyQ repeat in htt adopts a variety of conformations as a monomer77 and in aggregated species.78 It is plausible that a subset of these conformers mediate pathogenesis in HD, and in this regard, the monoclonal antibody 3B5H10 recognizes a species of polyQ that strongly predicts neuronal death in cell culture.79 If specific htt conformations play an important role in HD, understanding environmental factors that promote the relative abundance of disease relevant conformations may provide key insights into mechanisms associated with htt aggregation and toxicity. Lipid membranes represent such an environmental factor and have already been found to promote the formation of distinct aggregate species of htt fragments depending on the flanking sequences surrounding the polyQ domain.25 Here, we demonstrated that the accumulation and/or aggregation of htt-exon1 with an expanded polyQ domain can be further modulated by changing the cholesterol content in lipid bilayers. Computational studies have demonstrated that when a variety of model htt peptides directly interact with DOPC membranes that there is an increase in the β-sheet content within the peptide.76 Because of the lack of distinct htt aggregates observed on lipid bilayers enriched in cholesterol, it may be that cholesterol interferes with the ability of htt to transition to β-sheet rich structure that promotes aggregation.
The ability of protein aggregation to disrupt membrane integrity has been proposed as a potential mechanism associated with several neurodegenerative diseases.80 In this regard, the impact of htt on membrane integrity and mechanics was highly dependent on cholesterol content. Mechanical changes to lipid bilayers induced by htt exon1 could underlie membrane dysfunction observed in animal models,6,81 and it appears that increased levels of cholesterol may protect against these changes. However, it should be noted that, due to experimental convenience, the levels of exogenously added cholesterol used in this study were larger than would be typically observed in a cellular membrane. The development of globular htt aggregates on the surface was highly correlated with the development of regions of increased bilayer roughness observed upon exposure of pure TBLE bilayers to htt-exon1(51Q), while addition of cholesterol ameliorated these specific effects. Additionally, pure TBLE vesicles, when exposed to Nt17-Q35-P10-KK, had markedly higher permeability compared to those with exogenous cholesterol. This suggests that the formation of these globular htt aggregates may play a mechanistic role in htt-induced membrane permeabilization.
In the interaction of htt with lipid membranes, cholesterol appears to play a significant role. The affinity of htt for membranes decreased with increasing amounts of exogenously added cholesterol, and the addition of cholesterol significantly reduced the ability of htt to damage membranes. The reduced ability to damage membranes enriched in cholesterol may be attributed to the manner in which htt alters membrane morphology and aggregates on the membrane surface. Collectively, these observations suggest that alterations in cholesterol homeostasis associated with HD may play an important role in dictating the ability of htt to aggregate into specific species and damage cellular membranes. While the studies presented here demonstrate that cholesterol concentration has a dramatic effect on htt binding to and aggregation on lipid membranes, further structural studies may clarify the mechanisms underlying these observations.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institutes of Health Grant R15NS090380. P.J. was supported by the Honors Summer Undergraduate Research Experience program (The Honors College at West Virginia University). This work was supported, in part, by a grant to Gettysburg College from the Howard Hughes Medical Institute through the Precollege and Undergraduate Science Education Program. W.A.C. and S.L.F. thank Gettysburg College for financial support.
ABBREVIATIONS
- AFM
atomic force microscopy
- chol
choleseterol
- HD
Huntington disease
- HFIP
hexafluoroisopropanol
- htt
huntingtin
- GST
glutathione S-transferase
- LPLC
low pressure liquid chromatography
- LUVs
large unilamellar vesicles
- %CR
% colorimetric response
- PBS
phosphate-buffered saline
- PDA
polydiacetylene
- polyQ
polyglutamine
- polyP
polyproline
- Nt17
the first 17 amino acids of htt
- TBLE
total brain lipid extract
- TFA
trifluoroacetic acid
Footnotes
Author Contributions
J.L. and S.L.F. conceived and coordinated the study and wrote the paper. X.G. performed all in solution AFM experiments on htt-exon1(51Q) and lipid bilayers and purified GST-htt-exon1(51Q). W.A.C. performed Langmuir trough experiments and the vesicle leakage assays with Nt17-Q35-P10-KK. M.C. and P.J. performed the TBLE/PDA lipid binding assays and assisted in purifying GST-htt-exon1(51Q). A.E.L. performed cholesterol concentration assays and assisted with other control experiments. All authors analyzed the results and approved the final version of the manuscript.
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00900.
Additional supplemental data (PDF)
Supplemental movie 1 (MPG1)
Supplemental movie 2 (MPG2)
REFERENCES
- (1).The Huntington Disease Collaborative Research Group. MacDonald ME, et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- (2).DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- (3).Ravina B, Romer M, Constantinescu R, Biglan K, Brocht A, Kieburtz K, Shoulson I, McDermott MP. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov. Disord. 2008;23:1223–1227. doi: 10.1002/mds.21988. [DOI] [PubMed] [Google Scholar]
- (4).Kim YJ, Yi Y, Sapp E, Wang YM, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ratovitski T, Gucek M, Jiang H, Chighladze E, Waldron E, D’Ambola J, Hou Z, Liang Y, Poirier MA, Hirschhorn RR, Graham R, Hayden MR, Cole RN, Ross CA. Mutant Huntingtin N-terminal Fragments of Specific Size Mediate Aggregation and Toxicity in Neuronal Cells. J. Biol. Chem. 2009;284:10855–10867. doi: 10.1074/jbc.M804813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- (7).Landles C, Sathasivam K, Weiss A, Woodman B, Moffitt H, Finkbeiner S, Sun B, Gafni J, Ellerby LM, Trottier Y, Richards WG, Osmand A, Paganetti P, Bates GP. Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem. 2010;285:8808–8823. doi: 10.1074/jbc.M109.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- (9).Caviston JP, Holzbaur ELF. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Desmond CR, Maiuri T, Truant R. A multifunctional, multi-pathway intracellular localization signal in Huntingtin. Commun. Integr. Biol. 2013;6:e23318. doi: 10.4161/cib.23318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- (12).Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- (13).Kegel KB, Sapp E, Alexander J, Valencia A, Reeves P, Li X, Masso N, Sobin L, Aronin N, DiFiglia M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 2009;110:1585–1597. doi: 10.1111/j.1471-4159.2009.06255.x. [DOI] [PubMed] [Google Scholar]
- (14).Kegel KB, Sapp E, Yoder J, Cuiffo B, Sobin L, Kim YJ, Qin ZH, Hayden MR, Aronin N, Scott DL, Isenberg F, Goldmann WH, DiFiglia M. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- (15).Kegel KB, Schewkunow V, Sapp E, Masso N, Wanker EE, DiFiglia M, Goldmann WH. Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem. Biophys. Res. Commun. 2009;387:472–475. doi: 10.1016/j.bbrc.2009.07.039. [DOI] [PubMed] [Google Scholar]
- (16).Kegel-Gleason KB. Huntingtin Interactions with Membrane Phospholipids: Strategic Targets for Therapeutic Intervention? J. Huntington’s Dis. 2013;2:239–250. doi: 10.3233/JHD-130068. [DOI] [PubMed] [Google Scholar]
- (17).Qin ZH, Wang YM, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Suopanki J, Gotz C, Lutsch G, Schiller J, Harjes P, Herrmann A, Wanker EE. Interaction of huntingtin fragments with brain membranes - clues to early dysfunction in Huntington’s disease. J. Neurochem. 2006;96:870–884. doi: 10.1111/j.1471-4159.2005.03620.x. [DOI] [PubMed] [Google Scholar]
- (19).Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- (20).Vidal R, Caballero B, Couve A, Hetz C. Converging Pathways in the Occurrence of Endoplasmic Reticulum (ER) Stress in Huntington’s Disease. Curr. Mol. Med. 2011;11:1–12. doi: 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- (21).Atwal RS, Truant R. A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy. 2008;4:91–93. doi: 10.4161/auto.5201. [DOI] [PubMed] [Google Scholar]
- (22).Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LSB. Disruption of axonal transport by loss of huntingtin or expression of pathogenic PolyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- (23).Lee W-CM, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006;172:605–618. doi: 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Burke KA, Kauffman KJ, Umbaugh CS, Frey SL, Legleiter J. The Interaction of Polyglutamine Peptides With Lipid Membranes is Regulated by Flanking Sequences Associated with Huntingtin. J. Biol. Chem. 2013;288:14993–15005. doi: 10.1074/jbc.M112.446237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Michalek M, Salnikov, Evgeniy S, Bechinger B. Structure and Topology of the Huntingtin 1–17 Membrane Anchor by a Combined Solution and Solid-State NMR Approach. Biophys. J. 2013;105:699–710. doi: 10.1016/j.bpj.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Michalek M, Salnikov ES, Werten S, Bechinger B. Membrane Interactions of the Amphipathic Amino Terminus of Huntingtin. Biochemistry. 2013;52:847–858. doi: 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- (28).Burke KA, Hensal KM, Umbaugh CS, Chaibva M, Legleiter J. Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. Biochim. Biophys. Acta, Biomembr. 2013;1828:1953–1961. doi: 10.1016/j.bbamem.2013.04.025. [DOI] [PubMed] [Google Scholar]
- (29).Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- (30).Wilson JD, Lindsey CA, Dietschy JM. Influence of dietary cholesterol on cholesterol metabolism. Ann. N. Y. Acad. Sci. 1968;149:808–821. doi: 10.1111/j.1749-6632.1968.tb53837.x. [DOI] [PubMed] [Google Scholar]
- (31).Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell. Mol. Life Sci. 2000;57:1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 2000;2:42–49. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- (33).Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- (34).Serfis AB, Brancato S, Fliesler SJ. Comparative behavior of sterols in phosphatidylcholine-sterol monolayer films. Biochim. Biophys. Acta, Biomembr. 2001;1511:341–348. doi: 10.1016/s0005-2736(01)00291-7. [DOI] [PubMed] [Google Scholar]
- (35).Shamitko-Klingensmith N, Molchanoff KM, Burke KA, Magnone GJ, Legleiter J. Mapping the Mechanical Properties of Cholesterol-Containing Supported Lipid Bilayers with Nanoscale Spatial Resolution. Langmuir. 2012;28:13411–13422. doi: 10.1021/la302705f. [DOI] [PubMed] [Google Scholar]
- (36).Paratcha G, Ibáñez CF. Lipid rafts and the control of neurotrophic factor signaling in the nervous system: variations on a theme. Curr. Opin. Neurobiol. 2002;12:542–549. doi: 10.1016/s0959-4388(02)00363-x. [DOI] [PubMed] [Google Scholar]
- (37).Smaby JM, Brockman HL, Brown RE. Cholesterol’s Interfacial Interactions with Sphingomyelins and-Phosphatidylcholines: Hydrocarbon Chain Structure Determines the Magnitude of Condensation. Biochemistry. 1994;33:9135–9142. doi: 10.1021/bi00197a016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Valenza M, Cattaneo E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011;34:474–486. doi: 10.1016/j.tins.2011.06.005. [DOI] [PubMed] [Google Scholar]
- (39).Karasinska JM, Hayden MR. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011;7:561–572. doi: 10.1038/nrneurol.2011.132. [DOI] [PubMed] [Google Scholar]
- (40).Leoni V, Caccia C. The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2015;1851:1095–1105. doi: 10.1016/j.bbalip.2014.12.018. [DOI] [PubMed] [Google Scholar]
- (41).Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Valenza M, Carroll JB, Leoni V, Bertram LN, Bjorkhem I, Singaraja RR, Di Donato S, Lutjohann D, Hayden MR, Cattaneo E. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum. Mol. Genet. 2007;16:2187–2198. doi: 10.1093/hmg/ddm170. [DOI] [PubMed] [Google Scholar]
- (43).Valenza M, Leoni V, Tarditi A, Mariotti C, Bjorkhem I, Di Donato S, Cattaneo E. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2007;28:133–142. doi: 10.1016/j.nbd.2007.07.004. [DOI] [PubMed] [Google Scholar]
- (44).Ritch JJ, Valencia A, Alexander J, Sapp E, Gatune L, Sangrey GR, Sinha S, Scherber CM, Zeitlin S, Sadri-Vakili G, Irimia D, DiFiglia M, Kegel KB. Multiple phenotypes in Huntington disease mouse neural stem cells. Mol. Cell. Neurosci. 2012;50:70–81. doi: 10.1016/j.mcn.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet. 2006;15:3578–3591. doi: 10.1093/hmg/ddl434. [DOI] [PubMed] [Google Scholar]
- (46).Luthi-Carter R, Taylor DM, Pallos J, Lambert E, Amore A, Parker A, Moffitt H, Smith DL, Runne H, Gokce O, Kuhn A, Xiang Z, Maxwell MM, Reeves SA, Bates GP, Neri C, Thompson LM, Marsh JL, Kazantsev AG. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7927–7932. doi: 10.1073/pnas.1002924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).del Toro D, Xifro X, Pol A, Humbert S, Saudou F, Canals JM, Alberch J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010;115:153–167. doi: 10.1111/j.1471-4159.2010.06912.x. [DOI] [PubMed] [Google Scholar]
- (48).Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon I-JL, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chen SM, Wetzel R. Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 2001;10:887–891. doi: 10.1110/ps.42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Burke RW, Diamondstone BI, Velapoldi RA, Menis O. Mechanisms of the Liebermann-Burchard and Zak Color Reactions for Cholesterol. Clin. Chem. 1974;20:794–801. [PubMed] [Google Scholar]
- (52).Seelig A. Local anesthetics and pressure: a comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta, Biomembr. 1987;899:196–204. doi: 10.1016/0005-2736(87)90400-7. [DOI] [PubMed] [Google Scholar]
- (53).Anderson RL, Davis S. An organic phosphorus assay which avoids the use of hazardous perchloric acid. Clin. Chim. Acta. 1982;121:111–116. doi: 10.1016/0009-8981(82)90216-9. [DOI] [PubMed] [Google Scholar]
- (54).Rouser G, Fleischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- (55).Sokolovski M, Sheynis T, Kolusheva S, Jelinek R. Membrane interactions and lipid binding of casein oligomers and early aggregates. Biochim. Biophys. Acta, Biomembr. 2008;1778:2341–2349. doi: 10.1016/j.bbamem.2008.07.001. [DOI] [PubMed] [Google Scholar]
- (56).Zheng F, Wu Z, Chen Y. A quantitative method for the measurement of membrane affinity by polydiacetylene-based colorimetric assay. Anal. Biochem. 2012;420:171–176. doi: 10.1016/j.ab.2011.09.026. [DOI] [PubMed] [Google Scholar]
- (57).Melcher J, Carrasco C, Xu X, Carrascosa JL, Gomez-Herrero J, José de Pablo P, Raman A. Origins of phase contrast in the atomic force microscope in liquids. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13655–13660. doi: 10.1073/pnas.0902240106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).O’Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J. Lipid Res. 1965;6:537–544. [PubMed] [Google Scholar]
- (59).Groves JT, Ulman N, Boxer SG. Micropatterning fluid lipid bilayers on solid supports. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- (60).Legleiter J, Fryer JD, Holtzman DM, Kowalewski T. The Modulating Effect of Mechanical Changes in Lipid Bilayers Caused by ApoE-Containing Lipoproteins on A beta Induced Membrane Disruption. ACS Chem. Neurosci. 2011;2:588–599. doi: 10.1021/cn2000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Yates EA, Cucco EM, Legleiter J. Point Mutations in Aβ Induce Polymorphic Aggregates at Liquid/Solid Interfaces. ACS Chem. Neurosci. 2011;2:294–307. doi: 10.1021/cn200001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yates EA, Owens SL, Lynch MF, Cucco EM, Umbaugh CS, Legleiter J. Specific domains of Aβ facilitate aggregation on and association with lipid bilayers. J. Mol. Biol. 2013;425:1915–1933. doi: 10.1016/j.jmb.2013.03.022. [DOI] [PubMed] [Google Scholar]
- (63).Burke KA, Yates EA, Legleiter J. Amyloid-Forming Proteins Alter the Local Mechanical Properties of Lipid Membranes. Biochemistry. 2013;52:808–817. doi: 10.1021/bi301070v. [DOI] [PubMed] [Google Scholar]
- (64).Yip CM, Darabie AA, McLaurin J. A beta 42-peptide assembly on lipid Bilayers. J. Mol. Biol. 2002;318:97–107. doi: 10.1016/S0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- (65).Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membrane-associated Aβ-fibrillogenesis and neurotoxicity. J. Mol. Biol. 2001;311:723–734. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- (66).Burke KA, Godbey J, Legleiter J. Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods. 2011;53:275–284. doi: 10.1016/j.ymeth.2010.12.028. [DOI] [PubMed] [Google Scholar]
- (67).Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- (68).Leoni V, Mariotti C, Nanetti L, Salvatore E, Squitieri F, Bentivoglio AR, Bandettini del Poggio M, Piacentini S, Monza D, Valenza M, Cattaneo E, Di Donato S. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci. Lett. 2011;494:245–249. doi: 10.1016/j.neulet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- (69).Leoni V, Mariotti C, Tabrizi SJ, Valenza M, Wild EJ, Henley SMD, Hobbs NZ, Mandelli ML, Grisoli M, Björkhem I, Cattaneo E, Di Donato S. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain. 2008;131:2851–2859. doi: 10.1093/brain/awn212. [DOI] [PubMed] [Google Scholar]
- (70).Marullo M, Valenza M, Leoni V, Caccia C, Scarlatti C, De Mario A, Zuccato C, Di Donato S, Carafoli E, Cattaneo E. Pitfalls in the detection of cholesterol in Huntington’s disease models: Technical considerations in the detection of cholesterol in Huntington’s disease samples. PLoS Curr.: Huntington Dis. 2012;1:e9a1968. doi: 10.1371/505886e9a1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Orsini F, Cremona A, Arosio P, Corsetto PA, Montorfano G, Lascialfari A, Rizzo AM. Atomic force microscopy imaging of lipid rafts of human breast cancer cells. Biochim. Biophys. Acta, Biomembr. 2012;1818:2943–2949. doi: 10.1016/j.bbamem.2012.07.024. [DOI] [PubMed] [Google Scholar]
- (72).Yuan C, Furlong J, Burgos P, Johnston LJ. The Size of Lipid Rafts: An Atomic Force Microscopy Study of Ganglioside GM1 Domains in Sphingomyelin/DOPC/Cholesterol Membranes. Biophys. J. 2002;82:2526–2535. doi: 10.1016/S0006-3495(02)75596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99:179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- (74).Orr AL, Li S, Wang C-E, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li X-J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Côté S, Binette V, Salnikov, Evgeniy S, Bechinger B, Mousseau N. Probing the Huntingtin 1–17 Membrane Anchor on a Phospholipid Bilayer by Using All-Atom Simulations. Biophys. J. 2015;108:1187–1198. doi: 10.1016/j.bpj.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Nagarajan A, Jawahery S, Matysiak S. The Effects of Flanking Sequences in the Interaction of Polyglutamine Peptides with a Membrane Bilayer. J. Phys. Chem. B. 2014;118:6368–6379. doi: 10.1021/jp407900c. [DOI] [PubMed] [Google Scholar]
- (77).Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary Structure of Huntingtin Amino-Terminal Region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9679–9684. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, Yang XW, Masliah E, Thompson LM, Muchowski PJ, Weisgraber KH, Finkbeiner S. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Burke KA, Yates EA, Legleiter J. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 2013;4:17. doi: 10.3389/fneur.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Difiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat-brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.