

A patient-derived CTC culture assay for anticancer drug evaluation to guide therapy for personalized treatment.
Keywords: Breast cancer, circulating tumor cells, drug response, combinational therapy, microfluidics
Abstract
The lack of a robust anticancer drug screening system to monitor patients during treatment delays realization of personalized treatment. We demonstrate an efficient approach to evaluate drug response using patient-derived circulating tumor cell (CTC) cultures obtained from liquid biopsy. Custom microfabricated tapered microwells were integrated with microfluidics to allow robust formation of CTC clusters without pre-enrichment and subsequent drug screening in situ. Rapid feedback after 2 weeks promotes immediate intervention upon detection of drug resistance or tolerance. The procedure was clinically validated with blood samples (n = 73) from 55 patients with early-stage, newly diagnosed, locally advanced, or refractory metastatic breast cancer. Twenty-four of these samples were used for drug evaluation. Cluster formation potential correlated inversely with increased drug concentration and therapeutic treatment. This new and robust liquid biopsy technique can potentially evaluate patient prognosis with CTC clusters during treatment and provide a noninvasive and inexpensive assessment that can guide drug discovery development or therapeutic choices for personalized treatment.
INTRODUCTION
Progression of anticancer drug development into actual clinical utility remains hindered by the inherent heterogeneity of cancer. Furthermore, tumor biopsies often fail to reflect the complete cancer gene expression profile (1, 2). Current imaging techniques for cancer evaluation [for example, computed tomography (CT) scan] also suffer from technical limitations (3, 4), which lead to false-negative findings. These limitations affect the development of prognostic assays in aiding the development of robust drug regimens. At present, several drug screening systems available for cancer cell lines are often designed for use with multiwell plates (5) or robotics (6). However, the translational relevance of cell lines is often in question because prolonged culture and multiple passages lead to phenotypes that are no longer representative of the original tumor in terms of the cell’s epigenetics and gene expression.
In the past few years, several methods of in vitro cultures of circulating tumor cells (CTCs) were established (7–10). CTCs are primary cancer cells that originate from either primary or secondary tumors (11, 12). Their enumeration had led to the finding that, at least in metastatic patients, their prevalence correlates with shorter overall survival (13, 14). Previous attempts at CTC expansion led to the establishment of cell lines derived from CTCs of breast, colon, and prostate cancer patients. These cell lines were usually established after months in culture, requiring pre-enrichment procedures such as affinity binding, fluorescence-activated cell sorting, or negative selection (Table 1). The efficiency in obtaining CTC cultures using these methods was also low (<20%), and the need for pre-enrichment with each method resulted in a loss of CTCs.
Table 1. Comparison of the sensitivity of the CTC cluster assay and conventional CTC expansion techniques. N.D., not determined.
|
Cancer type |
Samples validated |
Duration | Culture type |
Pre- enrichment |
Efficiency in culture |
Correlation to treatment |
Reference |
| Breast | 36 | >6 months | Cell lines, long-term | Yes | 16.70% | N.D. | (8) |
| Breast | 8 | <1 month or ≧1 month |
Colonies (short-term) or cell lines (long-term) |
Yes | 37.50% | N.D. | (7) |
| Colon | 71 | >2 months | Cell lines, long-term | Yes | 2.80% | N.D. | (52) |
| Prostate | 17 | >6 months | Organoid lines, long-term | Yes | ~15–20% | N.D. | (9) |
| Breast | 73 | 2 weeks | Primary CTC cluster (short- term) |
No | 19.6–59.3% (depending on treatment time point) |
Yes | This work |
An efficient and reliable drug screening system for routine clinical use should consider the following factors: (i) minimal requirements for reagents and samples, (ii) cultures simulating in vivo conditions (15), and (iii) simplicity and robustness of the system. These factors are especially relevant in “real-world” circumstances, whereby clinicians are often provided with small “windows” of opportunity to make decisions for treatment based on the information gathered from tumor imaging and sample profiling. Because of the rate of acquiring drug resistance or tolerance, it is inevitable that a prolonged and continuous change of drug combinations will be required to match the ability of cancer cells to overcome treatment. Here, we demonstrate an efficient approach to evaluating patient drug response using patient-derived CTC cultures in an integrated microfluidic system incorporating microfabricated microwells. Our results showed that the assay can be used to rapidly evaluate the drug response of CTCs derived from blood of an individual patient throughout the time course of treatment. Rapid feedback of the median inhibitory concentration (IC50) values after 2 weeks promotes immediate intervention upon detection of drug resistance or tolerance. Our results demonstrated that the CTC cluster assay can be a potential tool for evaluating patient prognosis during treatment while guiding drug discovery development and providing therapeutic choices for personalized treatment at the same time.
RESULTS
Establishment of CTC assay for real-time evaluation of patient response
To realize the usage of CTCs in a clinical setting, we developed a method to rapidly evaluate patient drug response within 2 weeks based on a short-term primary CTC culture without the need for pre-enrichment (Fig. 1). This system uses a microfluidic assay integrated with two components: (i) a culture component comprising custom-designed tapered microwells and (ii) a drug assay component with a gradient generator to carry out drug screening of different concentrations simultaneously on the same patient-derived sample.
Fig. 1. Schematic overview depicting the procedure for anticancer drug screening via conventional methods and the CTC cluster method.

In the case of conventional methods, cancer cells are derived from commercialized cell lines or patient-derived CTCs and tumors. Establishment of CTC cell lines requires more than 6 months, and tumor sampling can only be carried out as a single sampling. In addition, pre-enrichment of CTCs is required before they can be cultured. Conversely, CTC clusters can be generated within 2 weeks, and the blood samples do not require pre-enrichment before culture. In this procedure, blood samples are first lysed briefly to remove red blood cells (RBCs), and the resultant nucleated cell fraction is seeded into an integrated microwell-based microfluidic assay. Drugs can be introduced directly in situ, and a microfluidic component helps to efficiently distribute a range of drug concentrations.
The microfluidic device comprised three polydimethylsiloxane (PDMS) layers. Each layer was obtained via a master mold, and the leak-free and permanent assembly was achieved by bonding via oxygen plasma surface activation (see Materials and Methods and fig. S1). The topmost layer contained the tree-like gradient generator (fig. S2), which enabled the mixing of two different chemicals to eight different resulting concentrations (16). The intermediate layer was the channel barrier, which prevented the fluids with different concentrations from mixing at the cell culture region. Finally, the bottommost layer contained the customized multimicrowell arrays. Each microwell had an elliptical top section of 250 × 150 μm and a depth of 150 μm.
As shown in Fig. 2A and fig. S1, the three PDMS layers produced from their master molds were assembled by using standard plasma treatment procedures. Performance stability of the gradient generator was ascertained by determining the concentration gradient generated using both actual runs with fluorescence dyes (Fig. 2B) and COMSOL-simulated flows. Deionized water and 100% dye solution were pumped into the integrated device at different flow rates. Both fluids mixed well in the serpentine channels and generated diluted dye solution under a range of concentrations. After the flow in the channels reached steady state, the fluids at the eight outlets were collected and measured for fluorescence intensity.
Fig. 2. Establishment of CTC cluster assay for routine drug screening.
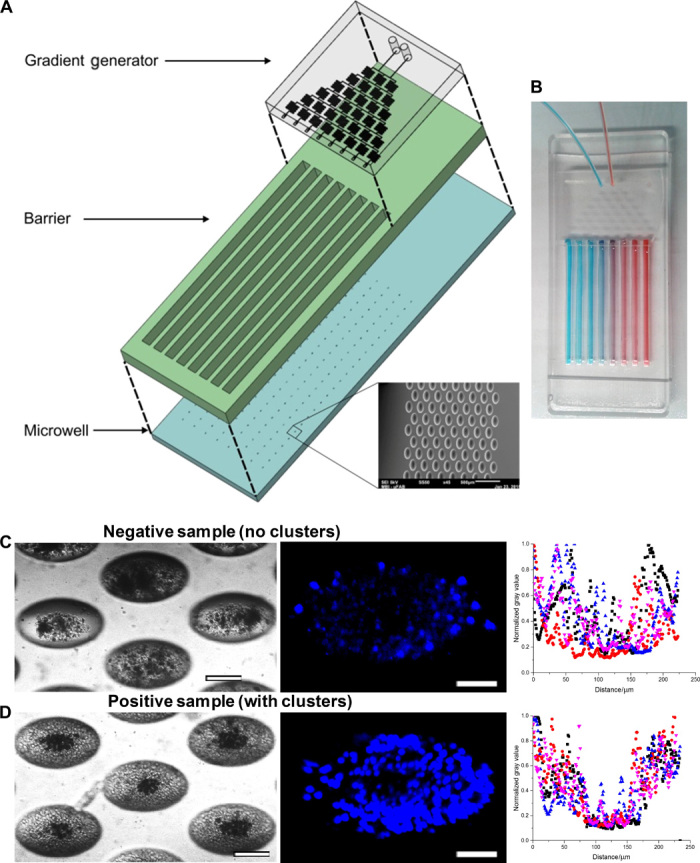
(A) Three-dimensional layout of drug assay displaying the layers for the gradient generator, barrier, and microwells. (B) Gradient distribution of input reagents demonstrated by blue and red dyes. (C) (Left) Representative images of negative and positive samples. Bright-field images of microwells comprising a negative sample at ×10 magnification. Scale bar, 100 μm. (Middle) Hoechst staining of clusters in situ. Negative samples generated debris with some residual white blood cells (WBCs). Scale bar, 50 μm. (Right) Combined scatterplots of gray values, which reflected the density of cells, across each microwell. Values were normalized to the highest count for a particular microwell. Microwells with sparse groups of cells or debris demonstrated high gray values within the microwell region. (D) (Left) Bright-field images of microwells comprising a positive sample at ×10 magnification. Scale bar, 100 μm. (Middle) Nuclei staining using Hoechst on cell clusters in situ. Positive samples generated clusters with some residual WBCs. Scale bar, 50 μm. (Right) Combined scatterplots of gray values, with values normalized to the highest count for a particular microwell. Microwells with dense cell clusters demonstrated consistently low gray values (<0.5) within the microwell region.
To verify that the gradient generation function was independent of dye concentration, two different dye concentrations (20 and 100 μM) were first tested under the same flow rate of 100 μl/min. On the basis of these calibration results (see Supplementary Materials and Methods and fig. S3A), we converted fluorescence intensity into dye concentration. Quantification of the relative fluorescein isothiocyanate (FITC) dye concentration in each of the eight channels confirmed that the gradient distribution was in line with the mathematical calculations, which assumed thorough mixing and negligible diffusion within the gradient generator (table S1). The trend was constant for five different flow rates (from 25 to 200 μl/min), suggesting that the device performed robustly under various flow rates (fig. S3B). In the case of two reagents, they would be mixed accordingly in the same pattern but opposing gradients, as illustrated by food dyes (Fig. 2B). Fluctuations from expected values were present but insignificant (P < 0.05). Subsequent experiments were then conducted at 100 μl/min as the flow profile generated was closest to that calculated, indicating that at this flow rate, (i) there was thorough mixing in the serpentine and (ii) there was negligible diffusion across the serpentines. These demonstrated that the integrated drug screening device was able to generate consistent concentration gradient under different flow rates and input concentrations. Using clinical samples for evaluation, we classified samples that only generated cell debris or sparse monolayers as negative (Fig. 2C), whereas samples that led to CTC-containing clusters in at least 50% of microwells (14) (~500 clusters established from 1.25 ml of blood) were determined to be positive (Fig. 2D). The determination of cluster morphology was standardized by obtaining plot profiles of gray values using image processing software (see Supplementary Materials and Methods).
Stability of microfluidic CTC cluster assay under perfusion
Because we intended to culture patient-derived primary cell samples in the integrated microfluidic device, we ensured that the shear rate and fresh medium perfusion rate were uniform across eight cell culture channels. To estimate the flow rate inside the eight cell culture channels, additional flow profiles were generated with simulated flow tests in a simplified gradient generator design using multiphysics modeling software (COMSOL) (fig. S4A). The simulation of a simplified channel version was shown, and flow rates were color-coded. The flow rate of eight outlets differs from each other at a large scale under this design because of the lack of serpentine channel for flow resistance balance. With proper serpentine channel design (fig. S2), the simulated flow rates of eight outlets for the real device were relatively constant (fig. S4B). The maximum flow rate was achieved at the center outlet, and the minimum flow rate was achieved at the side outlets. The difference between these two extreme flow rates was less than 10%. Therefore, we concluded that the deviation of shear and perfusion rates was negligible and that the device was suitable to culture cells in eight parallel channels.
To minimize the effects of evaporation (17) for long-term culture, the device was designed to fit into a 150-mm dish, which can be filled with a thin film of phosphate-buffered saline (PBS) or deionized water. The assay should also be maintained in a humidified chamber. To examine whether the gradient concentration in each channel remained constant over time, FITC dye was used to test for the presence of gradient shift. After stabilized generation of gradient (T = 0 hour), the assay was incubated in the dark. Channel contents were sampled at T = 0 and 24 hours. Under these conditions, we confirmed that the concentrations in each channel remained relatively constant over time (P < 0.05), demonstrating that any fluctuation of gradient was insignificant over time (fig. S5).
To determine cell conservation during solution exchange, we counted cells in specific microwells before and after the inward and outward flows (fig. S6). These microwells were selected at a consistent distance from the inward flow (middle of the channels). Cell counts were also obtained again after multiple solution exchanges. We observed that cell counts and cluster morphology were generally conserved under repeated inward or outward flow conditions (fig. S7A). An insignificant number of small cells from the microwells nearer to the inward flow source (upper portion of channels) were not attached to the cluster and may drift to an adjacent cluster under flow. Using syringe pumps at a constant infusion/withdrawal rate of 100 μl/min, the changes in cell count within microwells varied insignificantly (106.6 ± 9.5%, P = 0.204; Student’s t test), as opposed to when the solutions were exchanged via manual pipetting (88.1 ± 25.6%, P = 0.0261; Student’s t test) (fig. S7B).
An efficient CTC assay for a unique clinical application
The molds were produced with different strategies selected to meet the requirements for geometry, size, and tolerances of the features encoded (Fig. 3A). An overview illustration of the assay protocol is provided in movie S1. Each channel contained about 1000 microwells. To compare cluster formation in tapered and cylindrical microwells, ~50 MCF-7 cells per microwell were seeded into each channel of the assay. This concentration allows sufficient cluster formation to occur. Resultant cultures were contrasted in terms of morphology after 3 days of culture. It was observed that the MCF-7 culture in cylindrical microwells was only able to form multiple irregular small clusters of ~10 to 20 cells. In contrast, the MCF-7 culture in tapered microwells consistently formed a single large cluster comprising all ~50 cells at the center of each microwell (Fig. 3, B and C).
Fig. 3. Comparison of custom tapered microwells fabricated using diffuser back-side lithography for CTC cluster assay and conventional cylindrical microwells.
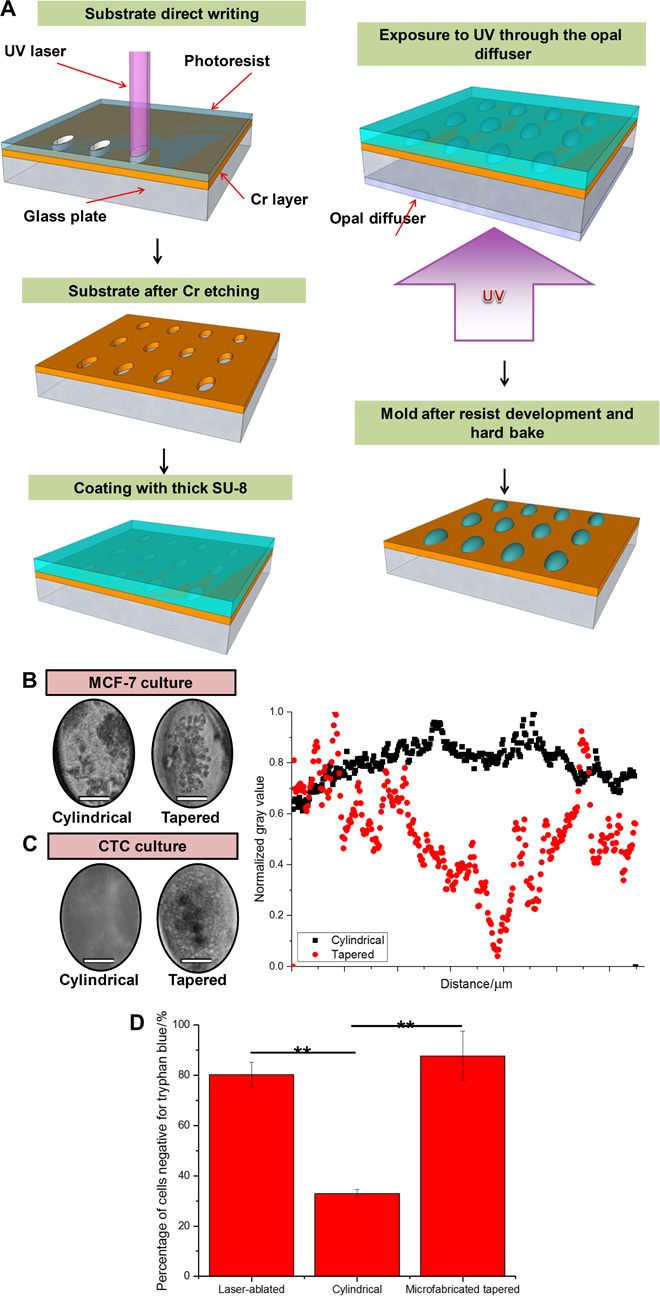
(A) Fabrication procedure: elliptical openings defining the size and position of the wells created on a soda-lime optical mask blank by laser direct writing. Subsequent Cr etching and stripping of the remaining resist were followed by coating with a layer of SU-8 2100 resist. Ultraviolet (UV) exposure to obtain pillar-like structures with the elliptical footprint and elliptical cross-shaped structures. Postbaking, ultrasound bath, and hard baking resulted in a template ready for PDMS molding. (B) (Left) Culture of MCF-7 in custom tapered microwells and cylindrical microwells. Single clusters were consistently established with tapered microwells. Scale bars, 50 μm. (C) (Left) Culture of clinical blood samples in custom tapered microwells and cylindrical microwells. Only debris was formed in cylindrical microwells. Scale bars, 50 μm. (Right) Combined scatterplots of gray values, with values normalized to the highest count for a particular microwell. Cylindrical microwells did not generate clusters, whereas tapered microwells led to a single dense cell cluster as observed from the region of low gray value. (D) Bar graph presenting results from a viability assay using trypan blue staining. Percentage of cells negative for trypan blue (viable cells) was significantly lower in the sample portion cultured in cylindrical microwells. All error bars represent SD of triplicate cultures from different samples. **P < 0.01.
We further validated the parameters with a clinical blood sample and similarly demonstrated consistent formation of a single large cell cluster only in tapered microwells at day 14 of culture (Fig. 3C). The cells within the cluster were heterogeneous and consisted of both CTCs and a residual portion of WBCs, as characterized in our previous publication (14). Trypan blue staining confirmed that the tapered PDMS microwells design retained the viability of the cells (88 ± 20%), in contrast to those cultured in cylindrical microwells (31.5 ± 3%) (Fig. 3D).
For actual studies, blood samples were acquired from breast cancer patients in a procedure termed “liquid biopsy” (18) (Fig. 1). Whole blood was mixed with RBC lysis buffer to retain only the nucleated cell fraction, which consisted of WBCs and CTCs. Nucleated cells were seeded evenly into the channels and cultured under optimized conditions (14). Drugs were introduced at day 11 of culture, and results were observed after 72 hours. We hypothesized that this assay may be a suitable platform for screening of combinational drug therapies on primary cancer cells obtained from cultures established with patient’s blood at various time points of treatment.
Screening of anticancer compounds in assay with cancer cell line
To validate the assay conditions, the assay was first screened by MCF-7 cultures. Initial stages of characterization involved evaluation of a range of cell seeding concentration for cluster formation of cancer cell lines. In the absence of surfactants such as bovine serum albumin, low concentrations of cancer cells adhered individually to the substrate instead of generating clusters (fig. S8). There were some variations in cluster-forming behavior between cancer cell lines and primary CTCs. Clusters only developed in uncoated microwells for cancer cell lines under high cell seeding concentrations. However, CTCs in clinical blood samples did not require surfactant coating to prevent cell adherence, possibly due to the “cushion” provided by nonadherent WBCs. Besides, cancer cell lines can also form clusters readily under both normoxic and hypoxic conditions (under high seeding concentrations). In contrast, clinical samples with primary CTCs can only develop clusters under hypoxic conditions similar to that of the in vivo tumor microenvironment (fig. S9). These highlight the intrinsic differences between immortalized cell lines and their tumor origins and the importance of validating research findings with primary clinical samples.
Subsequently, the drug screening protocol was evaluated by testing doxorubicin on MCF-7 breast cancer cell line clusters. Clusters were exposed to the doxorubicin gradient at day 3 of culture. The viability statistics (normalized to results obtained from samples in the last channel with the lowest drug concentration) of MCF-7 were obtained with LIVE/DEAD staining (calcein-AM/EtBr) after 72 hours of exposure to doxorubicin (Fig. 4A). Clusters under high drug concentrations were mostly nonviable (red), whereas clusters under low drug concentrations were mostly viable (green). The corresponding dose-response curve was plotted using a four-parameter logistic equation, and the IC50 value for MCF-7 cultures was obtained.
Fig. 4. Assay validation with controls.
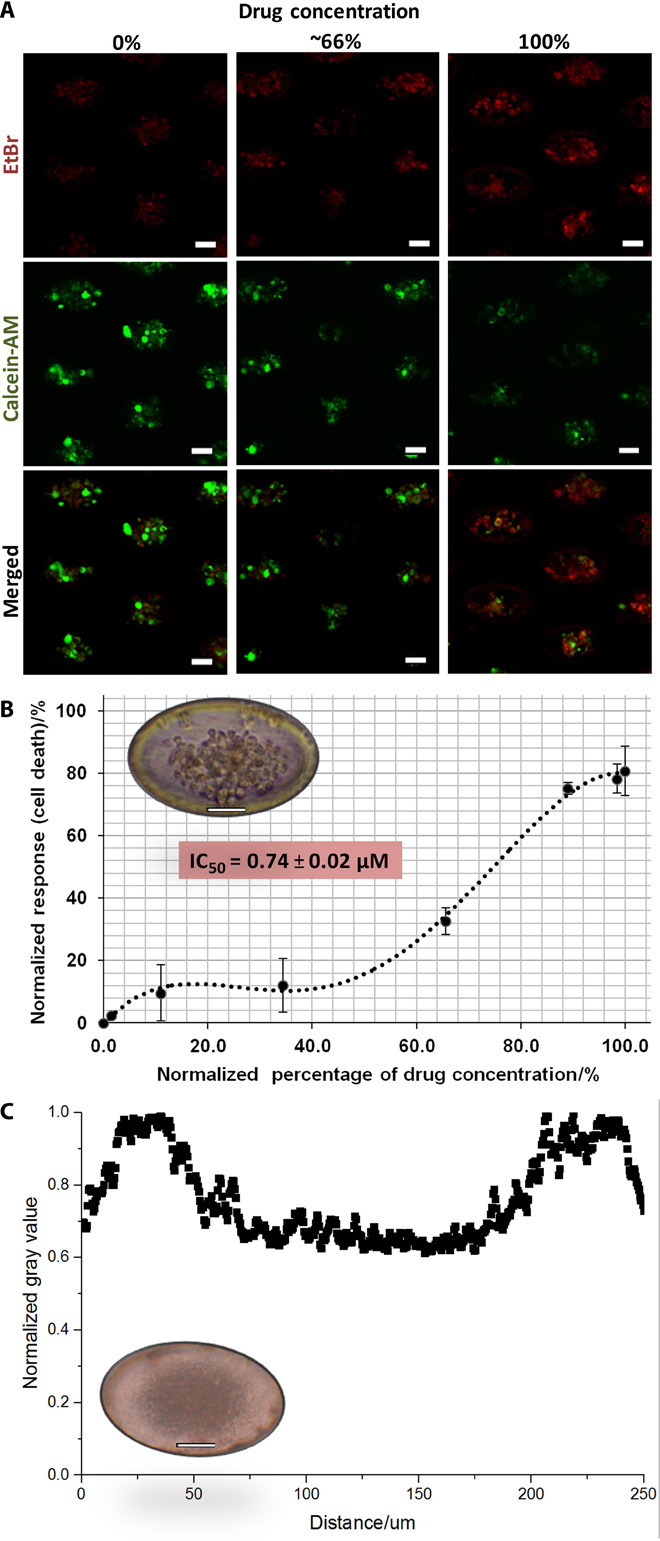
(A) Screening of doxorubicin in microwell assay using MCF-7 cancer cell line. Cultures were imaged in situ after staining with LIVE (calcein-AM; green) and DEAD [ethidium bromide (EtBr); red] under 72 hours of exposure to doxorubicin. Clusters under high drug concentrations are mostly nonviable (red), whereas clusters under low drug concentrations are mostly viable (green). (B) Dose-response curve and corresponding IC50 value (0.78 ± 0.02 μM) of MCF-7 generated from viability results. Representative image shows an MCF-7 cell cluster within a microwell (inset). Scale bars, 50 mm. (C) Scatterplot demonstrating overall high gray values that reflect the absence of clusters from cultures of blood from healthy volunteers. Representative image shows cell debris generated within a microwell from culture of a healthy sample (inset). Scale bars, 50 mm.
Using the microwell-based assay, we obtained an IC50 value of 0.78 ± 0.02 μM (Fig. 4B). Because of the multilayer nature of these clusters, the value obtained was slightly higher than that in previous studies done on monolayer cultures with lower cell counts (~0.5 μM, <10,000 cells/ml) (19). This observation could be due to heightened cell density in the presence of clusters or spheroids, which had been shown to reduce penetration of drugs (20). The evaluation of drug response on cancer cell clusters in vitro was generally more favorable than that on monolayer cultures, as it may better reflect the situation in vivo.
Screening of anticancer compounds in assay with clinical blood samples
Preprocessing steps of RBC lysis for the whole-blood sample were detailed in Materials and Methods. For clinical samples, cluster formation is also dependent on the density of CTCs to WBCs. We screened a number of samples for culture after processing using a label-free inertia-based microfluidic device (21) with a threshold of ~15 μm to remove a portion of WBCs. The results demonstrated that for samples that did not initially generate clusters before enrichment, they could lead to clusters after enrichment. Post-enriched samples were stained with calcein-AM and introduced back to a small count of WBCs from the initial sample in a 1:1 ratio (fig. S10). These post-enriched cells contain CTC-to-WBC counts in an approximately 1:100 to 1:1000 ratio. This puts a rough estimate of the minimum proportion of CTCs required for cluster formation at 0.05% (~1 CTC per microwell). The presence of cancer cells is likely to induce global, diffusion-based differentiation rules in the neighboring WBCs to induce the cluster phenotype. Cultures obtained from the blood of healthy volunteers did not generate clusters (Fig. 4C).
Forty-nine clinical samples from breast cancer patients were cultured with the microfluidic device as a preliminary validation of the procedure (table S2). Presence of cancer cells was validated with fluorescence in situ hybridization to identify cells with increased expression of breast cancer–associated markers TOP2A and CCND1 (fig. S11). We processed another four samples separately to correlate epithelial CTC counts (CK+) before culture with the presence of clusters. Four samples (1 ml each) were processed for CD45 (leukocyte marker) and pan-cytokeratin (CK; epithelial marker) immunostaining to identify epithelial CTC counts before culture, followed by screening with a conventional analysis method using flow cytometry. The rest of the samples were cultured and analyzed in a similar manner after 2 weeks to correlate epithelial CTC counts with cluster-forming potential. Overall, the proportion of epithelial CTCs after culture was found to correlate with cluster-forming potential (fig. S12). From these four samples, there appeared to be a lack of immediate correlation between postculture epithelial CTC counts and preculture epithelial cell counts.
Subsequently, 24 samples were cultured, and six positive samples that exhibited clusters at day 11 were eventually evaluated for drug screening (table S3). Samples were determined to be positive using the procedure discussed in the previous section (Fig. 2C, right). Samples were stained with LIVE/DEAD indicators (calcein-AM/EtBr) after 72 hours of exposure to doxorubicin, along with CD45-allophycocyanin (APC) staining, to identify viable non-WBCs (cells expressing green fluorescence) for determination of viability (Fig. 5A). The viability statistics (table S4) of clinical samples after doxorubicin treatment were obtained on day 14 after 72 hours of exposure. Viability data were normalized to results obtained from samples in the last channel (lowest drug concentration) to obtain the dose-response curve, which was plotted using a four-parameter logistic equation. Corresponding IC50 values and graphs of each cluster-positive sample (Table 2 and fig. S13) were obtained in this study.
Fig. 5. Screening of doxorubicin in microwell assay using clinical human primary cancer cells cultured from clinical samples at serial time points (before and after treatment).
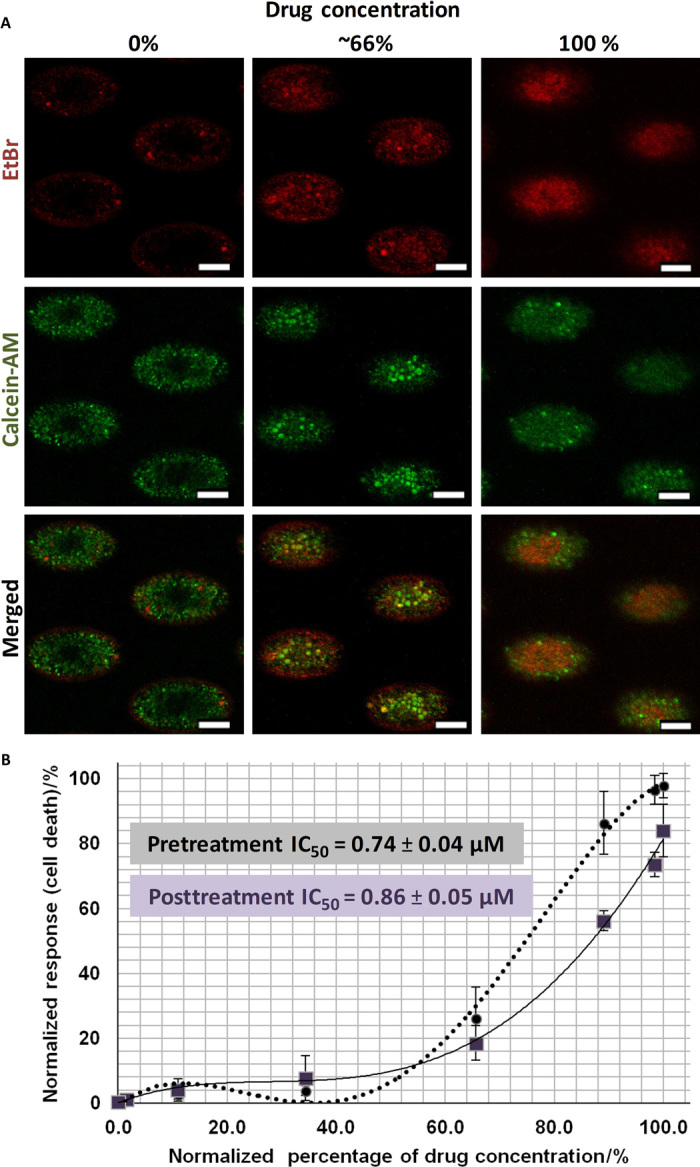
(A) Imaging of clusters generated from the pretreatment sample in situ after staining with LIVE (calcein-AM; green) and DEAD (EtBr; red) after 72 hours of exposure to doxorubicin. Clusters under high drug concentrations were mostly nonviable (red), whereas clusters under low drug concentrations were mostly viable (green). Scale bars, 100 μm. (B) Dose-response curve and corresponding IC50 value (0.94 ± 0.04 μM) of samples obtained from the same patient at different treatment time points. Monitoring IC50 values of a patient could reveal onset of drug tolerance or resistance. All error bars represent SD of triplicate cultures from different samples.
Table 2. IC50 values for the samples from breast cancer patients that yielded clusters.
Time point of blood withdrawal is provided.
| Sample | ID | Time point | IC50 value (μM) |
| 1 | CTB039 | Pretreatment | 0.85 |
| 2 | CES021 | Posttreatment | >1 |
| 3 | CES053 | Posttreatment | 0.34 |
| 4 | P2B28 | Posttreatment | >1 |
| 5 | P2B29 | Pretreatment | 0.94 |
| 6 | P2B29 | Posttreatment | 0.86 |
In accordance with our previous study, formation of CTC-containing clusters appeared to correlate negatively with patient response (14). Here, the percentage of microwells with clusters decreased with increasing concentration of doxorubicin (fig. S14A; average correlation coefficient, −0.71). In negative samples, the percentage of microwells with clusters remained constantly below 10%. As previously characterized, clusters also comprised a heterogeneous mixture of cells, including CK+/CD45− putative CTCs and residual blood cells such as macrophages (~33 ± 26%; fig. S15) (14). To evaluate a possible relationship between macrophage-like cell counts with drug concentration, the percentage of microwells with macrophage-like cells was evaluated (fig. S14B). The proportion of microwells with macrophage-like cells did not seem to correlate with drug concentration and were present both in samples with clusters and in those without. However, the number of macrophages per microwell varies across cultures obtained from serial samples of the same patient (fig. S14C). More specifically, macrophage-like cell counts per microwell were significantly lower in negative samples, as compared to positive samples with clusters [P = 0.02 for P2B29 (B) and P2B29 (Post S) cultures; P = 0.0003 for P2B29 (C1D8) and P2B29 (Post S) cultures; P = 0.02 for P2B28 (C2D1) and P2B28 (C3D1) cultures]. This suggests that macrophage-like cell counts could be another indicator for evaluating patient prognosis, an observation that has also been suggested in previous reports for tumor-associated macrophages (22).
Using the CTC cluster assay, we obtained the respective IC50 values for each of the three clinical samples that yielded clusters. Slight reduction of IC50 values in serial samples (before and after treatment with doxorubicin + sunitinib) was detected (P2B29; Fig. 5B), which could be due to increased drug sensitivity induced by a cycle of drug treatment with doxorubicin and sunitinib. One serial sample that yielded clusters only at a later time point gave a relatively higher IC50 value out of the 1 μM range (P2B28; Table 2).
These results, albeit preliminary, suggested that the assay could be used to monitor the drug response of a single patient over the treatment process. Three of these samples generated a positive culture in at least one time point and had available information on disease evaluation (table S5). Results suggest that cluster formation correlates with potential for worsened prognosis (no pathological complete response; 2 of 3, or 66.7%) (table S5). One sample with a negative culture at a posttreatment time point demonstrated partial response. Plans are in progress to continue monitoring the cohort of patients and to screen a larger sample cohort over several more months for correlation of in vitro drug testing parameters (cell viability and cluster formation) to actual patient survival and tumor response in vivo. This assay generates a two-pronged approach, which provides information on cluster formation potential as well as IC50 value variation during patient therapeutic treatment (Fig. 6). This integrated method allows efficient screening of anticancer drugs on primary breast cancer cells within 2 weeks, potentially allowing immediate intervention after early detection of drug resistance or tolerance.
Fig. 6. Proposed workflow of routine anticancer treatment evaluation with clinical human CTC cultures.

Cluster formation potential correlates inversely with overall patient survival, and increased IC50 values suggest possible onset of drug tolerance or resistance. The procedure can be completed within 2 weeks and will aid the clinician’s decision of maintaining or altering a patient’s drug regimen.
DISCUSSION
In vitro drug assays have led to the identification of novel drugs, some of which were further validated with in vivo models (23, 24). However, these drug regimens have not led to a breakthrough in cancer treatment because tumors acquire considerable and constantly evolving genomic heterogeneity, jeopardizing efforts in developing novel drug therapies (25–27). We presented a reliable label-free approach to efficiently monitor drug response of patients via serial sampling of patient-derived CTC cultures in an integrated microfluidic device. The CTC cluster assay was clinically validated with breast CTCs from patients with metastatic or early-stage cancer. Samples that led to clusters in ≥50% of microwells (n = 1000 microwells per channel; 1.25 ml of blood per channel) were selected for drug evaluation. Cluster formation reflects reduced overall patient survival; hence, positive samples with clusters were used for further analysis. The overall efficiency of positive cultures reported here is 34.2% (n = 73), but efficiency varies with the presence of therapeutic treatment (pretreatment samples: 59.3%, n = 27; posttreatment samples: 19.6%, n = 46). For the six positive samples that were subjected to drug screening, cluster formation also correlated negatively with drug concentration. Furthermore, culture from a sample obtained at a posttreatment time point (P2B29) also gave a relatively higher IC50 value as compared to the pretreatment sample, suggesting the onset of drug resistance. This integrated approach allows efficient screening of anticancer drugs on primary breast cancer cells within 2 weeks, potentially allowing immediate intervention after early detection of drug resistance or tolerance.
The ability to use heterogeneous CTCs (14, 28–32) from blood (33) for monitoring treatment efficacy is attractive because it does not require invasive procedures. Samples can thus be obtained rapidly on a regular basis, making it a promising strategy for early-stage cancer detection and monitoring treatment efficacy. CTCs can be previously enriched with surface antigen–coated (for example, epithelial cell adhesion molecule) substrates to trap specific subpopulations (12, 34, 35). However, the markers used for CTC detection are not applicable to all its subpopulations (32), partly due to the process of epithelial-to-mesenchymal transition (36). The epithelial (E) and mesenchymal (M) distributions in breast CTCs have been shown to vary across several parameters, with higher numbers of M cells correlating to the triple-negative subtype, poorer patient prognosis, or the presence of multicellular CTC aggregates (37). It is worth noting that at least for breast cancer, cells displaying intermediate E and M phenotypes are still highly prevalent in triple-negative cancer. However, some cancer cell lines derived from the triple-negative breast cancer subtype or others, which are characteristic of normal basal epithelial or adipose cells (32), have been reported to cease expression of any of the distinct markers used for CTC identification (38, 39). In view of these factors, others reach out to “label-free techniques” to selectively obtain CTCs by capitalizing on physical properties such as size (21, 40–42), density (43), or dielectrophoresis (44, 45). However, various groups reported the observation of small CTCs within the range of 10 to 20 μm, which coincides with the leukocyte size range (46). Other cells, such as normal-like breast cancer cells, do not bear resemblance to classic cancer phenotypes [for example, large cells with low nuclear-to-cytoplasmic (N/C) ratio] but still exhibit malignant properties. This heterogeneity severely limits the usefulness of existing enrichment methods. The exact frequency of CTCs in blood has been under debate, partly due to the varied detection sensitivity and recovery efficiency present with each enrichment assay. Previous experiments using mouse models with highly metastatic disease estimate the amount of shed carcinoma cells per day to be around 10,000 (47). A later study even suggested this number to be a million per day for each gram of tumor (48). Frequency of CTCs also varies with disease stage and presence of treatment or surgical procedures that may evoke heightened release of CTCs (47). Overall, current enrichment techniques are limited in sensitivity, depending on the method of selection and CTC counts presented by antigen-dependent methods (<100 CTCs per milliliter of blood) or techniques based on physical properties (hundreds to thousands of CTCs per milliliter of blood) (table S6). An efficient label-free method for establishing primary CTC cultures and subsequent drug evaluation is desired to achieve actual clinical utility.
Here, we describe a microfabricated assay that comprises custom-designed tapered microwells that allows for robust CTC expansion without pre-enrichment. Cultures are maintained as multilayered clusters, which may better reflect the in vivo state of a tumor because of heightened cell density in the presence of clusters or spheroids, which had been shown to reduce penetration of drugs (20). We demonstrated a similar observation with MCF-7 clusters, which yielded higher IC50 values as compared to studies on monolayer MCF-7 cultures. With clinical samples, the counts of macrophage-like cells within the clusters per microwell also correlated negatively with cluster formation, suggesting that macrophage-like cell counts could be another indicator for evaluating patient prognosis. This is a similar observation that has been suggested in previous reports for tumor-associated macrophages (22). Also, the formation of clusters may correlate with the potential of CTCs to form microemboli, which are associated with heightened metastatic potential and may participate directly in metastasis (49, 50).
Enrichment of CTCs by culture directly from blood prevents the loss of nonepithelial CTCs with antigen-based detection or smaller and more deformable CTCs using physical-based techniques. Although enrichment by culture will still select for the more proliferative and possibly more aggressive subtypes, these subpopulations are inevitably the ones that are crucial for our understanding of the metastatic cascade and are clinically relevant for the evaluation of patient prognosis. The underlying principle of the CTC cluster assay is the concentration of patient-derived blood cells as companion cells and hypoxia to mimic in vivo tumor conditions. Combination of these factors permits expansion of CTCs without pre-enrichment procedures or additional growth supplements, allowing the potential of maintaining most CTC subpopulations, including breast CTCs as well as CTCs from other cancer types.
The microwell-based device also delivers a robust design that enables comparative results to be drawn from samples obtained at different treatment time points. Molds for each device layer are produced with different strategies to accommodate the respective geometry, size, and tolerances of the features encoded. The integrated device showed robust performance with consistent generation of concentration gradients under various flow rates and input concentrations. Only a small volume of sample is required because of the integration of microfluidics. Deviation of shear and perfusion rates were negligible. The spread of concentrations in each channel remained stable over time, demonstrating the suitability of the device for long-term cultures. Manipulation of the device is straightforward, and fabrication is cost-effective, which promotes potential routine clinical usage for guiding therapeutics. Small sample volumes from clinical samples (10 ml) were required for each evaluation. This allows routine and rapid evaluation of drug treatment. The use of standardized microwells via microfabrication is important to allow even distribution of cells in each microwell, leading to the formation of clusters with consistent morphology. Tapered microwells are also preferred over conventional cylindrical microwells for cluster formation. This could be due to the inclination of the walls in tapered microwells, which allowed collected cells to settle in the center, forming a single cluster (instead of multiple small aggregates) to allow close interaction and “communication” between blood cells and CTCs. The development of this assay will promote noninvasive and inexpensive assessments of drug response, potentially guiding drug discovery development and therapeutic choices for personalized treatment.
Microfluidics is a valuable tool for biomedical applications as it provides flexibility in terms of design. Similarly, the assay described in this study can be further adapted to accommodate analysis of a wider range of drug gradients. Addition of inlets will enable two or more drugs to be evaluated as a combinatorial treatment method. This will also aid novel studies on the cumulative action of existing drugs when used in combination for therapy. Subsequent studies should focus on expanding the clinical cohort to include samples of later treatment time points and evaluate the IC50 values obtained with patient response. Availability of cultures from tumor biopsies will also enable comparison with primary cancer cell cultures after treatment to determine whether the variation in IC50 values could be due to increased drug tolerance or resistance. Analysis of IC50 values of samples from later treatment time points can be interpreted in terms of treatment efficacy. It will also be beneficial to increase the range of drug concentrations as two of the samples tested generated an IC50 value above the maximum range tested (1 μM). We believe that a routine, noninvasive, and inexpensive assessment of drug response on serial clinical samples can eventually pave the way for evaluation of novel drug combinations or implementation of personalized treatment.
MATERIALS AND METHODS
Fabrication of tapered microwells
Micropatterns were arranged in a densely packed array of ~1000 wells to maximize substrate use (fig. S16). For fabrication of the mold for the bottom layer comprising the array of microwells, we adapted from a process termed “diffuser back-side lithography” (51). The microwell array was fitted in the space given for each of the eight channels (2.3 mm × 56 mm, with a pitch of 4.7 mm), consisting of 250 μm × 150 μm elliptical wells with a tapered end and a depth of about 150 μm.
First, a soda-lime optical mask blank with the dense array of openings was created by laser direct writing (Heidelberg DWL 66fs tool, equipped with a Coherent I326C Ar laser) and subsequent Cr etching. After stripping the remaining resist, the mask was coated with a layer of SU-8 2100 resist (MicroChem Corp.) with a thickness exceeding the required depth for the wells. Here, we used a thickness of 300 μm for the resist layer, obtained by double spin-coating (60 s at 1500 rpm for both coatings, with a prebaking of 5 min at 65°C and 10 min at 95°C after the first coating and a final baking of 10 min at 65°C, followed by 3 hours at 95°C).
The resist was then exposed to UV light from the back of the mask and through an opal diffusing glass (Edmund Optics Inc.) placed in contact with the mask. According to the distribution of the scattered UV light and the exposure dose, different geometries can be realized, ranging from truncated conical polyhedrons to rounded domes. We used an MJB4 Suss MicroTec mask aligner as the exposure system, equipped with an Hg(Xe) arc lamp producing a power density of 12 mW/cm2 at 365 nm. With an experimentally optimized exposure time of 9 s, the final SU-8 dome-shaped structures with a height of 150 μm were obtained, with the elliptical 150 μm × 250 μm base defined by the openings on the mask.
After exposure, the sample was postbaked for 5 min at 65°C and 10 min at 95°C. Development using the SU-8 developer (MicroChem) was carried out in an ultrasound bath, which served to increase the development rate. Hard baking of the mold at 150°C for 5 min gives a mold ready for PDMS molding.
To preserve the lifetime of the master mold, we used a replica made of PDMS through double-casting as the working mold to produce the microwell array. PDMS (Sylgard 184, Dow Corning) in a prepolymer–to–curing agent ratio of 10:1 was poured on the master mold, degassed, and cured at 80°C for 1 hour; this first replica was with microwells at the place of the domes and was coated with an anti-sticking layer before further processing. Briefly, the PDMS surface was activated by oxygen plasma (60 W, 20 sccm of O2 at 5 mbar for 40 s) and immediately exposed to vapors of 1H,1H,2H,2H-perfluorooctyltrichlorosilane (Sigma-Aldrich) in a vacuum jar. Then, a second PDMS replica was produced by the same procedure as for the first, resulting in a PDMS working mold with the same features as in the master. This working mold was used for the production of the microwell array.
Fabrication of gradient generator and liquid barrier layer
The integrated device was made up of three PDMS layers (Fig. 2A) assembled with standard plasma treatment procedures. The mold for the gradient generator was fabricated via standard photolithographic procedures. Briefly, a (100) silicon wafer was coated with a 500-nm-thick SU-8 2000.5 resist (MicroChem Corp.), flood-exposed (30 mJ/cm2 at 365 nm), and postbaked. The exposed thin layer of SU-8 acts as an adhesion promoter for the following thick layer processing. SU-8 2050 was then spin-coated for 60 s at 1800 rpm, yielding a thickness of 100 μm after soft baking (5 min at 65°C plus 90 min at 95°C). The resist was UV-exposed (120 mJ/cm2 at 365 nm) through an optical mask to print the gradient generator pattern, which was finally revealed after postbaking (5 min at 65°C plus 10 min at 95°C) and development (10 min in the SU-8 developer; MicroChem Corp.). The mold was then ready for PDMS casting and curing, without any need for surface functionalization with the anti-sticking layer. For the production of the midlayer (barrier), which defines the eight channels, we used an aluminum mold fabricated by Whits Technologies, Singapore.
Cell culture of MCF-7 cancer cell line
MCF-7 (HTB-22TM, American Type Culture Collection), a human breast adenocarcinoma cell line, was first used to mimic the CTC cluster formation. Cell lines were maintained in supplemented high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 10% fetal bovine serum (FBS; Invitrogen) together with 1% penicillin-streptomycin (Invitrogen). Cultures were kept at 37°C in a humidified atmosphere containing 5% (v/v) CO2 until 80% confluence. Cells were cultured in sterile 25-cm2 flasks (BD Bioscience) and subcultivated two times a week with medium replaced every 48 hours. Subconfluent monolayers were dissociated using 0.01% trypsin and 5.3 mM EDTA solution (Lonza).
Processing of clinical samples
Blood samples were obtained from a total of 73 samples from 55 breast cancer patients (tables S2 and S3) enrolled in various anticancer therapeutic trials. This study was approved by our institutional review board and local ethics committee (DSRB reference 2012/00105, 2012/00979, 2010/00270, and 2010/00691). All patients gave their informed consent for inclusion in this study. Samples were collected from each patient either once or several times before and after treatment. Blood samples were stored in EDTA-coated vacutainer tubes (Becton-Dickinson). Blood samples were lysed within 10 hours after withdrawal using RBC lysis buffer (Life Technologies) for 3 to 5 min with mixing at room temperature and washed once with sterile PBS.
Cell seeding
For each clinical sample, cell suspension containing samples equivalent to 10 ml of whole blood was distributed evenly into each channel of the integrated assay. To minimize the variation on cell number between microwells and across channels, samples were diluted in 1.6 ml of fresh supplemented DMEM, which was evenly mixed before addition of 200 μl to each channel.
Estimation of cell loss
Cell loss was minimized by removing solution through the infusion/withdrawal modes of a syringe pump. To validate this hypothesis, the cells in specific microwells were marked and enumerated before and after three sets of infusion/withdrawal procedures at 100 μl/min using a syringe pump (NE-1000, New Era Pump Systems Inc.). Images were evaluated with ImageJ [National Institutes of Health (NIH)]. These results were compared with similar solution exchange performed manually by pipetting. Cell conservation rate was determined as “number of cells initially before flow”/“number of cells after flow” × 100%.
Calibration of fluorescent dye intensity
The fluorescence intensity increases with higher FITC dye concentration. Hence, fluorescence intensity of the dye was calibrated by preparing the dye (20 and 100 μM) at various concentrations (10 to 100%) and subsequently measuring their respective fluorescence intensity using a microplate reader. The values were fitted into an equation (linear for 20 μM and exponential for 100 μM) (fig. S1A).
Quantification of the dye concentration gradient generated at different flow rates
The device was plasma-treated and connected to a syringe via tubings. The setup was primed with ethanol manually using syringes. The primed device was checked to ensure that no air bubbles were trapped in the gradient generator. The device was then flushed with PBS once at 100 μl/min to remove the ethanol. Under dark conditions, 100% dye and deionized water were delivered using two syringe pumps at a range of flow rates (25, 50, 100, 150, and 200 μl/min). Triplicates of 60 μl of the sample at each respective outlet were collected every 2 to 5 min for seven time points into a 384-well plate. The data from the first time point were excluded to omit the dilution effects of existing ethanol in each well. Outliers due to the influence of instable flow or priming process were also excluded from the final concentration analysis. The device was washed thoroughly after the experiments and stored in a desiccator or oven to completely dry the channels.
COMSOL
We used the multiphysics modeling software COMSOL (COMSOL Inc.) to simulate the flow condition inside the gradient generator. A microplate reader (Tecan) and a 384-well plate (PerkinElmer) were used to determine the fluorescence intensity of dye solution.
Determination of culture morphology
Clear images of cultures (at least 24 bit) were obtained at high resolution and processed with image processing software (ImageJ). Compressed images may compromise the software’s ability to detect cell boundaries, leading to inaccurate outcomes. To evaluate cluster morphology, cultures should not have RBC contamination (such as when RBC lysis is incomplete), which will cover the cells and may compromise the ability of the software to distinguish a cluster from a noncluster phenotype (fig. S17). The plot profile across the maximum diameter of each cluster is obtained for each microwell to determine the gray values. Combined scatterplots of gray values, which reflect the density of cells across each microwell, were obtained. Values were normalized to the highest count for a particular microwell. Microwells with sparse groups of cells or debris demonstrated high gray values within the microwell region.
Maintenance of cultures on-chip
After cell seeding, the integrated assay was kept in a 150-mm dish filled with a thin layer of PBS and was incubated under humidified conditions. Devices were stored under hypoxia (1%) for clinical samples. MCF-7 cultures were maintained under normoxia (21%) to enable comparison of IC50 values with those previously reported. On every third day, 150 μl of medium from each channel was removed via either pump withdrawal action (at 100 μl/min) with two 10-ml BD Luer-Lok syringe (Becton, Dickinson and Company) or manual pipetting (for optimization studies), followed by introduction of 150 μl of fresh supplemented DMEM per channel. Supplemented DMEM was selected for culture because of previous optimization under various medium conditions (fig. S18). The closed system was then incubated at 37°C with 5% CO2 until drug treatment at day 3 and day 11 for MCF-7 cultures and clinical sample cultures, respectively.
Fluorescence in situ hybridization
Briefly, slides with cell spots were dehydrated in serial ethanol washes, followed by incubation with RNase/PBS (4 mg/ml; Sigma-Aldrich) for at least 45 min under optimal incubator conditions (37°C). Incubated slides were rinsed with 1× PBS/0.2% Tween 20 and treated with a mixture of 70% formamide and 2× saline sodium citrate solution (SSC; Sigma-Aldrich) for denaturation at 80°C. Denatured slides were further dehydrated and kept at 42°C until probe hybridization (CCND1, Empire Genomics; TOP2A, Abbott) and incubation overnight. Hybridized samples were washed thoroughly with a mixture of 50% formamide and 2×SSC, followed by 2×SSC, and were mounted with a coverslip using VECTASHIELD containing 4′,6-diaminido-2-phenylindole (DAPI) (Vector Laboratories).
Procedures for drug response profiling
Doxorubicin was used in this work to validate the assay. A stock solution was prepared in 100% dimethyl sulfoxide (DMSO) and was subsequently diluted in supplemented DMEM (1 μl to 1 ml of medium), resulting in ~0.1% of DMSO concentration (1 μM drug concentration), which has negligible effects on cells. Before addition of the drug, 150 μl of medium was withdrawn from each channel using a dual syringe pump connected to the common inlet. The device was primed briefly with fresh medium before introduction of medium containing the respective drugs at 100 μl/min. The loading of drugs should be carefully carried out to avoid influx of drugs upon reinsertion of tubings. The inward infusion rapidly generated a range of drug concentrations specific to each channel, which remained constant over time (as evaporation was limited by a humidified chamber).
After 72 hours of drug treatment, the viability of cells in each channel was determined by immunostaining. A cocktail comprising calcein-AM (green, 2 μM; Life Technologies), EtBr (red), and CD45-allophycocyanin (red) (1:100; Miltenyi Biotec Asia Pacific) was incubated with the cells in situ for 45 min. Samples were flushed gently with PBS and imaged with an Olympus inverted confocal microscope (Emission filters ET460/50m and ET535/50m; Olympus). Cell counts were obtained using ImageJ (NIH). Only microwells with clusters were considered for analysis.
To establish the IC50 values, Z stack images of 25 microwells from each channel were obtained with a confocal microscope. Images from each stack of 15 μm were compiled to obtain a merged image of maximum intensity. These images were individually preprocessed by cropping and thresholding to identify signals of 8 to 150 μm. Merged images were compared to rule out repeated signal counts. For consistency, the microwells considered for evaluation were obtained at the same distance from the assay inlets. For clinical samples, only CD45− cells (cells with green fluorescence) were considered for establishing the viability rate. Resultant viability percentages were normalized to those obtained from samples in the last channel (lowest drug concentration). A four-parameter logistic equation was used using Microsoft Excel for curve fitting analysis before determination of IC50 values. The IC50 value was obtained as the concentration value at which the curve passed through the 50% normalized response value corresponding to percentage of cell death (y axis).
Trypan blue assay for cell lines
Clusters were dissociated with pipetting following incubation for a maximum of 3 min at 37°C with 0.01% trypsin and 5.3 mM EDTA (Lonza) solution in PBS. Trypan blue–positive cells were then enumerated using an automated cell counter (TC20, Bio-Rad).
Statistical analysis
All error bars represented SD of triplicate cultures from different samples. Groups were compared using the Student’s t test to evaluate associations between independent variables, and the P values were obtained. Adjusted multivariate analyses for continuous independent variables (to other variables) required larger sample sizes and were not used in this study. Further Cox regression (investigation of multiple variables) was also not carried out because of the small sample size.
Supplementary Material
Acknowledgments
We thank C. Zhang and C. X. Wong from the Mechanobiology Institute (MBI) at the National University of Singapore for their effort in producing the video and schematics. We are grateful to the personnel, including T. Saunders, for the technical support and facility usage at the BioSystems and Micromechanics (BioSyM) Laboratory of Singapore–MIT Alliance for Research and Technology (SMART), MBI and Institute of Molecular and Cell Biology (IMCB) at A*STAR (Agency for Science Technology and Research). One or more authors have a pending patent related to this work. This work was supported by MBI and the National Research Foundation (NRF), Prime Minister’s Office, Singapore, under CREATE, SMART BioSyM IRG. Author contributions: B.L.K., G.G., C.T.L., and T.J. conceived and designed the experiments. B.L.K., G.G., T.J., and Y.B.L. performed the experiments. B.L.K., T.J., and Y.B.L. analyzed the data. C.T.L., G.G., S.C.L., J.P.T., and J.H. contributed reagents, materials, and/or analysis tools. B.L.K., G.G., T.J., Y.B.L., S.C.L., J.P.T., J.H., and C.T.L. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary materials for this article are available at http://advances.sciencemag.org/cgi/content/full/2/7/e1600274/DC1
fig. S1. Microfabricated molds for the assay.
fig. S2. Schematics of the gradient generator design.
fig. S3. Flow rate of the device and the dynamics within the channel.
fig. S4. Simulated flow conditions of the assay.
fig. S5. Consistency of gradient concentration in channels over time.
fig. S6. Estimation of cell counts after influence of flow.
fig. S7. Validation of integrated assay for proliferation with MCF-7 cell lines.
fig. S8. Optimization of cancer cell line culture parameters.
fig. S9. Phase-contrast images of 2-week cultures under different culture conditions.
fig. S10. Proportion of CTCs before culture affected the potential of cluster formation.
fig. S11. Fluorescence in situ hybridization of cultured cells.
fig. S12. Epithelial CTC counts before and after culture.
fig. S13. Screening of doxorubicin in microwell assay using clinical human primary cancer cells cultured from blood samples.
fig. S14. Characterization of cultures.
fig. S15. Representative images of residual WBC populations.
fig. S16. Scanning electron microscopy images demonstrating the densely packed array of microwells to maximize surface for capturing CTCs for culture.
fig. S17. Representative image of a microwell containing cells contaminated by RBCs due to inadequate RBC lysis.
fig. S18. Optimization of culture medium conditions.
table S1. Concentration of a single reagent at each serpentine.
table S2. Samples for preliminary validation of procedure.
table S3. Samples evaluated for drug screening.
table S4. Average percentage of viable CD45− cells in clinical samples.
table S5. Disease evaluation of three sets of serial samples with at least one positive culture generated.
table S6. CTC counts per milliliter as reported by notable CTC enrichment methods (non–culture-based).
movie S1. Illustration of seeding, growth, and drug flow.
REFERENCES AND NOTES
- 1.Kim C., Paik S., Gene-expression-based prognostic assays for breast cancer. Nat. Rev. Clin. Oncol. 7, 340–347 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Jain R. K., Duda D. G., Willett C. G., Sahani D. V., Zhu A. X., Loeffler J. S., Batchelor T. T.,Sorensen A. G., Biomarkers of response and resistance to antiangiogenic therapy. Nat. Rev. Clin. Oncol. 6, 327–338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambhir S. S., Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Gambarin-Gelwan M., Wolf D. C., Shapiro R., Schwartz M. E., Min A. D., Sensitivity of commonly available screening tests in detecting hepatocellular carcinoma in cirrhotic patients undergoing liver transplantation. Tests for HCG in cirrhoyics having OLT. Am. J. Gastroenterol. 95, 1535–1538 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Sundberg S. A., High-throughput and ultra-high-throughput screening: Solution- and cell-based approaches. Curr. Opin. Biotechnol. 11, 47–53 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Du G. S., Pan J.-Z., Zhao S.-P., Zhu Y., den Toonder J. M. J., Fang Q., Cell-based drug combination screening with a microfluidic droplet array system. Anal. Chem. 85, 6740–6747 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Zhang L., Ridgway L. D., Wetzel M. D., Ngo J., Yin W., Kumar D., Goodman J. C., Groves M. D., Marchetti D., The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 5, 180ra148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu M., Bardia A., Aceto N., Bersani F., Madden M. W., Donaldson M. C., Desai R., Zhu H., Comaills V., Zheng Z., Wittner B. S., Stojanov P., Brachtel E., Sgroi D., Kapur R., Shioda T., Ting D. T., Ramaswamy S., Getz G., Iafrate A. J., Benes C., Toner M., Maheswaran S., Haber D. A., Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao D., Vela I., Sboner A., Iaquinta P. J., Karthaus W. R., Gopalan A., Dowling C., Wanjala J. N., Undvall E. A., Arora V. K., Wongvipat J., Kossai M., Ramazanoglu S., Barboza L. P., Di W., Cao Z., Zhang Q. F., Sirota I., Ran L., MacDonald T. Y., Beltran H., Mosquera J.-M., Touijer K. A., Scardino P. T., Laudone V. P., Curtis K. R., Rathkopf D. E., Morris M. J., Danila D. C., Slovin S. F., Solomon S. B., Eastham J. A., Chi P., Carver B., Rubin M. A., Scher H. I., Clevers H., Sawyers C. L., Chen Y., Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maheswaran S., Haber D. A., Ex vivo culture of CTCs: An emerging resource to guide cancer therapy. Cancer Res. 75, 2411–2415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pantel K., Alix-Panabierès C., The clinical significance of circulating tumor cells. Nat. Clin. Pract. Oncol. 4, 62–63 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Nagrath S., Sequist L. V., Maheswaran S., Bell D. W., Irimia D., Ulkus L., Smith M. R., Kwak E. L., Digumarthy S., Muzikansky A., Ryan P., Balis U. J., Tompkins R. G., Haber D. A., Toner M., Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pantel K., Riethdorf S., Pathology: Are circulating tumor cells predictive of overall survival? Nat. Rev. Clin. Oncol. 6, 190–191 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Khoo B. L., Lee S. C., Kumar P., Tan T. Z., Warkiani M. E., Ow S. G. W., Nandi S., Lim C. T., Thiery J. P., Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget 6, 15578–15593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhadriraju K., Chen C. S., Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov. Today 7, 612–620 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Nguyen N.-T., Shaegh S. A. M., Kashaninejad N., Phan D.-T., Design, fabrication and characterization of drug delivery systems based on lab-on-a-chip technology. Adv. Drug Deliv. Rev. 65, 1403–1419 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Berthier E., Warrick J., Yu H., Beebe D. J., Managing evaporation for more robust microscale assays. Part 2. Characterization of convection and diffusion for cell biology. Lab Chip 8, 860–864 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alix-Panabières C., Pantel K., Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 59, 110–118 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Fang X. J., Jiang H., Zhu Y. Q., Zhang L. Y., Fan Q. H., Tian Y., Doxorubicin induces drug resistance and expression of the novel CD44st via NF-κB in human breast cancer MCF-7 cells. Oncol. Rep. 31, 2735–2742 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Jaganathan H., Gage J., Leonard F., Srinivasan S., Souza G. R., Dave B., Godin B., Three-dimensional in vitro co-culture model of breast tumor using magnetic levitation. Sci. Rep. 4, 6468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khoo B. L., Warkiani M. E., Tan D. S.-W., Bhagat A. A. S., Irwin D., Lau D. P., Lim A. S. T., Lim K. H., Krisna S. S., Lim W.-T., Yap Y. S., Lee S. C., Soo R. A., Han J., Lim C. T., Clinical validation of an ultra high-throughput spiral microfluidics for the detection and enrichment of viable circulating tumor cells. PLOS One 9, e99409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollard J. W., Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Guzman M. L., Rossi R. M., Neelakantan S., Li X., Corbett C. A., Hassane D. C., Becker M. W., Bennett J. M., Sullivan E., Lachowicz J. L., Vaughan A., Sweeney C. J., Matthews W., Carroll M., Liesveld J. L., Crooks P. A., Jordan C. T., An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 110, 4427–4435 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guzman M. L., Neering S. J., Upchurch D., Grimes B., Howard D. S., Rizzieri D. A., Luger S. M., Jordan C. T., Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98, 2301–2307 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Vogelstein B., Kinzler K. W., Cancer genes and the pathways they control. Nat. Med. 10, 789–799 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Fearon E. R., Vogelstein B., A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Hou H. W., Warkiani M. E., Khoo B. L., Li Z. R., Soo R. A., Tan D. S.-W., Lim W.-T., Han J., Bhagat A. A. S., Lim C. T., Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 3, 1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giordano A., Gao H., Anfossi S., Cohen E., Mego M., Lee B.-N., Tin S., De Laurentiis M., Parker C. A., Alvarez R. H., Valero V., Ueno N. T., De Placido S., Mani S. A., Esteva F. J., Cristofanilli M., Reuben J. M., Epithelial–mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol. Cancer Ther. 11, 2526–2534 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alix-Panabières C., Vendrell J.-P., Pellé O., Rebillard X., Riethdorf S., Müller V., Fabbro M., Pantel K., Detection and characterization of putative metastatic precursor cells in cancer patients. Clin. Chem. 53, 537–539 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Kasimir-Bauer S., Hoffmann O., Wallwiener D., Kimmig R., Fehm T., Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 14, R15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sieuwerts A. M., Kraan J., Bolt J., van der Spoel P., Elstrodt F., Schutte M., Martens J. W. M., Gratama J.-W., Sleijfer S., Foekens J. A., Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst. 101, 61–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A., Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Riethdorf S., Fritsche H., Müller V., Rau T., Schindlbeck C., Rack B., Janni W., Coith C., Beck K., Jänicke F., Jackson S., Gornet T., Cristofanilli M., Pantel K., Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 13, 920–928 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Stott S. L., Hsu C.-H., Tsukrov D. I., Yu M., Miyamoto D. T., Waltman B. A., Rothenberg S. M., Shah A. M., Smas M. E., Korir G. K., Floyd F. P. Jr, Gilman A. J., Lord J. B., Winokur D., Springer S., Irimia D., Nagrath S., Sequist L. V., Lee R. J., Isselbacher K. J., Maheswaran S., Haber D. A., Toner M., Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. U.S.A. 107, 18392–18397 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiery J. P., Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Yu M., Bardia A., Wittner B. S., Stott S. L., Smas M. E., Ting D. T., Isakoff S. J., Ciciliano J. C., Wells M. N., Shah A. M., Concannon K. F., Donaldson M. C., Sequist L. V., Brachtel E., Sgroi D., Baselga J., Ramaswamy S., Toner M., Haber D. A., Maheswaran S., Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallwiener M., Hartkopf A. D., Baccelli I., Riethdorf S., Schott S., Pantel K., Marmé F., Sohn C., Trumpp A., Rack B., Aktas B., Solomayer E.-F., Müller V., Janni W., Schneeweiss A., Fehm T. N., The prognostic impact of circulating tumor cells in subtypes of metastatic breast cancer. Breast Cancer Res. Treat. 137, 503–510 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Bidard F.-C., Peeters D. J., Fehm T., Nolé F., Gisbert-Criado R., Mavroudis D., Grisanti S., Generali D., Garcia-Saenz J. A., Stebbing J., Caldas C., Gazzaniga P., Manso L., Zamarchi R., de Lascoiti A. F., De Mattos-Arruda L., Ignatiadis M., Lebofsky R., van Laere S. J., Meier-Stiegen F., Sandri M.-T., Vidal-Martinez J., Politaki E., Consoli F., Bottini A., Diaz-Rubio E., Krell J., Dawson S.-J., Raimondi C., Rutten A., Janni W., Munzone E., Carañana V., Agelaki S., Almici C., Dirix L., Solomayer E.-F., Zorzino L., Johannes H., Reis-Filho J. S., Pantel K., Pierga J.-Y., Michiels S., Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 15, 406–414 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Di Carlo D., Irimia D., Tompkins R. G., Toner M., Continuous inertial focusing, ordering, and separation of particles in microchannels. Proc. Natl. Acad. Sci. U.S.A. 104, 18892–18897 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuntaegowdanahalli S. S., Bhagat A. A. S., Kumar G., Papautsky I., Inertial microfluidics for continuous particle separation in spiral microchannels. Lab Chip 9, 2973–2980 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Ozkumur E., Shah A. M., Ciciliano J. C., Emmink B. L., Miyamoto D. T., Brachtel E., Yu M., Chen P.-i, Morgan B., Trautwein J., Kimura A., Sengupta S., Stott S. L., Murat Karabacak N., Barber T. A., Walsh J. R., Smith K., Spuhler P. S., Sullivan J. P., Lee R. J., Ting D. T., Luo X., Shaw A. T., Bardia A., Sequist L. V., Louis D. N., Maheswaran S., Kapur R., Haber D. A., Toner M., Inertial focusing for tumor antigen–dependent and –independent sorting of rare circulating tumor cells. Sci. Transl. Med. 5, 179ra147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gertler R., Rosenberg R., Fuehrer K., Dahm M., Nekarda H., Siewert J. R., Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Recent Results Cancer Res. 162, 149–155 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Gascoyne P. R. C., Noshari J., Anderson T. J., Becker F. F., Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis 30, 1388–1398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta V., Jafferji I., Garza M., Melnikova V. O., Hasegawa D. K., Pethig R., Davis D. W., ApoStreamTM, a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics 6, 024133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alunni-Fabbroni M., Sandri M. T., Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods 50, 289–297 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Fidler I. J., Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-2’-deoxyuridine. J. Natl. Cancer Inst. 45, 773–782 (1970). [PubMed] [Google Scholar]
- 48.Butler T. P., Gullino P. M., Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 35, 512–516 (1975). [PubMed] [Google Scholar]
- 49.Alix-Panabières C., Pantel K., Challenges in circulating tumour cell research. Nat. Rev. Cancer 14, 623–631 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Aceto N., Toner M., Maheswaran S., Haber D. A., En route to metastasis: Circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 1, 44–52 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Lee J.-H., Lee H.-S., Lee B.-K., Choi W.-S., Choi H.-Y., Yoon J.-B., Simple liquid crystal display backlight unit comprising only a single-sheet micropatterned polydimethylsiloxane (PDMS) light-guide plate. Opt. Lett. 32, 2665–2667 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Cayrefourcq L., Mazard T., Joosse S., Solassol J., Ramos J., Assenat E., Schumacher U., Costes V., Maudelonde T., Pantel K., Alix-Panabières C., Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 75, 892–901 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials for this article are available at http://advances.sciencemag.org/cgi/content/full/2/7/e1600274/DC1
fig. S1. Microfabricated molds for the assay.
fig. S2. Schematics of the gradient generator design.
fig. S3. Flow rate of the device and the dynamics within the channel.
fig. S4. Simulated flow conditions of the assay.
fig. S5. Consistency of gradient concentration in channels over time.
fig. S6. Estimation of cell counts after influence of flow.
fig. S7. Validation of integrated assay for proliferation with MCF-7 cell lines.
fig. S8. Optimization of cancer cell line culture parameters.
fig. S9. Phase-contrast images of 2-week cultures under different culture conditions.
fig. S10. Proportion of CTCs before culture affected the potential of cluster formation.
fig. S11. Fluorescence in situ hybridization of cultured cells.
fig. S12. Epithelial CTC counts before and after culture.
fig. S13. Screening of doxorubicin in microwell assay using clinical human primary cancer cells cultured from blood samples.
fig. S14. Characterization of cultures.
fig. S15. Representative images of residual WBC populations.
fig. S16. Scanning electron microscopy images demonstrating the densely packed array of microwells to maximize surface for capturing CTCs for culture.
fig. S17. Representative image of a microwell containing cells contaminated by RBCs due to inadequate RBC lysis.
fig. S18. Optimization of culture medium conditions.
table S1. Concentration of a single reagent at each serpentine.
table S2. Samples for preliminary validation of procedure.
table S3. Samples evaluated for drug screening.
table S4. Average percentage of viable CD45− cells in clinical samples.
table S5. Disease evaluation of three sets of serial samples with at least one positive culture generated.
table S6. CTC counts per milliliter as reported by notable CTC enrichment methods (non–culture-based).
movie S1. Illustration of seeding, growth, and drug flow.