

Conspectus
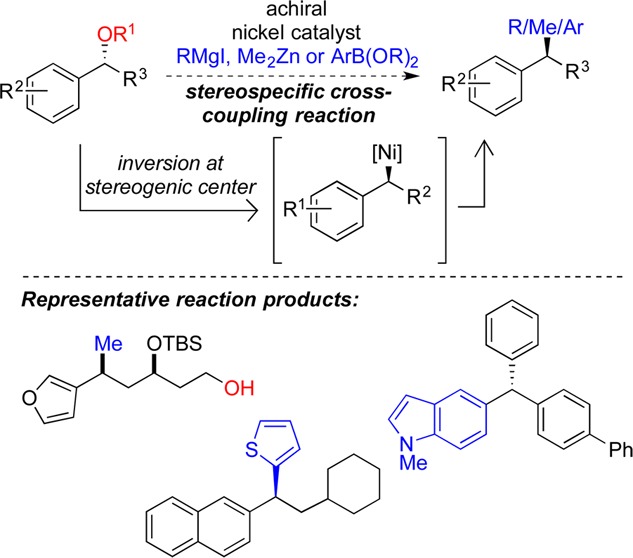
This Account presents the development of a suite of stereospecific alkyl–alkyl cross-coupling reactions employing nickel catalysts. Our reactions complement related nickel-catalyzed stereoconvergent cross-coupling reactions from a stereochemical and mechanistic perspective. Most reactions of alkyl electrophiles with low-valent nickel complexes proceed through alkyl radicals and thus are stereoablative; the correct enantioselective catalyst can favor the formation of one enantiomer. Our reactions, in contrast, are stereospecific. Enantioenriched ethers and esters are cleanly converted to cross-coupled products with high stereochemical fidelity. While mechanistic details are still to be refined, our results are consistent with a polar, two-electron oxidative addition that avoids the formation of radical intermediates. This reactivity is unusual for a first-row transition metal.
The cross-coupling reactions engage a range of benzylic ethers and esters, including methyl ethers, tetrahydropyrans, tetrahydrofurans, esters, and lactones. Coordination of the arene substituent to the nickel catalyst accelerates the reactions. Arenes with low aromatic stabilization energies, such as naphthalene, benzothiophene, and furan, serve as the best ligands and provide the highest reactivity. Traceless directing groups that accelerate reactions of sluggish substrates are described, providing partial compensation for arene coordination.
Kumada, Negishi, and Suzuki reactions provide incorporation of a broad range of transmetalating agents. In Kumada coupling reactions, a full complement of Grigard reagents, including methyl, n-alkyl, and aryl Grignard reagents, are employed. In reactions employing methylmagnesium iodide, ligation of the nickel catalyst by rac-BINAP or DPEphos provides the highest yield and stereospecificity. For all other Grignard reagents, Ni(dppe)Cl2 has emerged as the best catalyst. Negishi cross-coupling reactions employing dimethylzinc are reported as a strategy to increase the functional group tolerance of the reaction. We also describe Suzuki reactions using arylboronic esters. These reactions provided the first example in the series of a switch in stereochemical outcome. The reactions maintain stereospecificity, but reactions employing different achiral ligands provide opposite enantiomers of the product. Use of an N-heterocyclic carbene ligand, SIMes, provides inversion, consistent with our prior work in Kumada and Negishi coupling reactions. Use of the electron-rich phosphine PCy3, however, provides retention with stereospecificity, signaling a change in the mechanistic details.
Potential applications of the reported cross-coupling reactions include the synthesis of medicinal agents containing the 2-arylalkane and 1,1-diarylalkane moieties, which are pharmacophores in medicinal chemistry. These moieties are found in compounds with activity against a broad range of indications, including cancer, heart disease, diabetes, osteoporosis, smallpox, tuberculosis, and insomnia. We highlight representative examples of bioactive compounds that we have prepared with high enantioselectivity employing our methods, as well as the discovery of a new anti-cancer agent.
I. Introduction
Cross-coupling reactions have become an indispensable component of synthesis, particularly when bonds between sp2-hybridized carbons must be forged.1 Cross-coupling reactions that link sp3 centers using alkyl electrophiles and alkylorganometallic reagents are poised to impact synthesis.2 Strategic applications of cross-coupling reactions of primary alkyl halides and alkylmetal species en route to natural products have been reported. For example, in the synthesis of pyranicin, Griggs and Phillips3 employed an alkyl–alkyl cross-coupling reaction, using a catalyst developed by Fu, to stitch together the backbone of the molecule (Scheme 1). These disconnections have the advantage of being traceless, i.e., there are no telltale functional groups that dictate the placement of the disconnection, allowing synthetic chemists to break molecules at nonobvious positions.
Scheme 1. Alkyl–Alkyl Cross-Coupling Reaction en Route to Pyranicin.

Realizing the widespread application of alkyl cross-coupling reactions in the synthesis of natural products and medicinal agents will require further advances (Scheme 2).4 Incorporation of a broad range of secondary substrates with high stereoselectivity will be necessary. As with most synthetic transformations, alkyl cross-couplings will likely see the largest impact if both stereoconvergent (catalyst-controlled) and stereospecific (substrate-controlled) reactions are developed.5,6 The two strategies present different advantages and are complementary. For example, stereoconvergent reactions employing asymmetric catalysts often provide a strategic method for the installation of the first stereogenic center in a synthesis and exquisite control over the introduction of a remote stereogenic center.7 In contrast, stereospecific reactions proceed cleanly with inversion or retention, conserving stereochemical information present in the starting material without the need to identify a chiral catalyst. Such reactions are often valued in late-stage synthesis, since they provide predictable methods for elaboration of stereochemically complex intermediates.
Scheme 2. Strategies for Control of Stereochemistry in Alkyl–Alkyl Cross-Coupling Reactions.

Exciting advances in both stereoconvergent and stereospecific cross-coupling reactions have been reported. A rich literature describes the use of secondary alkylmetal reagents in stereospecific and stereoconvergent approaches.8,9 Stereoconvergent coupling reactions of secondary alkyl halides employing enantioselective nickel catalysts have been pioneered by the Fu group.10 This work has provided important lessons about the ability of weak directing groups to orchestrate enantioselective reactions11 and has been a springboard for creative ideas in photoredox catalysis.12 Our group has endeavored to develop the complementary approach, stereospecific cross-coupling reactions of secondary electrophiles.
II. Key Factors in Reaction Design
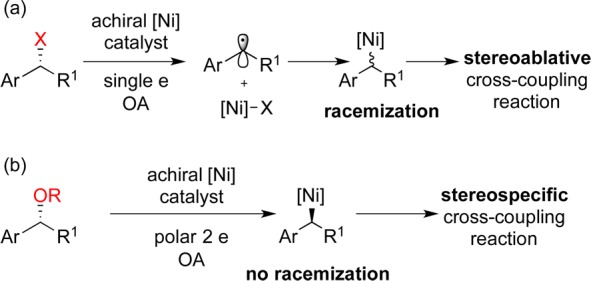
We began our investigation by focusing our efforts on the development of a nickel-catalyzed reaction. Complexes of the first-row transition metals nickel, iron, and cobalt are considered highly reactive toward oxidative addition and slow to undergo β-hydride elimination, which is ideal for alkyl–alkyl cross-coupling reactions.13−15 At the outset, we envisioned that the greatest challenge in the development of a stereospecific cross-coupling reaction would be to identify alkyl electrophiles that participate in polar, two-electron oxidative addition reactions with low-valent nickel complexes. This issue is one point of contrast between the precious metal palladium and the base metal nickel. Organopalladium complexes typically undergo stereospecific two-electron oxidative addition reactions.16,17 The corresponding nickel complexes frequently possess a greater density of states and can access one-electron pathways. Oxidative addition reactions of organonickel complexes with alkyl halides likely proceed through radical intermediates, and this reactivity has been critical for the development of catalyst-controlled, stereoablative reactions (Scheme 3a).18−20 To achieve a stereospecific reaction, we must suppress the inherent radical reactivity of the catalyst and favor a two-electron, polar oxidative addition (Scheme 3b), since 2° alkyl radicals undergo racemization with ΔG⧧ < 0.5 kcal/mol.21 We were optimistic that we could identify alkyl electrophiles that participate in robust two-electron reactions with nickel catalysts and that this step would initiate a stereospecific cross-coupling reaction.
Scheme 3. Influence of the Mechanism of Oxidative Addition on the Stereochemical Outcome.
Our choice of alkyl electrophile was guided by nickel-catalyzed allylic substitution reactions22 as well as contemporary developments in nickel-catalyzed coupling reactions of phenol derivatives.23 Our simple working hypothesis was that “hard” electrophiles would favor polar reactions and be less prone to radical reactions.24 In support of this hypothesis, since the 1980s there have been numerous reports of nickel-catalyzed reactions of allylic ethers and esters that are stereospecific and proceed with inversion (Scheme 4a).25 Furthermore, in 2008 Shi and co-workers reported nickel-catalyzed cross-coupling reactions of primary benzylic methyl ethers at elevated temperatures (Scheme 4b).26 They also reported a modest yield utilizing a secondary ether, but no stereochemical information was reported. On the basis of this rationale, we began our investigations with benzylic ethers (Scheme 4c).
Scheme 4. Nickel-Catalyzed Reactions of Allylic and Benzylic Ethers.

The choice of ethers and esters as starting materials provided a significant strategic and practical advantage in the development of stereospecific coupling reactions. By definition, any enantiospecific reaction will require synthesis of the starting material with high enantioselectivity. The requisite starting materials for our project were secondary alcohols, a functional group for which there are many outstanding enantioselective synthetic methods (Scheme 5).27 Therefore, we were confident at the outset that developing a method based on this functional group would stand on a solid foundation and dovetail nicely with modern synthetic planning.
Scheme 5. Enantioenriched Secondary Alcohols as Key Synthetic Intermediates.

III. Stereospecific Kumada Coupling Reactions
a. The Test Case: Benzylic Ethers with Methylmagnesium Iodide
Our investigation began with methylmagneisum iodide for several strategic reasons. The first was that this nucleophile does not include β-hydrogens, reducing the number of possible side reactions. The second was to provide synthetic methods for incorporation of benzylic methyl substituents, which can improve the bioavailability and activity of drug substances.28 Representative examples of medicinal agents bearing this moiety are shown in Figure 1.29
Figure 1.

Representative medicinal agents bearing benzylic methyl substituents.
As benzylic ethers are not highly activated electrophilic partners, we chose first to pair them with Grignard reagents, highly active nucleophiles, to develop Kumada-type cross-coupling reactions. Utilizing the secondary benzylic methyl ether reported by Shi and co-workers,26 we sought to find optimal conditions that would suppress the undesired competing β-hydride elimination reaction while furnishing the desired cross-coupling product with faithful transfer of stereochemical information from substrate to product. After a survey of reaction conditions, we found that using Ni(cod)2 with the bidentate phosphine ligand rac-BINAP provided 14 in 72% yield with 98% es at room temperature (Scheme 6).30 Enantiospecificity (es) is calculated to provide a metric of the stereochemical fidelity of the transformation, allowing direct comparison of reactions performed with starting materials of different ee.31
Scheme 6. Proof of Concept: Stereospecific Kumada Coupling Reactions of Benzylic Ethers.

With the optimized conditions in hand, we explored the scope of the transformation. A series of enantioenriched benzylic ethers were examined. Ethers activated by extended aromatic moieties underwent the desired stereospecific cross-coupling reaction in good yields with excellent transfer of stereochemical information (Scheme 6). This observation is consistent with coordination of the arene to the nickel catalyst, facilitating oxidative addition. We would need to address this limitation in subsequent generations of reaction design (vide infra). Higher yields were typically obtained using DPEphos in place of rac-BINAP for heterocycles (17 and 18).
To further challenge the Kumada coupling, we examined the influence of nearby stereogenic centers on the stereochemical course of the reaction. We chose to examine 2-aryltetrahydrofurans and tetrahydropyrans, as there are excellent established methods for their synthesis.32 The use of these scaffolds would take advantage of cyclic stereocontrol to easily set the relative configuration in the starting material, which would subsequently be translated to single diastereomers of acyclic products. Additionally, upon cross-coupling we would unveil an alcohol, which would allow for further manipulation and derivatization.
Both tetrahydropyrans and tetrahydrofurans react smoothly, unraveling to provide alcohols where the coupling proceeds with clean inversion at the benzylic stereogenic center (Table 1).33 In addition to benzofuran and benzothiophene, which successfully undergo the reaction with the use of DPEphos, we also found that simple 3-furyltetrahydropyran 23 affords 24 in high yield and dr.
Table 1. Scope of Stereospecific Ring Opening of Tetrahydropyrans and Tetrahydrofurans.


Importantly, comparison of reactions of diastereomeric starting materials demonstrated that additional stereogenic centers do not influence the stereochemical fidelity of the reaction. For example, cis-19 cleanly afforded only syn-20, while trans-19 provided anti-20. We found that the highest yields were obtained when the second stereogenic center was distal to the reactive center, with alkyl, aryl, and protected alcohol substitutents being well-tolerated. While the yields were generally diminished when the second substitutent was closer to the benzylic center, the stereochemical fidelity remained excellent (e.g., 26).
To further interrogate the potential for a match/mismatch effect when chiral ligands were employed, we subjected both diastereomers of tetrahydrofuran 29 to reactions employing (R)- and (S)-BINAP (Scheme 7). In all cases, we observed that the reaction proceeded in high yield with inversion at the benzylic center. These results indicate that the reaction is not influenced by the chirality of the catalyst and is robustly stereospecific.
Scheme 7. Lack of a Match/Mismatch Effect.

b. Expanding the Grignard Reagent Scope: Addressing β-Hydride Elimination
Transmetalating agents that contain β-hydrogen atoms pose a particular challenge in cross-coupling reactions, since they are prone to β-hydride elimination upon transmetalation to form alkylnickel intermediates. Indeed, such Grignard reagents performed poorly under our reported cross-coupling reaction conditions and were plagued by hydrogenolysis or β-hydride elimination (Scheme 8a). We reasoned that tuning of the steric and electronic properties of the ligand could provide a catalyst with a lower propensity for β-hydride elimination. Upon evaluation of a series of ligands, we found that dppe favored the desired coupling and suppressed the competing side reactions. Optimized conditions utilized commercially available, air-stable Ni(dppe)Cl2 with catalyst loadings as low as 2 mol % (Scheme 8b).34
Scheme 8. Ligand Tuning To Suppress β-Hydride Elimination.

We determined that for reactions employing n-alkyl or aryl Grignard reagents, Ni(dppe)Cl2 is the catalyst of choice (Scheme 9). Grignard reagents bearing trisubstituted olefin or trifluoromethyl groups are well-tolerated, allowing for installation of useful functional groups (e.g., 36 and 37). Branched alkyl Grignard reagents gave low yields of the desired cross-coupling products (e.g., 38) as a result of competitive β-hydride elimination, but the transfer of stereochemical information remained high. Both electron-rich and electron-deficient aryl Grignard reagents are tolerated in good yield and es (e.g., 39 and 40).
Scheme 9. Scope of Cross-Coupling Reactions with Alkyl and Aryl Grignard Reagents.
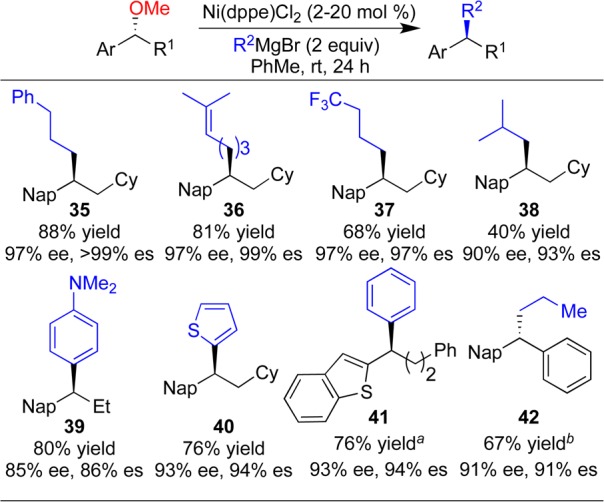
Ni(dppe)Cl2 was added in two aliquots of 10 mol %.
The reaction was run at 5 °C for 48 h.
These reaction conditions also translated smoothly to reactions of cyclic ethers with n-alkyl and aryl Grignard reagents (Table 2).33 Thus, Ni(dppe)Cl2 is the most general catalyst we have identified to date. The sole exception is in reactions employing MeMgX, where the BINAP- or DPEphos-ligated catalysts typically provide the highest yields.35
Table 2. Ring-Opening Reactions of Tetrahydrofurans with a Range of Grignard Reagents.


Over the course of these experiments, we noted an inverse correlation between the catalyst loading and reaction enantiospecificity.34 We hypothesized that, in analogy to palladium-catalyzed allylic and benzylic substitution reactions, at high catalyst loadings the key π-benzylnickel intermediate 48 racemizes by nucleophilic attack of a second nickel species (Scheme 10).17a,36 This information proved helpful in identifying modified reaction conditions to suit recalcitrant substrates. For example, substrates such as 41 with heterocyclic moieties generally were sluggish. To improve the yield without compromising the enantiospecificity, a second portion of Ni(dppe)Cl2 was added at 12 h to maintain a low catalyst concentration at all times. We also observed that for compounds that tend to undergo racemization, such as benzhydryl ethers, conducting the reaction at lower temperatures generally helps to maintain the stereochemical fidelity (e.g., 42).
Scheme 10. Mechanism for the Formation of Racemic Product.

c. Beyond Extended Aromatic Substituents: Accelerating Oxidative Addition
Stereospecific nickel-catalyzed alkyl–alkyl cross-coupling reactions were successful with a range of Grignard reagents, but the methodology required activation of the benzylic ether by an extended π system such as naphthalene, benzothiophene, or benzofuran. Preliminary mechanistic studies are consistent with rate-determining oxidative addition.37 This elementary step provides a π-benzylnickel complex; participation of the arene is critical in stabilizing the transition state.38 Substrates that present arenes with lower aromatic stabilization energies react more smoothly. For instance, in series of related tetrahydropyrans, we found that high yields were obtained for both the naphthyl- and 3-furyl-substituted tetrahydropyrans (Scheme 11). Both of these aromatic groups have relatively low aromatic stabilization energies.39 However, phenyl-substituted tetrahydropyran 50 does not undergo the cross-coupling reaction, even at elevated temperatures.
Scheme 11. Activation of Benzylic Ethers by Arenes with Low Aromatic Stabilization Energies.

To increase the reactivity of simple aromatic systems, a new strategy was required in order to accelerate the oxidative addition. Inspired by the use of directing groups in transition-metal-catalyzed reactions,7,40 we designed 2-methoxyethyl ether as a traceless directing group.41 We hypothesized that a five-membered chelate with magnesium salts present in the reaction would activate the C–O for oxidative addition. The directing group is traceless since it is cleaved over the course of the reaction. This strategy provided a substantial rate acceleration, such that benzhydryl alcohol derivatives that had previously resisted Kumada coupling reactions now provided good yields (cf. Scheme 12a vs Scheme 12b).
Scheme 12. Traceless Directing Group Strategy.

Good yields and excellent stereochemical fidelity were achieved with a variety of simple benzylic substrates (Scheme 13). Of particular interest, we found that N-heterocyclic aromatic groups, such as 3- and 4-substituted quinolines and pyridines, are well-tolerated and provide good yields with excellent es. This strategy has provided the foundation for our studies aimed at further expanding the scope of the electrophilic partner; improved advances will be required for the development of a truly general reaction with respect to the electrophilic partner.
Scheme 13. Cross-Coupling Reactions Facilitated by a Traceless Directing Group.

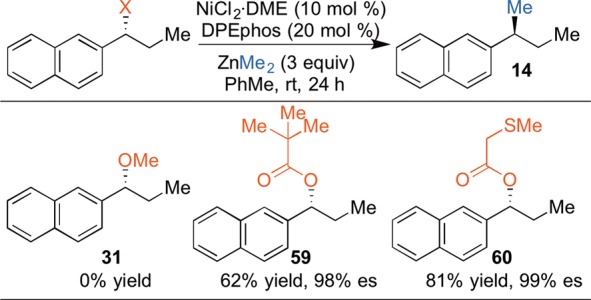
IV. Stereospecific Negishi-Type Cross-Coupling Reactions
Organozinc reagents are outstanding for a variety of applications since they have improved functional group compatibility compared with the corresponding Grignard reagents.42 Therefore, we sought to develop stereospecific Negishi-type coupling reactions. Initial investigations demonstrated that the simple benzylic ether 31 does not undergo nickel-catalyzed reaction with dimethylzinc (Scheme 14). We hypothesized that a more electrophilic substrate, such as an ester, would provide the requisite increase in reactivity. Indeed, for certain coupling reactions, simple pivalate esters (e.g., 59) provide reasonable yields. We found that the most robust strategy was to employ substrates incorporating a traceless directing group. These findings built on the work of Liebeskind40 and our previous use of the 2-methoxyethyl ether (Scheme 12). The pendant functional group would act by chelating the zinc reagent, weakening the C–O bond and accelerating oxidative addition, and also accelerate the transmetalation step. We found that esters with a chelating thioether (e.g., 60) provided the highest yields and excellent es.43 This directing group is easily installed via N,N′-dicyclohexylcarbodiimide (DCC) coupling of an enantioenriched benzylic alcohol with commercially available (methylthio)acetic acid.
Scheme 14. Identification of a Traceless Directing Group for Negishi Coupling Reactions.
With an optimal traceless directing group in hand, we examined the functional group tolerance of this reaction. As anticipated, a wide range of functional groups such as alkenes, alkynes, alcohols, and amines are tolerated and afford high yields and excellent es (Scheme 15). Indole-containing substrate 64, which was not tolerated under our previous Kumada conditions,44 also underwent a smooth and highly stereospecific cross-coupling reaction.
Scheme 15. Examples of Stereospecific Negishi Coupling Reactions.

We hypothesized that we could extend this methodology to include other activated esters.45 A series of enantioenriched aryl-substituted δ-valerolactones underwent stereospecific ring-opening reactions in good yields with excellent es (Scheme 16).33 We found that simple 3-furanyl lactones were amenable, providing enantioenriched carboxylic acids with functional group handles on both ends (e.g., 67). Many challenges remain, most notably expansion of the scope beyond dimethylzinc to include a range of alkyl- and arylzinc reagents as well as the use of alkylzinc halides.
Scheme 16. Ring Opening of δ-Valerolactones.

V. Stereospecific Nickel-Catalyzed Suzuki-Type Cross-Coupling Reactions
In our quest to utilize softer nucleophiles, we also examined arylboronic esters. From a practical perspective, such a reaction would fit nicely into the toolbox of a medicinal chemist, as it would engage the banks of arylboronic esters available for library synthesis. From an organometallic perspective, this reaction provided surprises with respect to the stereochemical outcome of oxidative addition (vide infra).
At the outset, in analogy to our development of the Negishi coupling, we chose to examine benzylic esters and electron-rich catalysts to facilitate oxidative addition. The best catalysts that emerged were indeed electron-rich, with ligation by SIMes or PCy3 (Schemes 17 and 18).46 Both systems provided high yields and es for cross-coupling reactions of a range of benzylic esters with arylboronic esters. Surprisingly, these two achiral catalyst systems provided opposite enantiomers of the product with high selectivity.47 The N-heterocyclic carbene (NHC) ligand provided cross-coupling with inversion, consistent with our previous findings in Kumada and Negishi-type coupling reactions. In contrast, PCy3 provided cross-coupling with retention at the site of oxidative addition. The origin of this change in selectivity is under investigation; our working hypothesis is that coordination of the ester to the phosphine-ligated catalyst serves to direct oxidative addition with retention. Consistent with our observations, Watson and co-workers determined that oxidative addition occurs with retention in a nickel-catalyzed elimination reaction of a benzylic ester using Ni(cod)2 in the presence of PCy3.48
Scheme 17. Stereospecific Suzuki Coupling with Inversion or Retention.

Scheme 18. Suzuki Coupling of Simple Benzhydryl Esters Using an NHC-Ligated Catalyst.

VI. Application in the Synthesis of Enantioenriched Bioactive Compounds
Application in target-oriented synthesis is typically the test of a new method’s practicality. To challenge our stereospecific cross-coupling reactions, we undertook the synthesis of compounds with a range of reported biological functions (Figure 2). By affecting cross-coupling at benzylic centers, these methods provide rapid access to the 1,1-diarylalkane pharmacophore, which is present in medicinal agents including Zoloft, tolterodine, lasofoxifene, and centchroman.49 Stereospecific cross-coupling reactions of benzylic ethers also provide a means of introducing benzylic methyl groups, a common practice in medicinal chemistry to improve drug bioavailability and potency.28 Our methodology allows us to utilize an uncommon disconnection to access these compounds as single enantiomers.
Figure 2.

Medicinal agents prepared by stereospecific cross-coupling reactions.
Our group has successfully synthesized single enantiomers of several bioactive compounds using Kumada, Negishi, and Suzuki-type coupling reactions (Figure 2). Diarylethane 76 is a combretastatin analogue with activity against colon cancer cell lines.50 With our methodology, a single enantiomer of 76 was obtained in 69% yield with excellent es.30 Similarly, sleep-inducing agent5177 was accessed in high ee, as installation of the tertiary stereogenic center was accomplished in 83% yield with good es.30 We prepared tamoxifen analogue5278 employing complementary Kumada or Suzuki reactions, giving direct access to either enantiomer of 78 from the same enantiomer of the intermediate benzylic alcohol.41b,53
The expansion of our methods to include Negishi-type coupling reactions has allowed the synthesis of bioactive compounds containing a variety of functional groups without resorting to protecting group manipulations. We prepared the retinoic acid receptor (RAR) ligand5480 and the fatty acid amide hydrolase (FAAH) inhibitor5581 with high enantiospecificity by means of Negishi-type reactions.43 Niacin receptor agonist5682 was prepared by Negishi-type ring opening of the requisite lactone.33 The previous synthesis required seven steps and chromatographic separation of the enantiomers; our synthesis requires two steps from the commercially available enantioenriched lactone and provides 98% ee.
In addition to preparing known targets as a synthetic challenge, we also sought to identify new leads for anti-cancer agents. Since the 1,1-diarylalkane scaffold is present in a range anti-cancer agents, we have begun to evaluate the new compounds that we prepare for activity against breast cancer cell lines. In preliminary studies, we have established that thioether 79 suppresses proliferation of the MCF-7 breast cancer line with an EC50 of 5 μM.34
VII. Conclusion
The field of stereoselective alkyl–alkyl cross-coupling reactions is still in its early stages, with many exciting advances on the horizon. We have described our contributions to the field in establishing nickel-catalyzed stereospecific cross-coupling reactions of secondary electrophiles. Our efforts have focused on benzylic ethers and esters because of the rate acceleration provided by the adjacent arene. These methods have provided a new strategy for the synthesis of enantioenriched medicinal agents with activities against a range of targets. Future advances will continue to expand the scope of these transformations as well as the development of related transformations that are initiated by a stereospecific oxidative addition event.
Acknowledgments
This work was supported by NIH NIGMS R01GM100212 and DOE PA200A120070 (GAANN Fellowship to L.E.H.).
Biographies
Emily J. Tollefson grew up in Tacoma, Washington, where she stayed to earn her undergraduate degree at Pacific Lutheran University in 2011. She worked in the lab of Neal Yakelis studying inverse-electron-demand Diels–Alder reactions. She also spent a summer with Joanne Romagni-Colvin at the University of Cádiz (Spain), where she conducted natural product isolation research in the laboratory of María Jesús Ortega and Eva Zubía. She is currently a fourth-year graduate student in the laboratory of Elizabeth R. Jarvo. Her doctoral research is focused on the development of stereospecific cross-coupling reactions of cyclic ethers and lactones.
Luke E. Hanna was born in Los Angeles, California, in 1986. He earned his B.Sc. in Biology and Biochemistry, working in the laboratories of Nilay Patel and Peter De Lijser. In 2011 he started graduate school at the University of California, Irvine, where is currently pursuing doctoral studies in the laboratory of Elizabeth R. Jarvo. His research interests include the development of new catalytic methods using the base metals nickel, cobalt, and iron.
Elizabeth R. Jarvo was born in Halifax, Nova Scotia, Canada, in 1975. She earned her B.Sc. (Honours) from Acadia University, working in the laboratory of Michael A. Kerr, and was a summer NSERC student at Concordia University with Youla Tsantrizos. She carried out her Ph.D. studies under the direction of Scott J. Miller at Boston College and postdoctoral studies with Eric N. Jacobsen at Harvard University. In 2005 she joined the faculty at the University of California, Irvine, where her research program focuses on the development of new catalytic reactions, including stereospecific cross-coupling reactions using nickel catalysts.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Earth Abundant Metals in Homogeneous Catalysis”.
References
- Johansson-Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- Geist E.; Kirschning A.; Schmidt T. sp3-sp3 Coupling Reactions in the Synthesis of Natural Products and Biologically Active Molecules. Nat. Prod. Rep. 2014, 31, 441–448. 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- Griggs N. D.; Phillips A. J. A Concise and Modular Synthesis of Pyranicin. Org. Lett. 2008, 10, 4955–4957. 10.1021/ol802041c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a transition-metal-free strategy, including the formation of quaternary stereocenters, see:; a Balieu S.; Hallett G. E.; Burns M.; Bootwicha T.; Studley J.; Aggarwal V. Toward Ideality: The Synthesis of (+)-Kalkitoxin and (+)-Hydroxyphthioceranic Acid by Assembly-Line Synthesis. J. Am. Chem. Soc. 2015, 137, 4398–4403. 10.1021/ja512875g. [DOI] [PubMed] [Google Scholar]; b Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Enantiospecific sp2-sp3 Coupling of Secondary and Tertiary Boronic Esters. Nat. Chem. 2014, 6, 584–589. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]
- a Nicolaou K. C.; Sorensen E. J.. Classics in Total Synthesis; Wiley-VCH: Weinheim, Germany, 1996; p 185. [Google Scholar]; b Carreira E. M.; Kvaerno L.. Classics in Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- For definitions and examples of stereospecific and stereoconvergent reactions, see:Eliel E. L.; Wilen S. H.. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994. [Google Scholar]
- Walsh P. J.; Kowzlowski M. C.. Fundamentals of Asymmetric Catalysis; University Science Books: Sausalito, CA, 2009. [Google Scholar]
- For reviews, see:; a Leonori D.; Aggarwal V. K. Stereospecific Couplings of Secondary and Tertiary Boronic Esters. Angew. Chem., Int. Ed. 2015, 54, 1082–1096. 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]; b Swift E. C.; Jarvo E. R. Asymmetric Transition Metal-Catalyzed Cross-Coupling Reactions for the Construction of Tertiary Stereocenters. Tetrahedron 2013, 69, 5799–5817. 10.1016/j.tet.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent example, see:; c Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. Stereospecific Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkylboron Nucleophiles and Aryl Chlorides. J. Am. Chem. Soc. 2014, 136, 14027–14030. 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Kanehira K.; Hioki T.; Kumada M. Kinetic Resolution of Racemic Grignard Reagents by Nickel-Catalyzed Asymmetric Grignard Cross-Coupling. Tetrahedron Lett. 1981, 22, 137–140. 10.1016/0040-4039(81)80169-4. [DOI] [Google Scholar]
- a Fischer C.; Fu G. C. Asymmetric Nickel-Catalyzed Negishi Cross-Couplings of Secondary α-Bromo Amides with Orangozinc Reagents. J. Am. Chem. Soc. 2005, 127, 4594–4595. 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]; b Arp F. O.; Fu G. C. Catalytic Enantioselective Negishi Reactions of Racemic Secondary Benzylic Halides. J. Am. Chem. Soc. 2005, 127, 10482–10483. 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]; For a lead reference, see:; c Schley N. D.; Fu G. C. Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a lead reference, see:; Wilsily A.; Tramutola F.; Owston N. A.; Fu G. C. New Directing Groups for Metal-Catalyzed Asymmetric Carbon–Carbon Bond-Forming Processes: Stereoconvergent Alkyl–Alkyl Suzuki Cross-Couplings of Unactivated Electrophiles. J. Am. Chem. Soc. 2012, 134, 5794–5797. 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Single-Electron Transmetallation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D.; Chu L.; Terrett J.; Doyle A. G.; MacMillan D. W. C. Merging Photoredox with Nickel Catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph A.; Lautens M. Secondary Alkyl Halides in Transition-Metal-Catalyzed Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2009, 48, 2656–2670. 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- a Tasker S. Z.; Standley E. A.; Jamison T. F. Recent Advances in Homogeneous Nickel Catalysis. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Giovannini R.; Stüdemann T.; Dussin G.; Knochel P. An Efficient Nickel-Catalyzed Cross-Coupling Between sp3 Carbon Centers. Angew. Chem., Int. Ed. 1998, 37, 2387–2390. . [DOI] [PubMed] [Google Scholar]
- a Sherry B. D.; Fürstner A. The Promise and Challenge of Iron-Catalyzed Cross-Coupling. Acc. Chem. Res. 2008, 41, 1500–1511. 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]; b Bauer I.; Knölker H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]; c Gosmini C.; Bégouin J.-M.; Moncomble A. Cobalt-Catalyzed Cross-Coupling Reactions. Chem. Commun. 2008, 3221–3233. 10.1039/b805142a. [DOI] [PubMed] [Google Scholar]; d Hess W.; Treutwein J.; Hilt G. Cobalt-Catalyzed Carbon-Carbon Bond-Formation Reactions. Synthesis 2008, 3537–3562. 10.1055/s-0028-1083210. [DOI] [Google Scholar]
- a Lau K. S. Y.; Fries R. W.; Stille J. K. Stereochemistry of Oxidative Addition of Alkyl Halides to Palladium(0) Complexes. J. Am. Chem. Soc. 1974, 96, 4983–4986. 10.1021/ja00822a044. [DOI] [Google Scholar]; b Netherton M. R.; Fu G. C. Suzuki Cross-Couplings of Alkyl Tosylates that Possess β Hydrogen Atoms: Synthetic and Mechanistic Studies. Angew. Chem., Int. Ed. 2002, 41, 3910–3912. . [DOI] [PubMed] [Google Scholar]
- For lead references on palladium-catalyzed cross-coupling reactions of alkyl electrophiles, see:; a Legros J.-Y.; Toffano M.; Fiaud J.-C. Palladium-Catalyzed Substitution of Esters of Naphthylmethanols, 1-Naphthylethanols, and Analogues by Sodium Dimethyl Malonate. Stereoselective Synthesis from Enantiomerically Pure Substrates. Tetrahedron 1995, 51, 3235–3246. 10.1016/0040-4020(95)00061-C. [DOI] [Google Scholar]; b He A.; Falck J. R. Stereospecific Suzuki Cross-Coupling of Alkyl α-Cyanohydrin Triflates. J. Am. Chem. Soc. 2010, 132, 2524–2525. 10.1021/ja910582n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Son S.; Fu G. C. Nickel-Catalyzed Asymmetric Negishi Cross-Coupling of Secondary Allylic Chlorides with Alkylzincs. J. Am. Chem. Soc. 2008, 130, 2756–2757. 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- a Hegedus L. S.; Miller L. L. Reactions of π-Allylnickel Bromide Complexes with Organic Halides. Stereochemistry and Mechanism. J. Am. Chem. Soc. 1975, 97, 459–460. 10.1021/ja00835a061. [DOI] [Google Scholar]; b Stille J. K.; Cowell A. B. The Oxidative Addition of Benzyl Halides to Tetrakis(triphenylphosphine)nickel (0). J. Organomet. Chem. 1977, 124, 253–261. 10.1016/S0022-328X(00)90972-0. [DOI] [Google Scholar]; c Tsou T. T.; Kochi J. K. Mechanism of Biaryl Synthesis with Nickel Complexes. J. Am. Chem. Soc. 1979, 101, 7547–7560. 10.1021/ja00519a015. [DOI] [Google Scholar]; d Jones G. D.; Martin J. L.; McFarland C.; Allen O. R.; Hall R. E.; Haley A. D.; Brandon R. J.; Konovalova T.; Desrochers P. J.; Pulay P.; Vicic D. A. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc. 2006, 128, 13175–13183. 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]; e Lin X.; Phillips D. L. Density Functional Theory Studies of Negishi Alkyl-Alkyl Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. J. Org. Chem. 2008, 73, 3680–3688. 10.1021/jo702497p. [DOI] [PubMed] [Google Scholar]; f Castaño A. M.; Echavarren A. M. Reactivity of a Nickelacycle Derived from Aspartic Acid: Alkylations, Insertions, and Oxidations. Organometallics 1994, 13, 2262–2268. 10.1021/om00018a020. [DOI] [Google Scholar]; g Phapale V. B.; Buñuel E.; García-Iglesias M.; Cárdenas D. J. Ni-Catalyzed Cascade Formation of C(sp3)–C(sp3) Bonds by Cyclization and Cross-Coupling Reactions of Iodoalkanes with Alkyl Zinc Halides. Angew. Chem., Int. Ed. 2007, 46, 8790–8795. 10.1002/anie.200702528. [DOI] [PubMed] [Google Scholar]; h Biswas S.; Weix D. J. Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Choi J.; Martín-Gago P.; Fu G. C. Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones. J. Am. Chem. Soc. 2014, 136, 12161–12165. 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin X.; Sun J.; Xi Y.; Lin D. How Racemic Secondary Alkyl Electrophiles Proceed to Enantioselective Products in Negishi Cross-Coupling Reactions. Organometallics 2011, 30, 3284–3292. 10.1021/om1012049. [DOI] [Google Scholar]
- Hu X. Nickel-Catalyzed Cross Coupling of Non-activated Alkyl Halides: A Mechanistic Perspective. Chem. Sci. 2011, 2, 1867–1886. 10.1039/c1sc00368b. [DOI] [Google Scholar]
- Hoffmann R. W. The Quest for Chiral Grignard Reagents. Chem. Soc. Rev. 2003, 32, 225–230. 10.1039/b300840c. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Kanno K.. Nickel-Catalyzed Cross-Coupling Reactions. In Modern Organonickel Chemistry; Tamaru Y., Ed.; Wiley-VCH: Weinheim, Germany, 2005; p 47. [Google Scholar]
- a Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Su B.; Cao Z.-C.; Shi Z.-J. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc. Chem. Res. 2015, 48, 886–896. 10.1021/ar500345f. [DOI] [PubMed] [Google Scholar]; c Yu D.-G.; Li B.-J.; Shi Z.-J. Exploration of New C–O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res. 2010, 43, 1486–1495. 10.1021/ar100082d. [DOI] [PubMed] [Google Scholar]; d Cornella J.; Zarate C.; Martin R. Metal-Catalyzed Activation of Ethers via C–O Bond Cleavage: A New Strategy for Molecular Diversity. Chem. Soc. Rev. 2014, 43, 8081–8097. 10.1039/C4CS00206G. [DOI] [PubMed] [Google Scholar]
- Fleming I.Molecular Orbitals and Organic Chemical Reactions, student ed.; John Wiley and Sons: Chichester, England, 2009; pp 97–102. [Google Scholar]
- a Consiglio G.; Morandini F.; Piccolo O. Stereochemical Aspects of the Nickel-Catalyzed Alkylation of Allylic Alcohols. J. Am. Chem. Soc. 1981, 103, 1846–1847. 10.1021/ja00397a048. [DOI] [Google Scholar]; b Kobayashi Y.; Ikeda E. Nickel-Catalysed Substitution Reactions of Allylic Carbonates with Aryl- and Alkenyl-Borates. J. Chem. Soc., Chem. Commun. 1994, 1789–1790. 10.1039/c39940001789. [DOI] [Google Scholar]; c Didiuk M. T.; Morken J. P.; Hoveyda A. H. Phosphine-Directed Stereo- & Regioselective Ni-Catalyzed Reactions of Grignard Reagents with Allylic Ethers. Tetrahedron 1998, 54, 1117–1130. 10.1016/S0040-4020(97)10212-5. [DOI] [Google Scholar]
- a Guan B.-T.; Xiang S.-K.; Wang B.-Q.; Sun Z.-P.; Wang Y.; Zhao K.-Q.; Shi Z.-J. Direct Benzylic Alkylation via Ni-Catalyzed Selective Benzylic sp3 C–O Activation. J. Am. Chem. Soc. 2008, 130, 3268–3269. 10.1021/ja710944j. [DOI] [PubMed] [Google Scholar]; b Tobisu M.; Yasutome A.; Kinuta H.; Nakamura K.; Chatani N. 1,3-Dicyclohexylimidazol-2-ylidene as a Superior Ligand for the Nickel-Catalyzed Cross-Couplings of Aryl and Benzyl Methyl Ethers with Organoboron Reagents. Org. Lett. 2014, 16, 5572–5575. 10.1021/ol502583h. [DOI] [PubMed] [Google Scholar]
- Jacobsen E. N.; Pfaltz A.; Yamamoto H.. Comprehensive Asymmetric Catalysis I–III; Springer: Berlin, 1999. [Google Scholar]
- a Barreiro E. J.; Kümmerle A. E.; Fraga C. A. M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]; b Schönherr H.; Cernak T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- a Aantaa R.; Kallio A.; Virtanen R. Dexmedetomidine, a Novel α2-Adrenergic Agonist. A Review of Its Pharmacodynamic Characteristics. Drugs Future 1993, 18, 49–56. 10.1358/dof.1993.018.01.198548. [DOI] [Google Scholar]; b Cheltsov A. V.; Aoyagi M.; Aleshin A.; Yu E. C.-W.; Gilliland T.; Zhai D.; Bobkov A. A.; Reed J. C.; Liddington R. C.; Abagyan R. Viccinia Virus Virulence Factor N1L is a Novel Promising Target for Antiviral Therapeutic Intervention. J. Med. Chem. 2010, 53, 3899–3906. 10.1021/jm901446n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mihalic J. T.; Chen X.; Fan P.; Chen X.; Fu Y.; Liang L.; Reed M.; Tang L.; Chen J.-L.; Jaen J.; Li L.; Dai K. Discovery of a Novel Series of Melanin-Concentrating Hormone Receptor 1 Antagonists for the Treatment of Obesity. Bioorg. Med. Chem. Lett. 2011, 21, 7001–7005. 10.1016/j.bmcl.2011.09.110. [DOI] [PubMed] [Google Scholar]
- Taylor B. L. H.; Swift E. C.; Waetzig J. D.; Jarvo E. R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Ethers: Enantioselective Synthesis of Diarylethanes. J. Am. Chem. Soc. 2011, 133, 389–391. 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Vogler T. Synthesis and Reactivity of Enantiomerically Enriched Thiiranium Ions. Chem. - Eur. J. 2009, 15, 11737–11745. 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Wolfe J. P.; Hay M. V. Recent Advances in the Stereoselective Synthesis of Tetrahydrofurans. Tetrahedron 2007, 63, 261–290. 10.1016/j.tet.2006.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pastor I. M.; Yus M. Focusd Update on the Prins Reaction and the Prins Cyclization. Curr. Org. Chem. 2012, 16, 1277–1312. 10.2174/138527212800564196. [DOI] [Google Scholar]
- Tollefson E. J.; Dawson D. D.; Osborne C. A.; Jarvo E. R. Stereospecific Cross-Coupling Reactions of Aryl-Substituted Tetrahydrofurans, Tetrahydropyrans, and Lactones. J. Am. Chem. Soc. 2014, 136, 14951–14958. 10.1021/ja5076426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonova I. M.; Johnson A. G.; Osborne C. A.; Moore C. E.; Morrissette N. S.; Jarvo E. R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Grignard Reagents and Identification of Selective Anti-Breast-Cancer Agents. Angew. Chem., Int. Ed. 2014, 53, 2422–2427. 10.1002/anie.201308666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We attribute the failure of Ni(dppe)Cl2 as the catalyst in reactions employing MeMgI to the stability of the Ni(dppe)Me2 complex, which is inactive. See ref (34).
- This mechanism was discounted to rationalize stereoablative reactions of benzylic bromides with nickel complexes. See ref (18b).
- a Greene M. A. Diastereoselective Synthesis of Seven-Membered Ring trans-Alkenes and Development of Stereospecific Nickel-Catalyzed Cross-Coupling Reactions. Ph.D. Thesis, University of California, Irvine, CA, May 2013. [Google Scholar]; b Erickson L. W.; Jarvo E. R.. 2015, in preparation.
- a Brauer D. J.; Krüger C. Bonding of Aromatic Hydrocarbons to Ni(0). Structure of Bistricyclohexylphosphine(1,2-η2-anthracene)nickel(0)-Toluene. Inorg. Chem. 1977, 16, 884–891. 10.1021/ic50170a033. [DOI] [Google Scholar]; b Hubig S. M.; Lindeman S. V.; Kochi J. K. Charge-Transfer Bonding in Metal-Arene Coordination. Coord. Chem. Rev. 2000, 200–202, 831–873. 10.1016/S0010-8545(00)00322-2. [DOI] [Google Scholar]
- Joule J. A.; Mills K.. Hetercyclic Chemistry, 5th ed.; John Wiley and Sons: Chichester, England, 2010; pp 6–7. [Google Scholar]
- Srogl J.; Liu W.; Marshall D.; Liebeskind L. Bio-organometallic Organosulfur Chemistry. Transition Metal-Catalyzed Cross-Coupling Using Coenzyme M or Thioglycolic Acid as the Leaving Group. J. Am. Chem. Soc. 1999, 121, 9449–9450. 10.1021/ja991654e. [DOI] [Google Scholar]
- a Greene M. A.; Yonova I. M.; Williams F. J.; Jarvo E. R. Traceless Directing Group for Stereospecific Nickel-Catalyzed Alkyl–Alkyl Cross-Coupling Reactions. Org. Lett. 2012, 14, 4293–4296. 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]; b Taylor B. L. H.; Harris M. R.; Jarvo E. R. Synthesis of Enantioenriched Triarylmethanes by Stereospecific Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2012, 51, 7790–7793. 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- Knochel P.; Leuser H.; Gong L.-Z.; Perrone S.; Kneisel F. F.. Polyfunctional Zinc Organometallics for Organic Synthesis. In Handbook of Functionalized Organometallics: Applications in Synthesis; Knochel P., Ed.; Wiley-VCH: Weinheim, Germany, 2005; p 251. [Google Scholar]
- Wisniewska H. M.; Swift E. C.; Jarvo E. R. Functional-Group-Tolerant, Nickel-Catalyzed Cross-Coupling Reaction for Enantioselective Construction of Tertiary Methyl-Bearing Stereocenters. J. Am. Chem. Soc. 2013, 135, 9083–9090. 10.1021/ja4034999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonova I. M. Stereospecific Nickel-Catalyzed Kumada Cross-Coupling Reactions of Benzylic Ethers. Ph.D. Thesis, University of California, Irvine, CA, November 2013. [Google Scholar]
- Sawama Y.; Shibata K.; Sawama Y.; Takubo M.; Monguchi Y.; Krause N.; Sajiki H. Iron-Catalyzed Ring-Opening Azidation and Allylation of O-Heterocycles. Org. Lett. 2013, 15, 5282–5285. 10.1021/ol402511r. [DOI] [PubMed] [Google Scholar]
- Harris M. R.; Hanna L. E.; Greene M. A.; Moore C. E.; Jarvo E. R. Retention or Inversion in Stereospecific Nickel-Catalyzed Cross-Coupling of Benzylic Carbamates with Arylboronic Esters: Control of Absolute Stereochemistry with an Achiral Catalyst. J. Am. Chem. Soc. 2013, 135, 3303–3306. 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awano T.; Ohmura T.; Suginome M. Inversion or Retention? Effects of Acidic Additives on the Stereochemical Course in Enantiospecific Suzuki–Miyaura Coupling of α-(Acetylamino)benzylboronic Esters. J. Am. Chem. Soc. 2011, 133, 20738–20741. 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Srinivas H. D.; Dasgupta S.; Watson M. P. Nickel-Catalyzed Cross-Couplings of Benzylic Pivalates with Arylboroxines: Stereospecific Formation of Diarylalkanes and Triarylmethanes. J. Am. Chem. Soc. 2013, 135, 3307–3310. 10.1021/ja312087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal F. S.; Panda G. Synthetic Methodologies of Achiral Diarylmethanols, Diaryl and Triarylmethanes (TRAMs) and Medicinal Properties of Diaryl and Triarylmethans- An Overview. RSC Adv. 2014, 4, 28317–28358. 10.1039/c4ra01341g. [DOI] [Google Scholar]
- a Messaoudi S.; Hamze A.; Provot O.; Tréguier B.; Rodrigo De Losada J.; Bignon J.; Liu J.-M.; Wdzieczak-Bakala J.; Thoret S.; Dubois J.; Brion J.-D.; Alami M. Discovery of Isoerianin Analogues as Promising Anticancer Agents. ChemMedChem 2011, 6, 488–497. 10.1002/cmdc.201000456. [DOI] [PubMed] [Google Scholar]; b Alami M.; Messaoudi S.; Hamze A.; Provot O.; Brion J.-D.; Liu J.-M.; Bignon J.; Bakala J.. Dihydro-iso-CA-4 and Analogues: Potent Cytotoxics, Inhibitors of Tubulin Polymerization. Patent WO/2009/147217 A1, Dec 10, 2009.
- Moree W. J.; Li B. F.; Jovic F.; Coon T.; Yu J. H.; Gross R. S.; Tucci F.; Marinkovic D.; Zamani-Kord S.; Malany S.; Bradbury M. J.; Hernandez L. M.; O’Brien Z.; Wen J. Y.; Wang H.; Hoare S. R. J.; Petroski R. E.; Sacaan A.; Madan A.; Crowe P. D.; Beaton G. Characterization of Novel Selective H-1-Antihistamines for Clinical Evaluation in the Treatment of Insomnia. J. Med. Chem. 2009, 52, 5307–5310. 10.1021/jm900933k. [DOI] [PubMed] [Google Scholar]
- Shagufta; Srivastava A. K.; Sharma R.; Mishra R.; Balapure A. K.; Murthy P. S. R.; Panda G. Substituted Phenanthrenes with Basic Amino Side Chains: A New Series of Anti-Breast Cancer Agents. Bioorg. Med. Chem. 2006, 14, 1497–1505. 10.1016/j.bmc.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Johnson A. G.; Tranquilli M. M.; Harris M. R.; Jarvo E. R. Selective Synthesis of Either Enantiomer of an Anti-Breast Cancer Agent via a Common Enantioenriched Intermediate. Tetrahedron Lett. 2015, 56, 3486–3488. 10.1016/j.tetlet.2015.02.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.-L.; Spinazze P.; Ostrowski J.; Currier S. J.; Pack E. J.; Hammer L.; Roalsvig T.; Honeyman J. A.; Tortolani D. R.; Reczek P. R.; Mansuri M. M.; Starrett J. E. Jr. Retinoic Acid Receptor β,γ-Selective Ligands: Synthesis and Biological Activity of 6-Substituted 2-Naphthoic Acid Retinoids. J. Med. Chem. 1996, 39, 2411–2421. 10.1021/jm9502293. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Ahn K.; Kesten S.; Lazerwith S. E.; Song Y.; Morris M.; Fay L.; Gregory T.; Stiff C.; Dunbar J. B. Jr.; Liimatta M.; Beidler D.; Smith S.; Nomanbhoy T. K.; Cravatt B. F. Benzothiophene Piperazine and Piperidine Urea Inhibitors of Fatty Acid Amid Hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2009, 19, 2865–2869. 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletti S. L.; Beresis R. T.; Chen W.; Tata J. R.; Shen H. C.; Marley D. M.; Deng Q.; Frie J. L.; Ding F.. Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment. WO 2006/052555 A2, May 18, 2006.