

Abstract
Hindbrain dorsal vagal complex A2 noradrenergic signaling represses the pre-ovulatory luteinizing hormone (LH) surge in response to energy deficiency. Insulin-induced hypoglycemia augments A2 neuron adenosine 5′-monophosphate-activated protein kinase (AMPK) activity and estrogen receptor-beta (ERβ) expression, coincident with LH surge suppression. We hypothesized that ERβ is critical for hypoglycemia-associated patterns of LH secretion and norepinephrine (NE) activity in key reproduction-relevant forebrain structures. The neural mechanisms responsible for tight coupling of systemic energy balance and procreation remain unclear; here, we investigated whether ERβ-dependent hindbrain signals also control glucose counter-regulatory responses to hypoglycemia. Gonadal steroid-primed ovariectomized female rats were pretreated by caudal fourth ventricular administration of the ERβ antagonist 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP) or vehicle at LH surge onset before insulin injection. Western blot analysis of laser-microdissected A2 neurons revealed hypoglycemic augmentation of 5′-monophosphate-activated protein kinase activity and dopamine-β-hydroxylase protein expression; the latter response was attenuated by PHTPP pretreatment. PHTPP regularized LH release, but not preoptic GnRH-I precursor protein expression in insulin-injected rats, and reversed hypoglycemic stimulation of glucagon and corticosterone secretion. Hypoglycemia caused PHTPP-reversible changes in NE and prepro-kisspeptin protein content in the hypothalamic arcuate (ARH), but not anteroventral periventricular nucleus. Results provide novel evidence for ERβ-dependent caudal hindbrain regulation of LH and counter-regulatory hormone secretion during hypoglycemia. That inhibition of LH likely involves mechanisms at the axon terminal that impede GnRH neurotransmission. Data also show that caudal hindbrain ERβ exerts site-specific control of NE activity in forebrain projection sites during hypoglycemia, including the ARH where prepro-kisspeptin may be a target of this signaling.
Keywords: estrogen receptor-beta, PHTPP, A2 noradrenergic neurons, GnRH-I, LH, arcuate nucleus
Introduction
Neural regulation of female reproductive function is tightly coupled with cellular energy metabolism, as substrate fuel shortage inhibits gonadotropin-releasing hormone (GnRH) output from the brain to anterior pituitary gonadotropes [Clarke et al., 1990; Chen et al., 1992]. Steroid positive-feedback activation of the GnRH-pituitary luteinizing hormone (LH) neuroendocrine axis triggers a critical mid-cycle signal to the ovary that controls oogenesis, ovulation, and corpus luteum function. Energy deficiency is a key cause of reduced frequency or cessation of ovulation in women [Crosignani, 2006]. GnRH regulation by extra-preoptic metabolic sensors is affirmed by hindbrain glucoprivation-induced reversal of steroid positive-feedback activation of GnRH neurons and induction of the LH surge [Briski and Sylvester, 1998]. Caudal dorsal vagal complex A2 noradrenergic neurons are able to integrate steroidal and metabolic stimuli as they express estrogen receptor-alpha (ERα) and -beta (ERβ) proteins, [Ibrahim et al., 2013] as well as molecular biomarkers for metabolic sensing, e.g. glucokinase, KATP, and the ultra-sensitive energy sensor, adenosine 5′-monophosphate-activated protein kinase (AMPK) [Briski et al., 2009; Cherian and Briski, 2011; Ibrahim et al., 2013].
Caudal dorsal vagal complex AMPK activity and ERβ protein expression strengthen in parallel during short-term food deprivation or simulation of local energy shortage by the AMP mimic 5-aminoimidazole-4-carboxamide-riboside (AICAR) [Ibrahim et al., 2015]. Insulin-induced hypoglycemia elevates A2 nerve cell phosphoAMPK (pAMPK) and ERβ protein profiles in steroid-primed ovariectomized (OVX) female rats, while ERα levels are unaffected [Shrestha and Briski, 2015]. These results support the possibility that ERβ protein responses to AMPK activity state may underlie A2 noradrenergic inhibitory metabolic signaling to the GnRH-pituitary LH axis. The present studies employed a pharmacological approach, involving intra-caudal fourth ventricular administration of the selective ERβ antagonist, 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP), to investigate the hypothesis that caudal hindbrain ERβ participates in hypoglycemic repression of steroid positive-feedback activation of the LH surge. Hypoglycemia reduces GnRH neuron GnRH-I precursor protein expression alongside reductions in circulating LH [Shrestha and Briski, 2015]. Here, micropunch-dissected rostral preoptic area (rPO) tissue was analyzed by high-sensitivity Western blot techniques to determine if PHTPP pretreatment alters hypoglycemic reduction of GnRH-I precursor protein content. In addition, micropunch samples of the rPO, AVPV, and other forebrain structures relevant to reproductive hormone secretion, e.g. medial preoptic nucleus (MPN) and hypothalamic arcuate nucleus (ARH), were evaluated for NE and reproductive neuropeptide content by ELISA and Western blot methods, respectively, to determine if caudal hindbrain ERβ – dependent mechanisms regulate local responses to hypoglycemia. AVPV and ARH kisspeptin levels were measured as distinctive Kiss1-expressing neurons in these loci mediate steroid positive- and negative-feedback action, respectively, on the GnRH-LH axis [Popa et al., 2008]. The ARH neurochemicals neuropeptide Y (NPY) and pro-opiomelanocortin (POMC) were also analyzed as NPY is obligatory for steroid positive-feedback induction of the LH surge [Kalra and Crowley, 1992], while the POMC peptide products β-endorphin (βEND) and α-melanocyte-stimulating hormone (MSH) respectively inhibit or stimulate LH release [Kalra, 1985; Murray et al., 2000].
Detection of brain metabolic deficiency elicits coordinated physiological and behavioral responses, including hyperphagia, glucose counter-regulatory hormone stimulation, and inhibition of reproductive hormone release, that increase systemic energy substrate supply via changes in intake, mobilization, and partitioning of metabolic fuel. Glucose anti-metabolite delivery to the hindbrain increases secretion of the counter-regulatory hormones glucagon and corticosterone [Andrew et al., 2007]. Our studies show that AICAR activation of caudal hindbrain AMPK intensifies release of these hormones in gonadal steroid - replaced, but not in non-steroid - treated OVX female rats [Ibrahim et al., 2013]. The current research addressed the premise that, similar to LH, hypoglycemic patterns of glucagon and corticosterone secretion are subject to control by ERβ - contingent hindbrain mechanisms.
Methods and Materials
Animals
Adult virgin female Sprague-Dawley rats (200–300 g bw) were maintained under a 14 hr-light: 10 hr-dark lighting schedule (light on at 05.00 h), and allowed ad-libitum access to standard rat chow (Harlan Teklad LM-485; Harlan industries, Madison, WI) and water. All animal protocols were conducted in accordance with NIH guidelines for care and use of laboratory animals, and approved by the ULM Institutional Animal Care and Use Committee. On day 1, rats were implanted with a PE-20 cannula into the caudal fourth ventricle (CV4) [coordinates: 0 mm lateral to midline; 13.3 mm posterior to bregma; 6.6 mm ventral to skull surface] under ketamine/xylazine anesthesia (0.1 mL/100 g bw i.p., 90 mg ketamine: 10 mg xylazine/mL Putney, Inc., Portland, ME; LLOYD laboratories Inc., Shenandoah, IO, USA). Animals were assigned to individual cages post-surgery. On day 7, animals were bilaterally OVX under ketamine/xylazine anesthesia. Rats were injected sc with estradiol benzoate (E; 10 μg/0.1 mL safflower oil) at 10.00 hr on days 14 and 17, and progesterone (P; 2.0 mg/0.2 mL safflower oil) at 11.00 hr on day 18 [Singh and Briski, 2004].
Experimental Design
At 14.00 hr on day 18, rats were injected subcutaneously (sc) with vehicle, sterile diluent, (V; Eli Lilly & Co., Indianapolis, IN; group 1, n=5) or neutral protamine Hagedorn insulin (I; 12.5 U/kg bw sc; Butler Schein, Melville, NY; groups 2 and 3, n=5 per group). Animals were pretreated between 13.40 and 13.45 hr by infusion into the caudal fourth ventricle (CV4) with the ERβ antagonist, PHTPP (10 μM, 200 nL [Saleh et al., 2013]; Tocris Bioscience, Bristol, U.K.; group 2), or the vehicle, dimethyl sulfoxide (DMSO; Sigma-Aldrich Co, LLC, St. Louis, MO; groups 1 and 3). Animals were sacrificed by decapitation at 16.00 hr for blood and brain tissue collection. Dissected brains were immediately snap-frozen in cooled isopentane and stored at −80°C. Plasma was stored at −20°C. All animals used in this study exhibited cerebrospinal fluid reflex from the cannula tip on day 18; postmortem histological examination confirmed accuracy of cannula placements within the CV4 [Alenazi et al., 2014].
Blood Glucose and Serum LH, Gucagon, Corticosterone, Estradiol, and Progesterone Analyses
Glucose values were measured with an ACCU-CHECK Aviva plus glucometer (Roche Diagnostics, Indianapolis, IN) (Kale et al., 2006). Serum LH, glucagon, and corticosterone concentrations were measured by radioimmunoassay [Singh et al., 2004; Paranjape and Briski, 2005]. Detection limits and coefficients of variation (CV) of these assays were as follows: LH: minimum and maximum sensitivity of 0.12 and 65.2 ng/mL, 6% intra- and 11% interassay CV; glucagon: minimum and maximum sensitivity of 18.5 and 400 pg/mL, 4% intra- and 12% interassay CV; corticosterone: minimum and maximum sensitivity of 7.7 and 1000 ng/mL, 6% intra- and 7% interassay CV.
Caudal Hindbrain A2 Noradrenergic Neuron Laser-Microdissection and Western Blot Analysis
Serial 10 μm-thick frozen sections were cut from each hindbrain, in a Leica 1860 cryostat, at rostro-caudal levels corresponding to A2 cell group location, e.g. −14.30 to −14.60 mm posterior to bregma, and mounted on polyethylene naphthalate (PEN) membrane slides (Carl Zeiss MicroImaging, Inc., Thornwood, NY). Sections were fixed with cold acetone (Sigma–Aldrich); blocked with 5% normal horse serum (Vectastain Elite ABC mouse IgG kit; Vector Laboratories, Inc., Burlingame, CA) diluted in 0.05 M Tris-buffered saline, pH 7.4, containing 0.05% Triton X-100 (Sigma-Aldrich) (TBS-Tx), and incubated with mouse monoclonal primary antiserum against tyrosine hydroxylase (TH) (1:2,000; ImmunoStar, Inc., Hudson, WI) diluted in TBS-Tx for 24 hr at 4°C. Tissues were sequentially incubated with Vectastain kit biotinylated secondary antibody, ABC reagent, and Vector DAB kit reagents (Vector Laboratories) to visualize TH-immunoreactive (-ir) neurons. Single TH-ir neurons exhibiting a visible nucleus and complete labeling of the cytoplasmic compartment were circumdissected with a Zeiss P.A.L.M. UV-A microlaser (Carl Zeiss Microimaging). For each treatment group, a minimum of four separate pools of n=50 TH-ir neuron lysates was created for each protein of interest prior to Western blot analysis. Proteins were denatured, electrophoresed in 10% gradient Tris-glycine gels, and transferred to 0.45μm polyvinyl difluoride (PVDF)-plus membranes (Osmonics Inc., Gloucester, MA) (Cherian and Briski, 2011, 2012). Membranes were treated with Pierce™ Western blot signal enhancer (ThermoFisherScientific, Rockford, IL), blocked with TBS containing 0.1% Tween-20 (Sigma-Aldrich) with 2% Bovine Serum Albumin (BSA; MP Biomedicals, Solon, OH) (TBS-T-BSA), then incubated overnight at 4°C with rabbit primary antisera against AMPKα1 (sc-19128), pAMPKα1/2 (Thr 172) (sc-33524), DβH (sc-15318), or Fos (sc-52) (Santa Cruz Biotechnology, Inc., Dallas, TX) diluted in TBS-T-BSA. The loading control protein, α-tubulin, was probed with a mouse monoclonal antiserum (sc-53646; Santa Cruz Biotechnol.). Membranes were then incubated with peroxidase-conjugated goat anti-rabbit or anti-mouse antisera (PerkinElmer, Waltham, MA) diluted TBS-T-BSA, followed by SuperSignal West Femto Maximum Sensitivity substrate (ThermoFisherSci.). Protein band chemiluminescence imaging and densitometric analysis was done using a G:Box Chemi and Genetool 4.01 software (Syngene USA, Frederick, MD). Protein optical density (O.D.) values were normalized to α-tubulin. Protein molecular weight markers were included in each western blot analysis.
Western Blot Analysis of rPO GnRH-I Protein
Serial 100 μm-thick frozen sections were cut through the preoptic area and hypothalamus of each brain. The Palkovits micropunch technique was employed to dissect the rPO [+0.48 mm − 0.00 mm relative to bregma; 0.51 mm needle diameter; Stoelting, Inc., Kiel, WI]; for each treatment group, lysate aliquots were combined from subjects to create four separate pools for Western blotting. Tissue lysate protein electrophoresis and transblotting was performed as described [Alenazi et al., 2014]. Membranes were blocked with TBS-T-BSA prior to overnight incubation at 4°C with rabbit primary anti-GnRH-I precursor (sc-32292; Santa Cruz Biotechnol.) or mouse primary anti-α-tubulin antibodies diluted in TBS-T-BSA. Chemiluminescence signal was developed by sequential incubation with peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antisera and SuperSignal West Pico chemiluminescent substrate (ThermoFisherSci.), imaged in a Syngene G:box Chemi, and quantified with Syngene Genetool 4.01 software. Protein optical densities (O.D.) were normalized to α-tubulin. Protein molecular weight markers were included in each analysis.
ELISA and Western Blot Analyses of rPO, AVPV, MPN, and ARH NE and Neuropeptide Content
NE ELISA measurements were performed on micropunch samples of rPO and AVPV [0.00 mm to − 0.26 mm; 0.29 mm needle diameter], medial preoptic nucleus [MPN; − 0.26 mm to − 0.60 mm; 0.51mm needle diameter], and hypothalamic arcuate nucleus [ARH; − 1.78 to −3.25 mm; 0.51 mm needle diameter]. For each rat, tissue punches of each dissected structure were collected in 100 μL of 0.01 N HCl containing 1 mM EDTA and 4 mM sodium metabisulfite; in each treatment group, aliquots from subjects were pooled to create triplicate samples for ELISA analysis of NE content [Shrestha et al., 2014]. For both the AVPV and ARH, aliquots of tissue lysate were combined within treatment groups to create four sample pools for Western blot analysis of prepro-kisspeptin protein levels, using a goat polyclonal antiserum (sc-18134; Santa Cruz Biotechnol.). In addition to prepre-kisspeptin, ARH samples were also pooled within treatment groups for evaluation of POMC and NPY proteins using described methods [Alenazi et al., 2014].
Statistical Analyses
Mean glucose, hormone, normalized Western blot O.D., and NE ELISA measures were evaluated by one-way ANOVA and Duncan’s multiple range test. Assessment of data in each analysis by the Shapiro-Wilks’ test confirmed normal distribution. Differences of p<0.05 were considered significant.
Results
Figure 1 depicts effects of insulin-induced hypoglycemia coincident with caudal fourth ventricular (CV4) administration of the ERβ antagonist PHTPP on A2 noradrenergic nerve cell AMPK (Figure 1.A), pAMPK (Figure 1.B), DβH (Figure 1.C), and Fos (Figure 1.D) protein expression. For each protein of interest, mean normalized O.D. measures and representative immunoblots are presented for laser-microdissected A2 neurons from groups of animals treated by 1) sc vehicle injection plus icv vehicle (DMSO) infusion (V/DMSO; white bars), 2) sc insulin (I) injection plus icv DMSO infusion (I/DMSO; gray bars), or 3) sc I injection plus icv PHTPP infusion (I/PHTPP; diagonal-striped gray bars). Data show that insulin-injected rats exhibited augmented pAMPK protein [F2,9 = 8.21, p=0.009; insulin effect: F = 14.73, p=0.004], but no change in net AMPK content [F2,9 = 1.04, p=0.39] of A2 neurons. A2 nerve cell AMPK and pAMPK profiles were equivalent in groups of I/DMSO and I/PHTPP rats. Insulin-induced hypoglycemia significantly increased A2 DβH enzyme proteins levels [F2,12 = 47.23, p<0.0001; insulin effect: F = 93.42, p<0.0001]. DβH content was significantly reduced in PHTPP- versus DMSO-pretreated hypoglycemic animals [PHTPP effect: F = 15.57, p=0.002]. A2 nerve cell Fos protein levels were refractory to hypoglycemia with or without intra-CV4 ERβ antagonist administration [F2,12 = 2.57; p=0.12].
Figure 1. Effects of Hindbrain Estrogen Receptor-Beta (ERβ) Antagonism on A2 Noradrenergic Nerve Cell Adenosine 5′-Monophosphate-Activated Protein Kinase (AMPK), PhosphoAMPK (pAMPK), Dopamine-β-Hydroxylase (DβH), and Fos Proteins in Insulin-Injected Steroid-Primed Ovariectomized (OVX) Female Rats.
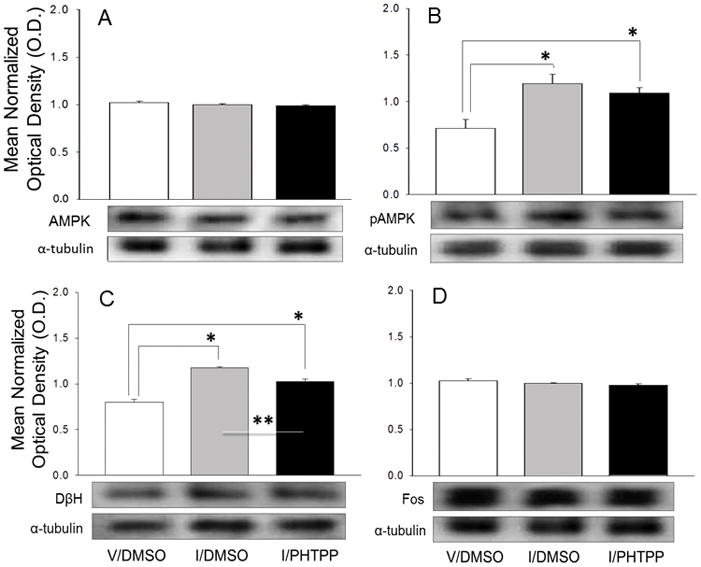
Individual tyrosine hydroxylase (TH)-immunoreactive (-ir) A2 neurons were harvested by laser-catapult microdissection from 10 μm-thick sections of the caudal dorsal vagal complex 2 hr after sc injection (14.00 hr; to) of vehicle (V; group 1; n=5) or neutral protamine Hagedorn Insulin (I; 12.5 U/kg bw; groups 2 and 3; n=5/group) of estradiol plus progesterone-primed OVX rats. Animals were pretreated at -20 min (13.40 hr) by caudal fourth ventricular (CV4) infusion of 200 nL of vehicle, dimethyl sulfoxide (DMSO; 200 nL; groups 1 and 2) or DMSO containing 10 uM 4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP; group 3). For each protein of interest, a minimum of four separate A2 nerve cell lysate pools were established for each treatment group (n=50 neurons per pool; n=10 cells per rat) for Western blot analysis. Protein band optical densities (O.D.) were quantified with Syngene Genetool 4.01 software and expressed relative to α-tubulin. Panels depict mean normalized O.D. values ± S.E.M. for A2 neuron AMPK (Figure 1.A), pAMPK (Figure 1.B), DβH (Figure 1.C), and Fos (Figure 1.D) proteins for the following treatment groups: V/DMSO (white bars), I/DMSO (gray bars), and I/PHTPP (black bars). Each panel also contains a representative immunoblot of the protein of interest and α-tubulin. *p < 0.05, versus V/DMSO; **p < 0.05, I/PHTPP versus I/DMSO.
As illustrated in Figure 2, the insulin plus DMSO group showed significant diminishment of rPO GnRH-I precursor protein content relative to V/DMSO controls [F2,12 = 18.05, p=0.0002; insulin effect: F = 16.75, p=0.002]. PHTPP pretreatment prior to insulin injection did not modify this inhibitory response to hypoglycemia.
Figure 2. PHTPP Does Not Reverse Hypoglycemic Suppression of Rostral Preoptic Area Gonadotropin-Releasing Hormone (GnRH) GnRH-I Precursor Protein Expression.
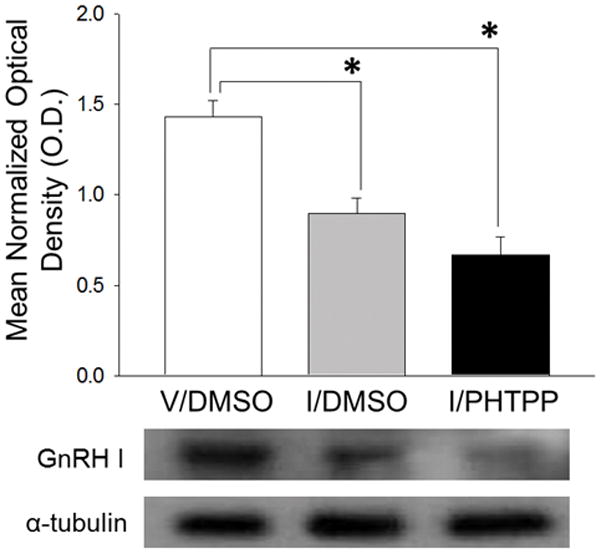
GnRH-I precursor Western blot analysis was performed on rostral preoptic area (rPO) tissue obtained by calibrated hollow needle micropunch dissection 2 hr after sc injection of V (group 1) or I (groups 2 and 3). Twenty min before injections, animals were pretreated by delivery of DMSO alone (groups 1 and 2) or DMSO containing PHTPP (group 3) to the CV4. For each treatment group, lysate aliquots from individual subjects were combined to create four individual pools for Western blot analysis. Bars represent mean normalized GnRH-I precursor protein O.D. measures ± S.E.M. for V/DMSO (white bar), I/DMSO (gray bar), and I/PHTPP (black bar) groups. Typical GnRH-I and α-tubulin Western immunoblots are shown below the graph. *p <0.05, versus V/DMSO.
Figure 3 illustrates effects of CV4 administration of PHTPP on hypoglycemia-associated patterns of NE activity in the rPO (Figure 3.A), AVPV (Figure 3.B), MPN (Figure 3.C), and ARH (Figure 3.D). Results show that DMSO-pretreated hypoglycemia animals exhibited increased tissue NE levels in the rPO [F2,6 = 7.01, p=0.03; insulin effect: F = 8.40, p=0.03], AVPV [F2,6 = 26.27, p=0.002; insulin effect: F = 48.87, p=0.001], MPN [F2,6 = 10.92, p=0.01; insulin effect: F = 16.51, p=0.007], and ARH [F2,6 = 50.44, p=0.005; insulin effect: F = 87.41, p=0.002; PHTPP effect: F = 61.68, p=0.004] relative to V/DMSO controls. Intra-CV4 PHTPP administration reversed hypoglycemic augmentation of NE levels in the ARH, but not other structures.
Figure 3. Site-Specific Effects of PHTPP on Norepinephrine (NE) Content in Insulin-Injected Steroid-Primed OVX Female Rats.
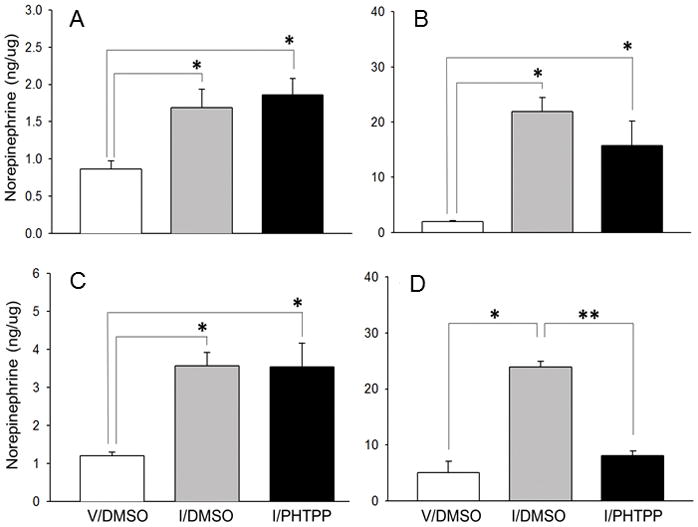
NE ELISA analysis was carried out on rostral preoptic area (rPO; Figure 3.A), anteroventral periventricular nucleus (AVPV; Figure 3.B), medial preoptic nucleus (MPN; Figur3.C), and hypothalamic arcuate nucleus (ARH; Figure 3.D) tissue samples obtained by bilateral micropunch dissection. For each structure, sample aliquots were combined from subjects to create three individual pools per treatment group for analysis. Bars depict mean NE content ± S.E.M. for V/DMSO (white bars), I/DMSO (gray bars), and I/PHTPP (black bars) groups. *p <0.05, versus V/DMSO; **p < 0.05, I/PHTPP versus I/DMSO.
Figure 4 depicts effects of insulin-induced hypoglycemia with or without PHTPP pretreatment on AVPV and ARH prepro-kisspeptin and ARH POMC and NPY protein levels. The data show that AVPV prepro-kisspeptin content (Figure 4.A) was unaltered during hypoglycemia [F2,9 = 11.17, p=0.004; PHTPP effect: F = 8.79, p=0.02], but was significantly diminished in the I/PHTPP group. ARH prepro-kisspeptin levels (Figure 4.B) were elevated in DMSO-pretreated hypoglycemic animals, but normalized in PHTPP-pretreated insulin-injected rats [F2,9 = 5.39, p=0.03; insulin effect: F = 10.60, p=0.01]. ARH POMC (Figure 4.C) and NPY (Figure 4.D) levels were respectively decreased [F2,6 = 11.59, p=0.01; insulin effect: F=11.07, p=0.02] or increased [F2,9 = 4.91, p=0.03; insulin effect: F = 5.22, p=0.04] by hypoglycemia. PHTPP pretreatment did not modify either response.
Figure 4. Site Specificity of PHTPP Effects on Prepro-Kisspeptin Protein Responses to Insulin-Induced Hypoglycemia area.
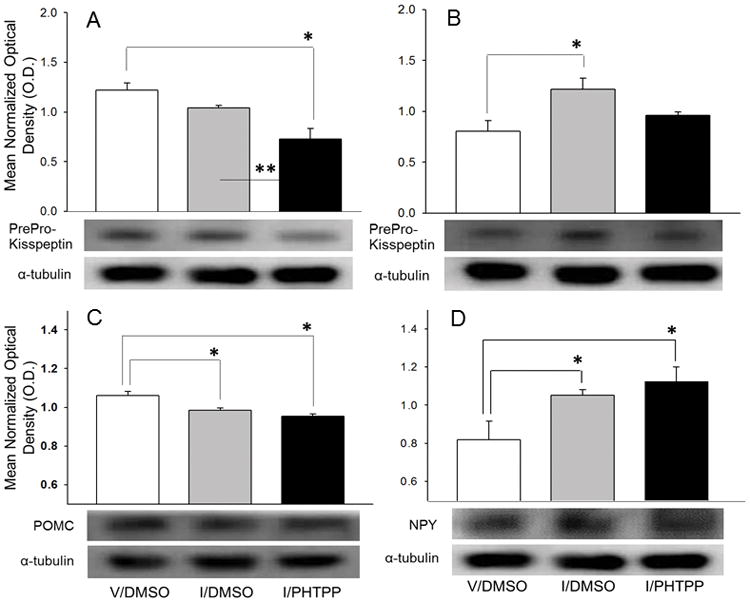
Micropunch samples of the AVPV [prepro-kisspeptin] and ARH [prepro-kisspeptin, pro-opiomelanocortin (POMC), neuropeptide Y (NPY)] from V/DMSO (white bar), I/DMSO (gray bar), and I/PHTPP (diagonal-striped gray bar) groups were analyzed by immunoblot. For each protein of interest, lysate aliquots were combined from subjects within treatment groups to create three or more independent pools per treatment group. Bars represent mean normalized protein O.D. measures ± S.E.M. for V/DMSO (white bars), I/DMSO (gray bars), and I/PHTPP (black bars) groups. Typical neuropeptide and α-tubulin Western immunoblots are shown below each graph. *p <0.05, versus V/DMSO; **p < 0.05, I/PHTPP versus I/DMSO.
As shown in Figure 5.A, blood glucose levels were significantly reduced in response to insulin injection [F2,12 = 633.85, p<0.0001; insulin effect: F = 911.35, p<0.0001]. Values for glucose were equivalent in DMSO- versus PHTPP-pretreated hypoglycemic rats. The I/DMSO treatment group exhibited diminished plasma luteinizing hormone (LH) levels (Figure 5.B) [F2,12 = 85.92, p<0.0001], and elevated circulating glucagon (Figure 5.C) [F2,12 = 71.35, p<0.0001] and corticosterone (Figure 5.D) [F2,12 = 64.48, p<0.0001] concentrations compared to V/DMSO controls. Pretreatment with PHTPP normalized patterns of secretion of each hormone in hypoglycemic rats.
Figure 5. PHTPP Alters Glucose, Luteinizing Hormone (LH), Glucagon, and Corticosterone Responses to Insulin-Induced Hypoglycemia.
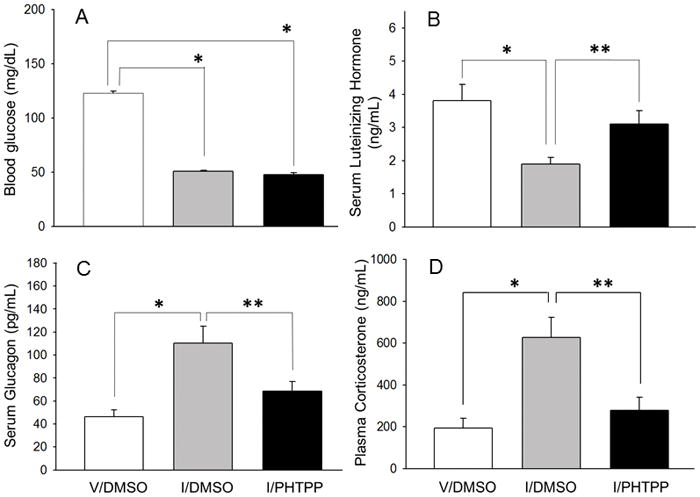
Data depict mean circulating glucose (Figure 4.A), LH (Figure 4.B, glucagon (Figure 4.C), and corticosterone levels ± S.E.M. (n=5 rats/group) 2 hr in V/DMSO (white bars), I/DMSO (gray bars), and I/PHTPP (black bars) treatment groups. *p<0.05; versus V/DMSO; **p<0.05; I/PHTPP versus I/DMSO.
Discussion
The mid-cycle LH surge is triggered by gonadal steroid positive-feedback amplification of GnRH release to the anterior pituitary, but is repressed by caudal hindbrain detection of acute energy shortfall. Tandem augmentation of caudal dorsal vagal complex pAMPK (a measurable molecular indicator of ATP deficiency) and ERβ protein profiles by the AMP mimic 5-aminoimidazole-4-carboxamide-riboside (AICAR) or food deprivation suggests that this ER variant may be crucial for regulatory effects of local AMPK activation [Ibrahim and Briski, 2015]. The present studies show that the selective ER β antagonist PHTPP reversed insulin-induced hypoglycemic inhibition of LH secretion, but not GnRH nerve cell GnRH-I precursor protein expression, and prevented hypoglycemic stimulation of counter-regulatory hormone output. PHTPP pretreatment normalized ARH, but not AVPV NE and prepro-kisspeptin content in hypoglycemic rats. Current results implicate ERβ in caudal hindbrain mechanisms governing LH and counter-regulatory hormone release during hypoglycemia. Discordant GnRH-I precursor protein and LH secretory responses to PHTPP pretreatment suggest that ERβ-controlled hindbrain signaling may inhibit hypothalamic GnRH output to the pituitary by action on neurotransmitter exocytosis and/or degradation within axon terminals. PHTPP normalization of hypoglycemia-associated patterns of ARH, but not rPO, AVPV, or MPN NE content implies that during hypoglycemia, caudal hindbrain ERβ exerts site-specific control of NE activity in forebrain projection sites. Further research is needed to identify caudal hindbrain ERβ-sensitive neurons in the ARH that impose metabolic restraint of LH release.
Caudal dorsal vagal complex ERβ protein content is increased by AICAR, suggesting that AMPK activation augments this receptor [Ibrahim et al., 2015]. Hypoglycemia elicits concurrent up-regulation of A2 pAMPK and ERβ proteins [Shrestha and Briski, 2015]. Current findings that PHTPP does not alter hypoglycemic patterns of A2 pAMPK expression, but attenuates hypoglycemic stimulation of DβH protein imply that ERβ function downstream of AMPK to enact sensor activation effects on A2 neurotransmitter production. Alternatively, ERβ action upstream of AMPK during hypoglycemia to regulate AMPK reactivity to that condition would be consistent with outcomes showing PHTPP-mediated normalization of sensor activity in insulin-injected animals. A2 neurons provide inhibitory metabolic input to the GnRH-LH axis, as neurotoxin decimation of these cells abolishes hindbrain glucoprivic inhibition of the LH surge [Ibrahim and Briski, 2014]. PHTPP reversal of hypoglycemic suppression of LH implicates caudal hindbrain ERβ in metabolic restraint of the LH surge. We presume that A2 neurons are a direct target for this ERβ action [Ibrahim and Briski, 2014], but cannot discount the possibility that this receptor may act through indirect mechanisms involving afferent input to A2 cells. Rat brain GnRH neurons that react to hindbrain metabolic signaling reside in the rPO [Briski and Sylvester, 1998]. In the present study, PHTPP did not reverse hypoglycemic suppression of rPO GnRH-I precursor protein content. GnRH is transported within axons that project from the rPO to the hypothalamic median eminence, where this neurochemical is stored and released from axon terminals near hypothalamo-hypophyseal portal capillaries. Our results suggest that ERβ-driven hindbrain stimuli may not inhibit GnRH neuropeptide synthesis, but might instead act distal to the GnRH cell body to regulate axon terminal neurotransmitter storage, metabolism, and/or release. Data here imply that separate ERβ-independent and -dependent mechanisms occur in parallel to repress the LH surge by actions at distinct neuroanatomical sites (i.e. GnRH cell body versus axon terminal) that impact different aspects of GnRH neuro-secretion.
A2 neurons mediate estrogen negative- and positive-feedback regulation of LH secretion, responding to these dualistic signals by release of norepinephrine into the medial preoptic area at basal versus elevated rates [Demling et al., 1985; Mohankumar et al., 1994; Szawka et al., 2013]. Metabolic restraint of the LH surge involves diminished hindbrain NE signaling to the preoptic AVPV, as administration of the alpha-adrenergic receptor agonist methoxamine to that site reverses hindbrain glucoprivic suppression of GnRH neuron Fos immuno-labeling and pituitary LH release [Ibrahim and Briski, 2014]. Here, hypoglycemia elevated NE levels in the AVPV and other structures relevant to reproduction, e.g. rPO, MPN, and ARH, results that concur with prior reports of hypoglycemic enhancement of hypothalamic NE activity [Bellin and Ritter; 1981; Beverly et al., 2001]. Taken together, our studies suggest that hypoglycemia may simultaneously increase and decrease NE inputs aimed at different subsets of AVPV targets, and that the latter earmarks substrates that govern GnRH. As the caudal dorsal vagal complex is characterized by a high concentration of neurons that increase or decrease electrical activity in response to glucose [Mizuno and Oomura; 1984], we speculate that distinct subsets of A2 neurons may likewise increase or decrease neurotransmission in response to metabolic shortfall. It is not known if bimodal NE signals are also directed to the rPO, MPH, and ARH during hypoglycemia. It is noted that measures of steady-state NE tissue content do not yield conclusive insight on how hypoglycemia impacts the ratio of NE synthesis/metabolism/release within individual neural loci. Observations here of amplified NE tissue levels are construed to likely indicate enhanced neurotransmitter influx, but might alternatively reflect, in part, diminished NE breakdown or release. Normalization of ARH, but not rPO, AVPV, or MPN NE content suggests that caudal hindbrain ERβ exert site-specific control of NE accumulation in forebrain projection sites during hypoglycemia. Concurrent adjustments in ARH NE content and LH release due to PHTPP suggest that caudal hindbrain ERβ control of NE processing, uptake, and/or packaging in the ARH is crucial for this LH response. In the context of GnRH-LH axis regulation, our data intimate that ERβ stimulate hypoglycemic-driven enhanced NE input to the ARH (which we presume to inhibit axonal GnRH release), but do not induce concurrent suppression of steroid feedback-induced signaling to the AVPV (which is surmised to impair GnRH neuropeptide synthesis).
The current studies yield novel proof for integrated hindbrain regulation of reproductive and counter-regulatory hormone responses to hypoglycemia. Several ARH neuropeptides are candidates for unified control of reproduction and energy homeostasis, including NPY and the POMC derivatives β-END and α-MSH [Beck, 2000; Williams et al., 2000]. NPY is critical for steroid positive-feedback induction of the LH surge [Kalra and Crowley, 1992], while β-END inhibits and α-MSH stimulates LH release [Kalra, 1985; Murray et al., 2000]. NPY and β-END stimulate food intake, whereas α-MSH inhibits feeding [Kanatani et al., 1996; Silva et al., 2001; Vergoni and Bertolini, 2000]. NPY increases insulin, glucagon, and glucocorticoid secretion [Marke and Waite; 1997; Parikh and Marks, 1997], whereas β-END augments adrenomedullary epinephrine and NE release [Van Loon et al., 1980; Knudtzon, 1986]. Present data show that hypoglycemia alters ARH NPY and POMC protein profiles during ovarian steroid positive-feedback in female rats. As these neuropeptide responses are not reliant on hindbrain ERβ function, they are not a likely outcome of ERβ-associated augmentation of ARH NE and, moreover, may regulate LH and counter-regulatory hormone secretion via mechanisms that do not involve that receptor. These results are of interest in comparison to earlier reports that insulin-induced hypoglycemia did not alter ARH NPY and POMC gene expression in OVX rats replaced with estradiol at basal or tonic negative-feedback levels [Nedungadi and Briski, 2007]; it is thus intriguing to speculate whether NPY and POMC responses to hypoglycemia are modulated by ovarian hormone concentration. Because neuropeptide mRNA expression was not analyzed here, we do not know if hypoglycemic augmentation of these proteins involves transcriptional and/or translational/posttranslational mechanisms.
Present data show that AVPV and ARH prepro-kisspeptin was respectively unresponsive to or elevated by hypoglycemia. Previous studies similarly noted that hypoglycemia elevated ARH, but not medial preoptic area KiSS1 gene expression in rats [Kinsey-Jones et al., 2008], and increased Fos expression in ARH, but not preoptic area KiSS1 neurons in sheep [Fergani et al., 2014]. Yet, AVPV Kiss1 gene expression is decreased after prolonged (48 hours) fasting [Kalamatianos et al., 2008], which suggests that ARH and AVPV kisspeptin neurons may impose effects of acute versus persistent metabolic imbalance on GnRH. Recent studies show that ARH kisspeptin/neurokinin B/dynorphin (KNDy) neurons may regulate the LH surge via dynorphin signaling to the AVPV [Helena et al., 2015]. The present work introduces the novel premise that these cells may mediate rapid hypoglycemic repression of the LH surge. Hypoglycemic amplification of ARH kisspeptin precursor levels may reflect diminished cleavage of this parent molecule to mature, biologically active peptide transmitters; however, this notion is speculative as effects of hypoglycemia on ARH Gpr54 ligand levels were not assessed here. PHTPP-mediated attenuation of hypoglycemic effects on ARH prepro-kisspeptin content signifies that ARH kisspeptin neurons are targets of hindbrain ERβ-driven signaling. Our assumption that elevated ARH NE conveys that signal to ARH prepro-kisspeptin neurons requires further investigation. Kisspeptin is implicated in neural regulation of energy homeostasis [Crown et al., 2007; Navarro and Tena-Sempere, 2011; DeBond and Smith, 2014; Roa and Tena-Sempere, 2014], but effects of this neuropeptide on counter-regulatory hormone secretion are unclear. Exogenous kisspeptin delivery to the brain is reported to stimulate corticosterone release [Csabafi et al., 2013], whereas glucagon reactivity to this neuropeptide has not yet been investigated.
The current work does not address the possibility that observed PHTPP effects on peripheral hormone secretion may reflect, in part, action on extra-brain targets. A possible route of transfer from cerebrospinal fluid to blood involves the hindbrain area postrema, a circumventricular organ that contains ‘leaky’ fenestrated capillaries. A plausible scenario is that direct drug action on pituitary gonadotropes may contribute to augmented LH secretion. However, the role of ERβ in regulating gonadotrope function in our model is controversial as ERβ mRNA levels are reportedly decreased by steroid positive-feedback [Schreihofer et al., 2000], and negative- and positive effects of estradiol on gonadotropin secretion are ER-alpha-dependent [Lindzey et al., 2006]. Likewise, the chance that PHTPP might directly regulate pancreatic islet glucagon and/or adrenal corticosterone secretion also cannot be overlooked.
In summary, current studies provide novel evidence for ERβ-dependent caudal hindbrain regulation of LH and counter-regulatory hormone secretion during hypoglycemia in steroid-primed OVX female rats [Figure 6.]. Discrepant GnRH-I precursor protein and LH secretory responses to PHTPP pretreatment suggest that ERβ-controlled hindbrain signaling may inhibit hypothalamic GnRH output to the pituitary by action on neurotransmitter exocytosis and/or degradation within axon terminals. Results also show that caudal hindbrain ERβ exerts site-specific control of NE activity in forebrain projection sites during hypoglycemia, including the ARH, where prepro-kisspeptin may be a target of this signaling.
Figure 6. Model for Hindbrain ERβ Regulation of the LH Surge during Insulin-Induced Hypoglycemia.
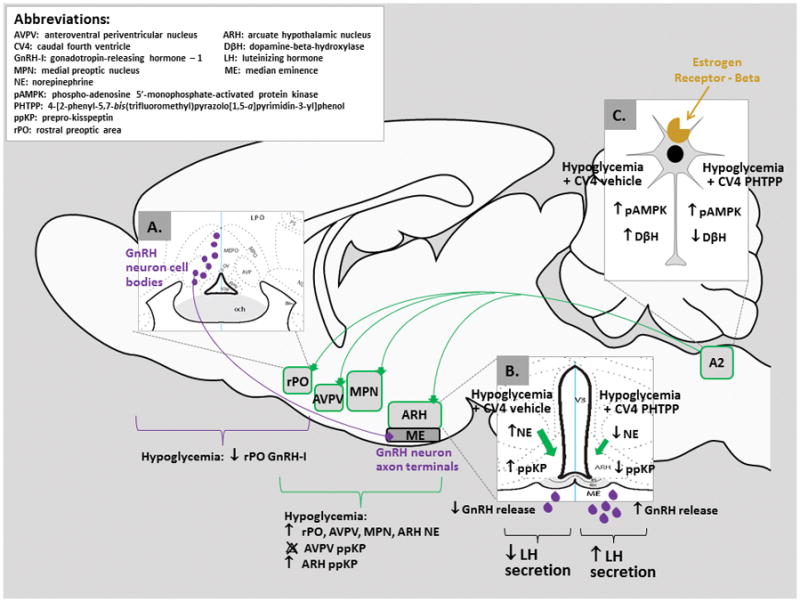
Estrogen- and metabolic-sensitive hindbrain A2 neurons [Insert C] exhibit singular reactivity to hypoglycemia among medullary catecholamine cell groups, and exhibit elevated ERβ protein during this metabolic stress [Ibrahim and Briski, 2015]. Current data show that hypoglycemic augmentation of A2 DβH protein expression and concurrent suppression of the LH surge are each reversed by intra-CV4 administration of the ERβ antagonist PHTPP. GnRH-I content of the rPO, where perikarya of glucoprivic-responsive GnRH neurons reside [Insert A] was diminished by hypoglycemia, but was not normalized in PHTPP- plus insulin-treated rats. Augmenting effects of hypoglycemia on rPO, AVPV, MPN, and ARH NE levels were reversed only in the latter site by PHTPP, where prepro-kisspeptin content was also normalized by that pretreatment [Insert B]. These results suggest that ERβ-dependent mechanisms may repress the LH surge through mechanisms that target axon terminal neurotransmitter storage, metabolism, and/or release.
Highlights.
Hypoglycemia elevates estrogen receptor-beta (ERβ) protein in A2 metabolo-sensory neurons
Hindbrain ERβ antagonism normalizes LH release but not preoptic GnRH-I protein content
The ERβ antagonist PHTPP attenuates counter-regulatory hormone output during hypoglycemia
PHTPP prevents hypoglycemic augmentation of NE in the ARH, but not other forebrain loci
PHTPP normalizes ARH, but not AVPV preprokisspeptin protein profiles in hypoglycemic rats
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alenazi FSH, Ibrahim BA, Briski KP. Estradiol regulates effects of hindbrain AICAR administration on hypothalamic AMPK activity and metabolic neurotransmitter mRNA and protein expression. J Neurosci Res. 2014 doi: 10.1002/jnr.23520. [DOI] [PubMed] [Google Scholar]
- Alenazi FSH, Ibrahim BA, Briski KP. Estradiol regulates effects of hindbrain AICAR administration on hypothalamic AMPK activity and metabolic neurotransmitter mRNA and protein expression. J Neurosci Res. 2014;93:651–659. doi: 10.1002/jnr.23520. [DOI] [PubMed] [Google Scholar]
- Andrew SF, Dinh TT, Ritter S. Localized glucoprivation of hindbrain sites elicits corticosterone and glucagon secretion. Amer J Physiol. 2007;292:R1792–1798. doi: 10.1152/ajpregu.00777.2006. [DOI] [PubMed] [Google Scholar]
- Beck B. Neuropeptides and obesity. Nutrition. 2000;16:916–923. doi: 10.1016/s0899-9007(00)00410-x. [DOI] [PubMed] [Google Scholar]
- Bellin SI, Ritter S. Insulin-induced elevation of hypothalamic norepinephrine turnover persists after glucorestoration unless feeding occurs. Brain Res. 1981;217:327–337. doi: 10.1016/0006-8993(81)90008-1. [DOI] [PubMed] [Google Scholar]
- Beverly JL, De Vries MG, Bouman SD, Arseneau LM. Noradrenergic and GABAergic systems in the medial hypothalamus are activated during hypoglycemia. Amer J Physiol Regul Integr Comp Physiol. 2001;280:R563–569. doi: 10.1152/ajpregu.2001.280.2.R563. [DOI] [PubMed] [Google Scholar]
- Briski KP, Koshy Cherian A, Genabai NK, Vavaiya KV. In situ coexpression of glucose and monocarboxylate transporter mRNAs in metabolic-sensitive dorsal vagal complex catecholaminergic neurons: transcriptional reactivity to insulin-induced hypoglycemia (IIH) and caudal hindbrain glucose or lactate repletion during IIH. Neuroscience. 2009;164:1152–1160. doi: 10.1016/j.neuroscience.2009.08.074. [DOI] [PubMed] [Google Scholar]
- Briski KP, Sylvester PW. Effects of the glucose antimetabolite, 2-deoxy-D-glucose (2-DG), on the LH surge and Fos expression by preoptic GnRH neurons in ovariectomized, steroid-primed rats. J Neuroendocrinol. 1998;10:769–776. doi: 10.1046/j.1365-2826.1998.00262.x. [DOI] [PubMed] [Google Scholar]
- Chen MD, O’Byrne KT, Chiappini SE, Hotchkiss J, Knobil E. Hypoglycemic ‘stress’ and gonadotropin-releasing hormone pulse generator activity in the rhesus monkey: role of the ovary. Neuroendocrinology. 1992;56:666–673. doi: 10.1159/000126291. [DOI] [PubMed] [Google Scholar]
- Cherian A, Briski KP. Quantitative RT PCR and immunoblot analyses reveal acclimated A2 noradrenergic neuron substrate fuel transporter, glucokinase, phospho-AMPK, and dopamine-beta-hydroxylase responses to hypoglycemia. J Neurosci Res. 2011;89:1114–1124. doi: 10.1002/jnr.22632. [DOI] [PubMed] [Google Scholar]
- Clarke IJ, Horton RJE, Doughton BW. Investigation of the mechanism by which insulin-induced hypoglycemia decreases luteinizing hormone secretion in ovariectomized ewes. Endocrinology. 1990;127:1470–1476. doi: 10.1210/endo-127-3-1470. [DOI] [PubMed] [Google Scholar]
- Crosignani PG. Nutrition and reproduction in women. The ESHRE Capri Workshop Human Reproduction Update. 2006;12:193–207. doi: 10.1093/humupd/dmk003. [DOI] [PubMed] [Google Scholar]
- Csabafi K, Jaszberenyi M, Bagosi Z, Liptak N, Telegdy G. Effects of kisspeptin-13 on the hypothalamic-pituitary-adrenal axis, thermoregulation, anxiety, and locomotor activity in rats. Behav Brain Res. 2013;241:56–61. doi: 10.1016/j.bbr.2012.11.039. [DOI] [PubMed] [Google Scholar]
- Crown A, Clifton DK, Steiner RA. Neuropeptide signaling in the integration of metabolism and reproduction. Neuroendocrinology. 2007;86:175–182. doi: 10.1159/000109095. [DOI] [PubMed] [Google Scholar]
- De Bond JA, Smith JT. Kisspeptin and energy balance in reproduction. Reproduction. 2014;147:R53–R63. doi: 10.1530/REP-13-0509. [DOI] [PubMed] [Google Scholar]
- Demling J, Fuchs E, Baumert M, Wuttke W. Preoptic catecholamine, GABA, and glutamate release in ovariectomised and ovariectomised estogen-primed rats utilising a push-pull cannula technique. Neuroendocrinology. 1985;41:212–218. doi: 10.1159/000124180. [DOI] [PubMed] [Google Scholar]
- Fergani C, Routly JE, Jones DN, Pickavance LC, Smith RF, Dobson H. Kisspeptin, c-Fos and CRFR type 2 co-expression in the hypothalamus after insulin-induced hypoglycaemia. Reprod Domest Anim. 2014;49:433–440. doi: 10.1111/rda.12293. [DOI] [PubMed] [Google Scholar]
- Gujar AD, Ibrahim BA, Tamrakar P, Briski KP. Hypoglycemia differentially regulates hypothalamic glucoregulatory neurotransmitter gene and protein expression: Role of caudal dorsomedial hindbrain catecholaminergic input. Neuropeptides. 2013;47:139–147. doi: 10.1016/j.npep.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Helena CV, Toporikova N, Kalil B, Stathopoulos AM, Pogrebna VV, Carolino RO, Anselmo-Franci JA, Bertram R. KNDy neurons modulate the magnitude of the steroid-induced luteinizing hormone surges in ovariectomized rats. Endocrinology. 2015;156:4200–4213. doi: 10.1210/en.2015-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim BA, Briski KP. Role of dorsal vagal complex A2 noradrenergic neurons in hindbrain glucoprivic inhibition of the luteinizing hormone surge in the steroid-primed ovariectomized female rat: Effects of 5-thioglucose on A2 functional biomarker and AMPK activity. Neuroscience. 2014;269:199–214. doi: 10.1016/j.neuroscience.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim BA, Alenazi FSH, Briski KP. Energy status determines hindbrain signal transduction pathway transcriptional reactivity to AMPK in the estradiol-treated ovariectomized female rat. Neuroscience. 2015;284:888–899. doi: 10.1016/j.neuroscience.2014.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim BA, Tamrakar P, Gujar AD, Koshy Cherian A, Briski KP. Caudal fourth ventricular administration of the AMPK activator 5-aminoimiazole-4-carboxamide-riboside regulates glucose and counterregulatory hormone profiles, dorsal vagal complex metabolosensory neuron function, and hypothalamic Fos expression. J Neurosci Res. 2013;91:1226–1238. doi: 10.1002/jnr.23230. [DOI] [PubMed] [Google Scholar]
- Kalamatianos T, Grimshaw SE, Poorun R, Hahn JD, Coen CW. Fasting reduces KiSS-1 expression in the anteroventral periventricular nucleus (AVPV): effects of fasting on the expression of KiSS-1 and neuropeptide Y in the AVPV or arcuate nucleus of female rats. J Neuroendocrinol. 2008;20:1089–1097. doi: 10.1111/j.1365-2826.2008.01757.x. [DOI] [PubMed] [Google Scholar]
- Kalra SP, Crowley WR. Neuropeptide Y: a novel neuroendocrine peptide in the control of pituitary hormone secretion, and its relation to luteinizing hormone. Front Neuroendocrinol. 1992;13:1–46. [PubMed] [Google Scholar]
- Kalra SP. Neural circuits involved in the control of LHRH secretion: a model for estrous cycleregulation. J Steroid Biochem. 1985;23:733–742. doi: 10.1016/s0022-4731(85)80009-1. [DOI] [PubMed] [Google Scholar]
- Kanatani A, Ishihara A, Asahi S, Tanaka T, Ozaki S, Ihara M. Potent neuropeptide Y Y1 receptor antagonist, 129U91: blockade of neuropeptide Y-induced and physiological food intake. Endocrinology. 1996;137:3177–3182. doi: 10.1210/endo.137.8.8754736. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Senba E. Electrophysiological and morphological characterization of cytochemically-defined neurons in the caudal nucleus of tractus solitarius of the rat. Neuroscience. 1999;89:1347–1355. doi: 10.1016/s0306-4522(98)00393-5. [DOI] [PubMed] [Google Scholar]
- Kinsey-Jones JS, Li XF, Knox AM, Wilkinson ES, Zhu XL, Chaudhary AA, Milligan SR, Lightman SL, O’Byrne KT. Down-regulation of hypothalamic kisspeptin and its receptor, Kiss1r, mRNA expression is associated with stress-induced suppression of luteinising hormone secretion in the female rat. J Neuroendocrinol. 2009;21:20–29. doi: 10.1111/j.1365-2826.2008.01807.x. [DOI] [PubMed] [Google Scholar]
- Knudtzon J. Effects of pro-opiomelanocortin-derived peptides on plasma levels of glucagon, insulin, and glucose. Horm Metab Res. 1986;18:579–583. doi: 10.1055/s-2007-1012379. [DOI] [PubMed] [Google Scholar]
- Lindzey J, Jayes FI, Yates MM, Couse JF, Korach KS. The bi-modal effects of estradiol on gonadotropin synthesis and secretion in female mice are dependent upon estrogen receptor-alpha. J Endocrinol. 2006;191:309–317. doi: 10.1677/joe.1.06965. [DOI] [PubMed] [Google Scholar]
- Mohankumar PS, Thyagarajan S, Quadri SK. Correlations of catecholamine release in the medial preoptic area with proestrous surge. Endocrinology. 1994;135:119–126. doi: 10.1210/endo.135.1.8013343. [DOI] [PubMed] [Google Scholar]
- Marke JL, Waite K. Intracerebroventricular neuropeptide Y acutely influences glucose metabolism and insulin sensitivity in the rat. J Neuroendocrinol. 1997;9:99–103. doi: 10.1046/j.1365-2826.1997.00554.x. [DOI] [PubMed] [Google Scholar]
- Murray JF, Adan RA, Walker R, Baker BI, Thody AJ, Nijenhuis WA, Yukitake J, Wilson CA. Melanin-concentrating hormone, melanocortin receptors and regulation of luteinizing hormone release. J Neuroendocrinol. 2000;12:217–223. doi: 10.1046/j.1365-2826.2000.00440.x. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 1984;307:109–116. doi: 10.1016/0006-8993(84)90466-9. [DOI] [PubMed] [Google Scholar]
- Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci. 2009;29:11859–11866. doi: 10.1523/JNEUROSCI.1569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM, Tena-Sempere M. Neuroendocrine control by kisspeptins: role in metabolic regulation of fertility. Nat Rev Endocrinol. 2011;8:40–53. doi: 10.1038/nrendo.2011.147. [DOI] [PubMed] [Google Scholar]
- Nedungadi TP, Briski KP. Effects of estradiol on acute and recurrent insulin-induced hypoglycemia-associated patterns of arcuate neuropeptide Y, proopiomelanocortin, and co-caine- and amphetamine related-transcript gene expression in the ovariectomized rat. Neuroendocrinology. 2007;86:270–276. doi: 10.1159/000109678. [DOI] [PubMed] [Google Scholar]
- Paranjape SA, Briski KP. Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neuroscience. 2005;130:957–970. doi: 10.1016/j.neuroscience.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Parikh R, Marks JL. Metabolic and orexigenic effects of intracerebroventricular neuropeptide Y are attenuated by food deprivation. J Neuroendocrinol. 1997;9:789–795. doi: 10.1046/j.1365-2826.1997.00648.x. [DOI] [PubMed] [Google Scholar]
- Popa SM, Clifton DK, Steiner RA. The role of kisspeptins and GPR54 in the neuroendocrine regulation of reproduction. Annu Rev Physiol. 2008;70:213–238. doi: 10.1146/annurev.physiol.70.113006.100540. [DOI] [PubMed] [Google Scholar]
- Roa J, Tena-Sempere M. Connecting metabolism and reproduction: roles of central energy sensors and key molecular mediators. Mol Cell Endocrinol. 2014;397:4–14. doi: 10.1016/j.mce.2014.09.027. [DOI] [PubMed] [Google Scholar]
- Saleh MC, Connell BJ, Saleh TM. Resveratrol induced neuroprotection is mediated via both estrogen receptor subtypes, ERα and ERβ. Neurosci Lett. 2013;548:217–221. doi: 10.1016/j.neulet.2013.05.057. [DOI] [PubMed] [Google Scholar]
- Schreihofer DA, Stoler MH, Shupnik MA. Differential expression and regulation of estrogen receptors (ERs) in rat pituitary and cell lines: estrogen decreases ERalpha protein and estrogen responsiveness. Endocrinology. 2000;141:2174–2184. doi: 10.1210/endo.141.6.7505. [DOI] [PubMed] [Google Scholar]
- Shrestha PK, Tamrakar P, Ibrahim BA, Briski KP. Hindbrain medulla catecholamine cell group involvement in lactate-sensitive hypoglycemia-associated patterns of hypothalamic norepinephrine and epinephrine activity. Neuroscience. 2014;278:20–30. doi: 10.1016/j.neuroscience.2014.07.033. [DOI] [PubMed] [Google Scholar]
- Shrestha PK, Briski KP. Hindbrain lactate regulates preoptic gonadotropin-releasing hormone (GnRH) neuron GnRH-I protein but not AMPK responses to hypoglycemia in the steroid-primed ovariectomized female rat. Neuroscience. 2015;298:467–474. doi: 10.1016/j.neuroscience.2015.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RM, Hadjimarkou MM, Rossi GC, Pasternak GW, Bodnar RJ. Beta-endorphin-induced feeding: pharmacological characterization suing selective opioid antagonists and antisense probes in rats. J Pharmacol Exp Ther. 2001;297:590–596. [PubMed] [Google Scholar]
- Singh SR, Briski KP. Septopreoptic mu opioid receptor mediation of hindbrain glucoprivic inhibition of reproductive neuroendocrine function in the female rat. Endocrinology. 2004;145:5322–5331. doi: 10.1210/en.2004-0130. [DOI] [PubMed] [Google Scholar]
- Szawka RE, Poletini MO, Leite CM, Bernuci MP, Kalil BK, Mendonca LBD, Carolino ROG, Helena CVV, Bertram R, Franci CR, Anselmo-Franci JA. Release of norepinephrine in the preoptic area activates anteroventral periventricular nucleus neurons and stimulates the surge of luteinizing hormone. Endocrinology. 2013;154:363–374. doi: 10.1210/en.2012-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamrakar P, Ibrahim BA, Gujar AK, Briski KP. Estrogen regulates energy metabolic pathway and upstream AMPK kinase and phosphatase enzyme expression in dorsal vagal complex metabolo-sensory neurons during glucostasis and hypoglycemia. J Neurosci Res. 2015;93:321–332. doi: 10.1002/jnr.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loon GR, Appel NM. Beta-endorphin-induced increases in plasma dopamine, norepinephrine, and epinephrine. Res Commun Chem Pathol Pharmacol. 1980;27:607–610. [PubMed] [Google Scholar]
- Vergoni AV, Bertolini A. Role of melanocortins in the central control of feeding. Eur J Pharmacol. 2000;405:25–32. doi: 10.1016/s0014-2999(00)00538-0. [DOI] [PubMed] [Google Scholar]
- Williams G, Harrold JA, Cutler DJ. The hypothalamus and the regulation of energy homeostasis: lifting the lid on a black box. Proc Nutr Soc. 2000;59:385–396. doi: 10.1017/s0029665100000434. [DOI] [PubMed] [Google Scholar]