

Abstract
Purpose of review
Calcific aortic valve stenosis (AS) affects over 1 million patients in the US. Effective medical therapies do not exist. With the aging of the population and increase in incidence of AS, improved understanding of this disease and novel therapies to reduce the progression of AS and need for aortic valve replacement (AVR) are urgently needed.
Recent findings
Lipoprotein(a) [Lp(a)] is the only monogenetic risk factor for AV calcification and AS. Elevated Lp(a) levels are a strong, causal, independent risk factor for AS, as demonstrated in epidemiological, genome-wide association studies and Mendelian randomization studies. Lp(a) is the major lipoprotein carrier of oxidized phospholipids (OxPL) which are pro-inflammatory and promote calcification of vascular cells, two key pathophysiological drivers of AS. Moreover, Lp(a)-associated enzymes and lipids derived from breakdown of OxPL have been implicated in the pathogenesis of AS. These mechanistic insights likely explain the recent findings that elevated plasma Lp(a) and OxPL on apoB containing lipoproteins (OxPL-apoB) predict progression of pre-existing AS and need for AVR. The failure of the statin trials in AS may be partially explained by the fact that statins, as monotherapy or in combination with ezetimibe, have neutral or Lpa(a) raising effects, as has been shown most recently in the ASTRONOMER trial. Antisense oligonucleotides targeted to apo(a) are in Phase 2 clinical development and shown to lower both Lp(a) and OxPL-apoB.
Summary
Lp(a) and OxPL are key therapeutic targets in AS. Strategies aimed at potent Lp(a) lowering to normalize levels and/or suppress the pro-inflammatory effects of OxPL may be beneficial for preventing progression of AS and need for AVR.
Keywords: lipoprotein(a), apo(a), oxidized phospholipids, aortic stenosis, animal models
Introduction
Calcific aortic valve stenosis (AS) is a progressively debilitating and potentially fatal disease that is currently treated with surgical aortic valve replacement (AVR) in patients who can tolerate surgery or with transcatheter aortic valve replacement in those with high surgical risk. It is estimated that there are over 1 million patients with AS in the US and over 2.5 million worldwide [1]. Affected aortic valve (AV) leaflets are characterized by progressive fibrosis, thickening and most importantly, calcification [2, 3], resulting in decreased leaflet mobility and increased obstruction of blood outflow from the left ventricle. Progressive worsening of AS occurs in the majority of patients, eventually leading to symptoms, such as angina, syncope and heart failure requiring AVR. Approximately 50% of patients are not eligible for AVR due to co-morbidities, and generally develop progressive heart failure and death. Effective medical therapies do not exist, and statins have failed to reduce progression of AVR in four randomized trials [4–7]. Novel therapies to reduce the progression of AS are urgently needed, since as the population ages there will be a corresponding progressive increase in incidence and prevalence of AS. We review the literature examining the role of Lp(a) as a risk factor for AS in clinical studies, as well as evidence describing the mechanisms by which its oxidized phospholipids (OxPL) content may mediate progression of AS.
Lipoprotein(a) is a risk factor for AS
Epidemiology Studies
Lipoprotein(a) [Lp(a)] was initially identified as a risk factor for AV disease based on the results of several epidemiologic and observational studies (summarized in Table 1). Cross-sectional studies have found enrichment of individuals with elevated Lp(a) in those diagnosed with AV sclerosis or stenosis by echocardiography [8, 9]. Case-control studies revealed elevated Lp(a) was an independent risk factor for AS among patients who had AVR [10] and for AV calcification in consecutive patients who had outpatient echocardiograms [11]. Data from the Copenhagen City Heart Study (CCHS) and Copenhagen General Population Study (CGPS) showed an observational hazard ratio (HR) of aortic stenosis of 1.4 (95% confidence interval (CI) 1.2–1.7) for a 10-fold increase in Lp(a) plasma levels [12]. Another analysis of the same cohorts examined both causal and observational associations between AS and Lp(a) and reported an observational HR for AS of 1.23 (CI 1.06–1.41) for each SD increase in Lp(a) [13]. The European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk study found that after adjusting for sex, age, smoking, and low density lipoprotein cholesterol levels, Lp(a) ≥ 50 mg/dL (~125 nmol/L) was associated with a HR of 1.98 (CI 1.25–3.09) for risk of AS, compared with Lp(a) < 50 mg/dL(~125 nmol/L) [14]. In patients with heterozygous familial hypercholesterolemia, Lp(a) was a predictor of AV calcification with an odds ratio per 10 mg/dL (~25 nmol/L) increase in Lp(a) of 1.11 (CI 1.01–1.20) after multivariate analysis including lifelong LDL-C burden [15]. A recent secondary analysis of the prospective Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin (ASTRONOMER) trial involving patients with mild to moderate AS identified elevation of Lp(a) within the top tertile (> 58.5 mg/dL or ~146 nmol/L) as an independent risk factor for the rate of progression of AS and occurrence of AS related events (AVR, death), especially in patients ≤ 57 years [16]. In summary, multiple epidemiological studies provide strong support that elevated Lp(a) levels are associated with AS. Epidemiologically, the risk of AS seems to start at Lp(a) levels of ~30 mg/dL (~75 nmol/L) but substantial risk does not accrue until Lp(a) levels are ~>60 mg/dL (~>150 nmol/L).
Table 1.
Epidemiologic Studies of Lp(a) as a Risk Factor for Aortic Valve Disease
| Author | Year | Study Design | Size | Key Findings |
|---|---|---|---|---|
| Gotoh et al. [8] | 1995 | Cross-Section | 784 | AV sclerosis present in 36.1% of those with Lp(a) > 30 mg/dL and 12.7% of those with Lp(a) < 30 mg/dL. |
| Stewart [9] | 1997 | Cross-Section | 5,201 | Lp(a) elevation is a risk factor for AV sclerosis or stenosis (OR 1.23, CI 1.14–1.32). |
| Glader [10] | 2003 | Case-Control | 101 cases, 101 controls | Lp(a) ≥ 48 mg/dL present in 21/101 cases of AS resulting in AVR vs 5/101 controls (OR 5, CI 1.7–14.6). |
| Bozbas [11] | 2007 | Case-Control | 285 (112 with AV calcification) | Lp(a) levels are higher among those with AV calcification (AVC) (27.4 mg/dL vs 19.9 mg/dL without AVC; p = 0.033). |
| Kamstrup [12] | 2014 | Cohort | 77,680 | Lp(a) levels > 95th percentile (> 90 mg/dL), associated with HR for AS of 2.9 (CI 1.8–4.9); overall observational HR of AS 1.4 (CI 1.2–1.7) for a 10-fold increase in Lp(a) plasma levels. |
| Arsenault [14] | 2014 | Cohort, with case-control replication | 17,553 | Lp(a) ≥ 50 mg/dL is associated with increased risk of AV stenosis, adjusted HR 1.98 (CI 1.25–3.09, p = 0.002), compared with Lp(a) < 50 mg/dL. |
| Capoulade [16] | 2015 | Cohort | 220 | Lp(a) elevation > 58.5 mg/dL is an independent risk factor for increased rate of AS progression, especially in those ≤ 57 years. |
| Langsted [13] | 2015 | Cohort | 100,578 | Observational HR for AV stenosis for an increase in 1 SD of Lp(a): 1.23 (CI 1.06–1.41). |
| Vongpromek [15] | 2015 | Cross-Section | 129 (HeFH) | OR (per 10 mg/dL increase in Lp(a)) of AV calcification: 1.11 (CI 1.01–1.20). |
Genetic Studies
In addition to epidemiologic studies describing a relationship between Lp(a) and AS, a genetically determined relationship exists between Lp(a) levels and calcific AV disease (summarized in Table 2). The single nucleotide polymorphism (SNP) rs10455872 in the gene LPA, encoding for apo(a), was found in a Genome Wide Association Study (GWAS) to be strongly linked to AV calcification as determined by computed tomography. This finding was reported by the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) consortium using data, in the discovery phase of their analysis, from three large cohorts: The Framingham Heart Study (FHS), the Age, Gene/Environment Susceptibility-Reykjavik Study (AGES-RS), and white European participants from the Multi-Ethnic Study of Atherosclerosis (MESA), a total of 6,942 participants. After multivariate adjustments, the SNP rs10455872 (risk allele (G)) was associated with an increased risk of AV calcium with an odds ratio of 2.05 (CI 1.63–2.57). Mendelian randomization analysis within the same study, demonstrated that the LPA SNP rs10455872, associated with elevated plasma Lp(a) and AV calcification in whites and blacks, was a genetically determined risk factor for AV calcification (OR 1.62 (CI 0.94–2.81) per log-nmol/L increase in Lp(a)) and that this risk was likely mediated by the biological activity of Lp(a). Using data from the Malmö Diet and Cancer Study (MDCS) (n = 28,193) and CHS (n = 10,400), an independent association between rs10455872 and incident AS was also demonstrated, with a HR of 1.68 (CI 1.32–2.15) per risk allele reported for the MDCS cohort and a HR of 1.6 (CI 1.12–2.28) for the presence of at least one risk allele reported for the CCHS cohort. Importantly, additional analysis demonstrated that the association of rs10455872 with AV calcium was independent of coronary artery calcium and clinical coronary artery disease [17].
Table 2.
Genetic Studies of Lp(a) as a Risk Factor for Aortic Valve Disease
| Author | Year | Study Design | Size | Key Findings |
|---|---|---|---|---|
| Thanassoulis [17] | 2013 | GWAS, cohort | 6,942 (AVC analysis), 28,193 (MDCS analysis of AS), 10,400 (CCHS analysis of AS) | SNP rs10455872 in LPA is associated with AVC (OR per G allele 2.05 (CI 1.63–2.57, p = 9.0×10−10)) and with incident AS: HR 1.68 per risk allele (CI 1.32–2.15; p = 3×10−5) in MDCS, HR 1.6 for presence of at least one risk allele (CI 1.12–2.28, p = 0.01) in CCHS. |
| Kamstrup [12] | 2014 | Cohort | 77,680 | Relative risk of AV stenosis of 1.6 (95% CI 1.2–2.1) for a 10-fold increase in genetically determined Lp(a) values (includes rs10455872, rs3798220, and KIV-2 repeats). |
| Arsenault [14] | 2014 | Cohort, with case-control replication | 17,553 | Risk of AV stenosis increases with number of rs10455872 G alleles: 1 allele, unadjusted HR 1.78 (CI 1.11–2.87); 2 alleles, unadjusted HR 4.83 (CI 1.77–13.20). Case-control: rs10455872 associated with AV stenosis, OR 1.57 (95% CI 1.10–2.26). |
| Langsted [13] | 2015 | Cohort | 100,578 | Causal risk ratio (CRR) for AS associated with LPA SNPs (rs10455872 and rs3798220) for 1 SD increase in Lp(a): 1.38 (CI 1.23–1.55). For LPA KIV-2, CRR for AS for 1 SD increase in Lp(a): 1.21 (CI 1.06–1.40). |
A Mendelian randomization analysis with LPA SNPs was performed using clinical data containing individual diagnoses for AS from the CGPS and CCHS (n = 77,680). Median Lp(a) levels varied with SNP rs10455872 carrier status (11 mg/dL or ~27.5 nmol/L for non-carriers, 60 mg/dL or ~150 nmol/L for heterozygotes, and 108 mg/dL or ~270 nmol/L for homozygotes). Lp(a) levels were also increased in carriers of SNP rs3798220 and with decreasing numbers of LPA kringle IV type 2 (KIV-2) repeats. Carriers of SNP rs10455872 had a multivariable adjusted HR of 1.6 (CI 1.2–2.0) for heterozygotes and 1.5 (CI 0.5–4.8) for homozygotes (trend, p < 0.001) compared to non-carriers. The rs3798220 SNP was also associated with elevated Lp(a), but its prevalence was too low to rule out whether an association was also present with AS. When including all LPA genotypes (i.e. rs10455872, rs3798220, and KIV-2 percentile group), the authors reported a relative risk of AS of 1.6 (CI 1.2–2.1) for a 10-fold increase in genetically determined Lp(a) values [12]. Using a multidirectional Mendelian randomization design, a study with 100,578 subjects reported a causal risk ratio for AS of 1.38 (CI 1.23–1.55). for each SD increase in Lp(a) based on LPA SNPs (rs10455872 and rs3798220) and 1.21 (CI 1.06–1.40) for each SD increase in Lp(a) associated with LPA KIV-2 genotype [13]. Within the EPIC-Norfolk study (n= 17,553) described above, carriers with one rs10455872 G allele had an unadjusted HR for AS of 1.78 (CI 1.11–2.87), and those with two G alleles had an unadjusted HR for AS of 4.83 (CI 1.77–13.20), compared to non-carriers [14].
In summary, 9 unique clinical studies demonstrated the predictive value of Lp(a) towards risk of developing AS and 4 large genetic studies using Mendelian randomization demonstrated that Lp(a) is a genetically determined, and therefore likely causal mediator of AS. With this in mind, reduction of Lp(a) concentrations, unlike reduction of low-density lipoprotein–cholesterol (LDL-C) with statins, is a promising novel therapeutic approach for AS.
Statins and Lp(a)
Though LDL-C is also a risk factor for AS, LDL-C lowering by statins has not been successful in altering the natural history of the disease. A recent Mendelian randomization analysis of 6942 subjects from the CHARGE consortium and the MDCS demonstrated that a higher LDL-C genetic risk score (GRS), consisting of SNPs specifically associated with LDL-C and weighed by each SNP’s correlation to plasma LDL-C levels, was associated with increased risk of incident AS [18]. Although the LDL-C GRS did not correlate with Lp(a) mass, all conventional measurements “LDL-C” contains the content of Lp(a)-cholesterol (Lp(a)-C), which can be 30–45% of Lp(a) mass [19], and may be a confounding variable in this analysis. Despite the role of LDL-C as a risk factor for AS, and small (n=121) open-label trial demonstrating reduced progression of AS in patients treated with rosuvastatin [20], four randomized placebo-controlled LDL-C lowering trials have been conducted in well established AS with rosuvastatin (ASTRONOMER [6] and Tyrolean Aortic Stenosis Study (TASS) [20]), atorvastatin (Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) [4]), and simvastatin/ezetimibe (Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) [5]). These collectively enrolled 2344 patients ages 58–68 years with mild-moderate AS (ASTRONOMER, TASS, and SEAS) or moderate-severe AS (SALTIRE), and all failed to show reduction in AS progression [21].
One possible explanation for the ineffectiveness of statins in these well-conducted trials is that LDL-C lowering must be initiated at an earlier age or stage of the disease (i.e. aortic sclerosis) in order to prevent AS [22]. However, it is also likely that Lp(a), instead of LDL-C, is the major modifiable risk factor in AS. Using the latter paradigm, it is not surprising that statins were not beneficial for AS. Lp(a), compared to LDL, is relatively refractory to statin therapy. The LPA SNP rs10455872, which is associated with higher Lp(a) levels, has been associated with poorer response to LDL-C lowering by statins [23–25]. As the Lp(a) mass rises, the contribution of Lp(a)-C to measured LDL-C also increases [19], therefore Lp(a) represents a pool of “LDL-C” which is resistant to statin therapy and contributing to AS risk and can confound interpretation of AS risk due to LDL-C. Studies on the precise effects of statins on Lp(a) are controversial, with differences in assay quality compromising the ability to perform robust meta-analyses [26, 27]. In certain statin trials, including those with rosuvastatin, atorvastatin, pravastatin, pitavastatin, and simvastatin/ezetimibe, Lp(a) increases 10–20% with statin therapy [28] (Figure 1). Specifically, in the ASTRONOMER trial Lp(a) increased by 20% (p<0.05) from baseline levels in the 112 out of 220 patients randomized to rosuvastatin and treated for 1 year [16]. Lp(a) increased by 23% (p<0.01) from baseline levels in hyperlipidemic patients treated with simvastatin/ezetimibe [28], which was also the therapeutic intervention studied in SEAS [5]. These findings suggest that the benefits of LDL-C lowering by statins towards aortic stenosis may be offset by a deleterious increase in Lp(a).
Figure 1. Effects of therapeutic interventions on OxPL-apoB and Lp(a) levels.
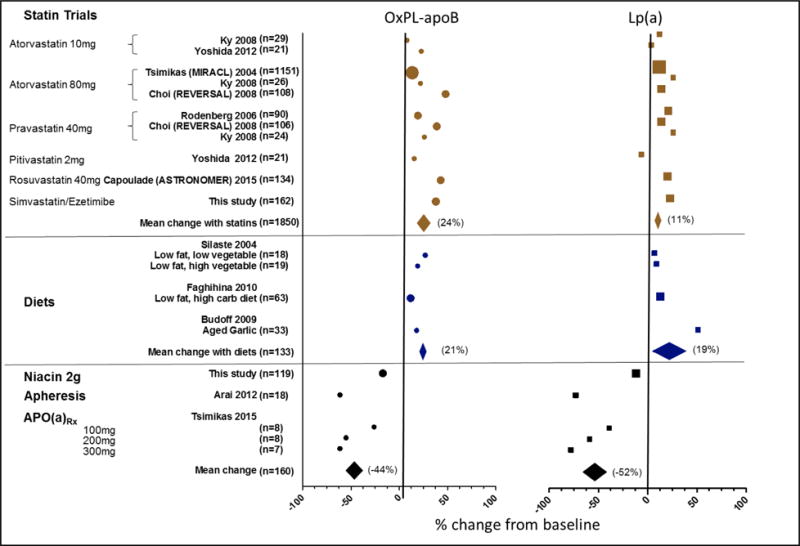
Systematic review of trials with OxPL-apoB and Lp(a) levels following intervention with statins (brown), beneficial diets (blue), and Lp(a) lowering therapies (black). Each filled symbol represents the mean percent change, or the delta mean percent change between the intervention and placebo group where available, from each respective trial. Diamond symbols represent the mean change within each respective interventional category and span the 95% confidence interval. Data from trials with larger of subjects are represented with larger symbol. Reprinted with permission from J Clinical Lipidology [28].
Mechanisms for Lp(a) and OxPL towards AV calcification and the pathogenesis of AS
Early lesions on AV leaflets in AS contain oxidized lipids, apoB and apo(a), macrophages and T-cells [29–32]. Apo(a), via its lysine binding domains can bind to fibrin on denuded or injured endothelium [33–35], such as that on AVs subjected to mechanical stress in vivo, as a likely mechanism for intravasation of Lp(a) into the valve. Thereafter, OxPL present on Lp(a) can mediate pro-inflammatory and pro-calcific pathways potentiating AS (Summarized in Figure 2).
Figure 2. Potential mechanisms for the causal role of Lp(a) and OxPL in AS.
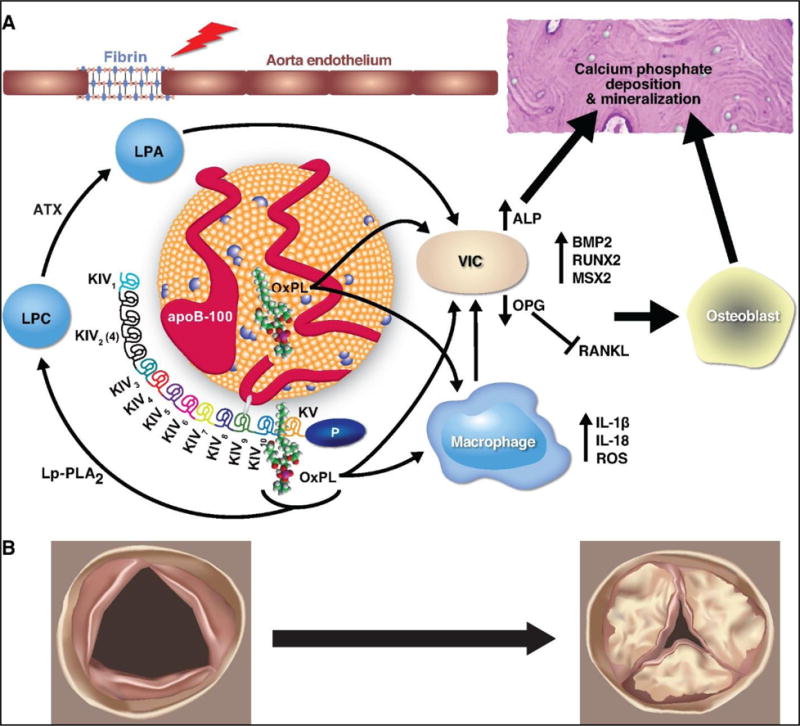
(A) Molecular changes involved in progression of AS. Fibrin is exposed at sites of aorta endothelial injury (depicted by lightning bolt) and can bind to Lp(a), leading to its retention in the valve. Pro-inflammatory lipids on Lp(a), such as OxPL, can promote calcification and bone formation via VIC directly or via up-regulation of ROS and pro-inflammatory cytokines in macrophages. (B) Anatomic changes with progression of a normal valve (left) to a severely stenotic valve (right).
Abbreviations: ATX = autotaxin; Lp-PLA2 = Lipoprotein-associated phospholipase A2; LPC = lysophosphatidylcholine; LPA = lysophosphatidic acid; OxPL = oxidized phospholipids; Lp(a) = lipoprotein(a); ROS = reactive oxygen species; VIC = vascular interstitial cell; ALP = alkaline phosphatase
OxPL is covalently bound to the apo(a) moiety of Lp(a), but is also present in the lipid-phase of the particle [36]. In humans, more than 85% of plasma lipoprotein-associated OxPL are bound to Lp(a) while the remaining exist primarily on apoB containing lipoproteins [37–39]. A second large and independent pool of OxPL present on other proteins is covalently linked to plasminogen, which has no other lipid content [40, 41]. Consistent with the role of this apoB-100 containing lipoprotein as the preferential lipoprotein carrier of OxPL, OxPL on plasma apoB (OxPL-apoB) levels correlate with Lp(a) levels in the population as a whole. However, this is dependent on genetics and underlying apo(a) isoform composition, irrespective of race [42–45]. Further genetic evidence that Lp(a) levels determine plasma OxPL levels include the high genetic covariance of Lp(a) and OxPL-apoB in a study examining these parameters in monozygotic versus dizygotic twins [43], and that the LPA SNPs rs3798220 [45] and rs10455872 [43] are also associated with elevated levels. Lastly, in trials with pharmacologic [16, 38, 46–49] or dietary [50–52] interventions that raise Lp(a), OxPL-apoB is increased as well. Conversely, lipid apheresis [37], and antisense oligonucleotides to apo(a) [53] and niacin [28], all lower OxPL-apoB.
OxPL play a central role in the development of atherosclerosis, which shares a similar pathogenesis with AS, particularly in pro-inflammatory pathways [54, 55]. Using E06, a monoclonal IgM natural antibody that binds to the PC head group of oxidized but not native PC-containing phospholipids [56–58], we have developed and clinically validated an ELISA that detects OxPL on plasma apoB [57, 59], which primarily detects OxPL on Lp(a). Because most of the OxPL in human plasma is bound to Lp(a), the OxPL-apoB assay thus detects the most inflammatory and atherogenic Lp(a). In over 40 publications, we have shown that elevated OxPL-apoB levels predict death/MI/stroke in unselected populations followed prospectively [60, 61], correlate with endothelial dysfunction and progression of coronary calcification [51, 62], predict the progression of femoral/carotid disease [63], coronary artery disease (CAD) [64], and are elevated in patients with ACS [65] and following PCI [66].
Oxidized lipoproteins, which are highly enriched in OxPL [67–69], have been implicated in promoting inflammation, valvular ectopic calcification and bone formation, features that are pronounced in severe AS. OxLDL potentiates macrophage-derived reactive oxygen species (ROS) and cytokine production (including CXCL1, CXCL2, CCL9, CCL5, and IL-1β) via toll-like receptors TLR-2/4 and NFκB signaling, leading to increased oxidative stress and inflammation that can accelerate AS [70–73]. Specifically, monocytes exposed to OxPL on apo(a) up-regulate expression of the inflammatory cytokine IL-8 [74].
Bone formation within the diseased AV is driven by the differentiation of vascular cells into osteoblasts [29] via bone morphogenic protein (BMP) signaling and up-regulation of osteoblastic transcription factors including RUNX2 and MSX2. BMP2 [69, 75–77] as well as RUNX2 [78] and MSX2 [79] expression in vascular cells, including AV derived vascular interstitial cells (VIC), are up-regulated following exposure to OxLDL. OxLDL exposure also suppresses osteoprotegerin [80], an inhibitor of vascular calcification via the RANK-L pathway [81, 82]. Finally, exposure to OxLDL in vitro stimulates extracellular matrix calcium deposition by vascular cells [78, 79, 83, 84] as well as up-regulation of alkaline phosphatase (ALP) [67, 79, 83, 85, 86] promoting mineralization.
In addition to OxPL, Lp(a) associated enzymes and lipids have been implicated in the pathogenesis of calcific AS. Lipoprotein-associated phospholipase A2 (Lp-PLA2), enriched on Lp(a) [87] hydrolyzes OxPL to yield a free oxidized fatty acid and lysophosphatidylcholine (LPC). Lp-PLA2 expression and activity levels are higher in mineralized AVs compared to controls [88] and Lp-PLA2 co-localizes with Lp(a) in human calcific AVs [89]. Moreover, LPC promotes mineralization of VICs in culture [89]. Autotaxin (ATX), another enzyme present on Lp(a), hydrolyzes LPC to form lysophosphatidic acid (LPA), and has also been found in diseased valves in tight proximity to apo(a) [89]. Administration of exogenous LPA to a mouse model of AS resulted in higher peak velocities across the AV and more AV calcification compared with controls [89]. These findings consistently support the role of Lp(a) as a vehicle for harmful substrates that promote AS. Whether Lp(a), its associated OxPL or its metabolites will be viable therapeutic targets to mitigate AS remains to be determined.
Lp(a) and OxPL are Associated with Increased AS progression
Elevated Lp(a) and OxPL-apoB were predictive of a worse outcome in the ASTRONOMER trial, originally designed to evaluate the role of rosuvastatin in preventing the progression of AS in those with mild-moderate AS followed for 3.5 years [6]. In a prospective analysis of 220/269 subjects from the original cohort with baseline Lp(a) and OxPL-apoB measurements, those with the highest tertile of Lp(a) (>58.5 mg/dl or ~146 nmol/L) had AS which progressed ~1.5 times faster (average peak velocity ± SD) (0.26 m/s/yr ± 0.03 vs 0.17 m/s/yr ± 0.02) [Figure 3] and had ~2 fold increased risk of a composite outcome of AVR (n = 47 in the cohort) and cardiac death (n = 2 in the cohort) [16]. This relationship was the same between individuals with tricuspid or bicuspid valves (consisting of 48% of subjects in this trial), but the risk of high Lp(a) on AS progression and AVR was more pronounced in those younger than 57 years of age. The risk of AS progression conveyed by elevated Lp(a) levels in this cohort of subjects starting out with mild-moderate disease actually more closely reflects individuals with moderate-severe AS (Figure 3). Of great relevance, those with the highest tertile of OxPL-apoB (>5.50 nM) as well as OxPL on apo(a) [OxPL-apo(a)] (>33.5 nM) had increased risk of AS progression and AVR which was nearly identical to those with the highest tertile of Lp(a), consistent with the thesis that OxPL carried by Lp(a) is responsible with the biological activity of the particle.
Figure 3. Rates of AS progression in stain trials.
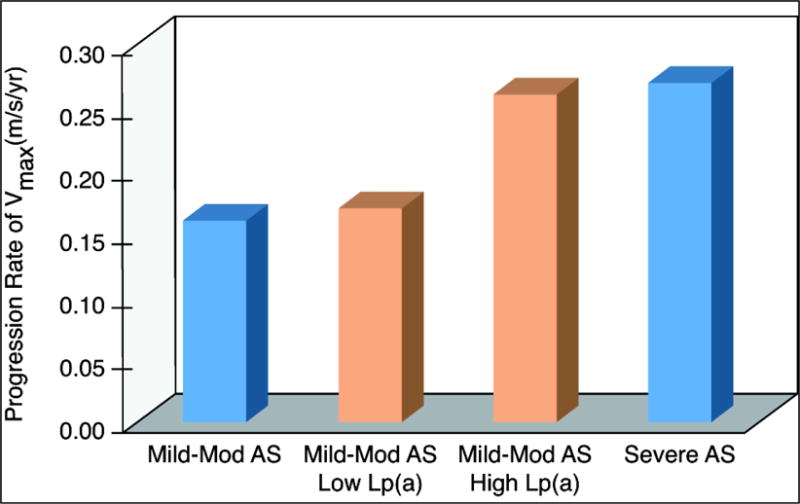
Annual rates of progression in those with mild-moderate (mod) AS (composite estimate using data from ASTRONOMER [6], SEAS [5], and TASS [20]), those with mild-mod AS and low Lp(a) and high Lp(a) [orange bars] (data from Capoulade et al. [16]), and those with severe AS (estimated using data from SALTIRE [4]).
Future directions
There is a growing body of literature describing the role of Lp(a) as a genetically determined, causal risk factor for AS. However, two major questions linger: 1) what are the precise mechanisms linking Lp(a) to progression of AS? and, 2) will Lp(a) lowering attenuate AV calcification and the progression of AS?
We are only beginning to understand the mechanisms by which Lp(a) promotes the progression of AS. The relative contributions of lipids associated with Lp(a) implicated in AS, namely OxPL, PLC, and LPA remain to be characterized. One approach towards this question would be to quantify and compare OxPL, PLC, and LPA levels in human stenotic AVs and from human AVs without significant disease (i.e. from explanted hearts from patients undergoing cardiac transplant) using liquid chromatography-mass spectrophotometry (LC-MS). This could be complemented by quantification of OxPL, PLC, and LPA on purified plasma Lp(a) in comparison to other lipoproteins such as LDL, also feasible by LC-MS, from subjects with AS enrolled in long term prospective studies tracking progression of the disease.
Whether Lp(a)-associated OxPL or its metabolites are directly responsible for calcific AS can be tested in vivo using animal models. To date, the only animal models that develop clinically significant calcific AS are aged transgenic mice on an apoB100 only/LDLR−/− background, fed a high cholesterol diet [89–91]. Based on these existing models, we have generated transgenic hyperlipidemic Lp(a) mice which express human apoB-100 and apo(a) [92] on an apoB100 only/LDLR−/− background. These models, along with transgenic models resulting in varying OxPL and/or Lp(a) levels in plasma may be useful in studying mechanisms of calcific AS development and progression.
Trials evaluating the effect of Lp(a) lowering on AS progression are currently underway. Niacin [93], CETP inhibitors such as anacetrapib [94], PCSK9 inhibitors including alirocumab and evolocumab [19] all lower plasma Lp(a), though only moderately (by 20–40%), and have diverse metabolic effects including alterations in LDL-C, HDL-C and triglycerides. Participants are currently being recruited for a randomized, double-blind placebo control trial designed to assess the effect of extended release niacin (1.5–2 g/day) on aortic valve disease progression over 2 years in those > 50 years and < 85 years with aortic sclerosis or mild AS and Lp(a) > 50 mg/dL (~125 nmol/L). The primary outcome is AV calcium score progression by cardiac CT, although the rate of hemodynamic progression of AS will be determined by echocardiography at 1 and 2 years [95]. Antisense oligonucleotides (ASO) targeted against mRNA encoding apo(a) [IONIS-APO(a)Rx] (previously called ISIS-APO(a)Rx), specifically lowered Lp(a) and its associated OxPL by up to 89% and 93%, respectively, in a recent Phase I trial [53]. Results from clinical trials in subjects with existing AS randomized to antisense to apo(a) or placebo will add valuable knowledge regarding the mechanism and role of Lp(a) and OxPL in AS and the efficacy of this novel therapeutic approach.
Conclusion
Lp(a) is a prevalent, genetically determined causal risk factor for calcific AS and its pro-inflammatory and pro-calcific lipid cargo, including OxPL, are likely mechanistically linked to the development of calcific AS. Elevated Lp(a) (>58.5 mg/dl or ~146 nmol/L) and OxPL-apoB (>5.50 nM) levels predict faster rate of AS progression as well as need for AVR. Whereas LDL-C lowering by statins has not been effective in altering the natural progression of AS, trials with potent Lp(a) lowering therapies including IONIS-APO(a)-LRx represent a promising novel therapeutic approach for AS.
Key Points.
Lp(a) is a genetically determined, causal, independent risk factor for calcific AS.
Lp(a) is the major lipoprotein carrier of OxPL, a set of bioactive lipids which likely contribute to AV inflammation and calcification in AS.
Elevated Lp(a) [>58.5 mg/dl)] and OxPL-apoB (>5.50 nM) levels predict faster rate of progression of AS and need for AVR.
Statins, which may raise plasma Lp(a) and OxPL-apoB, have not been shown to alter the natural progression of AS in clinical trials.
Antisense oligonucleotides targeted to apo(a) potently reduce Lp(a) and OxPL-apoB levels and can be tested to reduce the progression of AS and need for AVR.
Acknowledgments
We thank Tracy Reigle of Ionis Pharmaceuticals for preparation of the figures.
Financial support and sponsorship
None
Footnotes
Conflicts of interest
ST is a co-inventor of and receives royalties from patents or patent applications owned by the University of California San Diego and has a dual appointment at UCSD and Ionis Pharmaceuticals, Inc. The other authors report no conflicts.
References
- 1.Yutzey KE, Demer LL, Body SC, et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. 2014;34:2387–2393. doi: 10.1161/ATVBAHA.114.302523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Otto CM, Prendergast B. Aortic-valve stenosis–from patients at risk to severe valve obstruction. N Engl J Med. 2014;371:744–756. doi: 10.1056/NEJMra1313875. [DOI] [PubMed] [Google Scholar]
- 3##.Pawade TA, Newby DE, Dweck MR. Calcification in Aortic Stenosis. The Skeleton Key. J Am Coll Cardiol. 2015;66:561–577. doi: 10.1016/j.jacc.2015.05.066. Excellent review article on the role of calcification in aortic stenosis. [DOI] [PubMed] [Google Scholar]
- 4.Cowell SJ, Newby DE, Prescott RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 5.Rossebo AB, Pedersen TR, Boman K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 6.Chan KL, Teo K, Dumesnil JG, et al. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 7.Dichtl W, Alber HF, Feuchtner GM, et al. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg) Am J Cardiol. 2008;102:743–748. doi: 10.1016/j.amjcard.2008.04.060. [DOI] [PubMed] [Google Scholar]
- 8.Gotoh T, Kuroda T, Yamasawa M, et al. Correlation between lipoprotein(a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study) Am J Cardiol. 1995;76:928–932. doi: 10.1016/s0002-9149(99)80263-x. [DOI] [PubMed] [Google Scholar]
- 9.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 10.Glader CA, Birgander LS, Soderberg S, et al. Lipoprotein(a), Chlamydia pneumoniae, leptin and tissue plasminogen activator as risk markers for valvular aortic stenosis. Eur Heart J. 2003;24:198–208. doi: 10.1016/s0195-668x(02)00385-8. [DOI] [PubMed] [Google Scholar]
- 11.Bozbas H, Yildirir A, Atar I, et al. Effects of serum levels of novel atherosclerotic risk factors on aortic valve calcification. J Heart Valve Dis. 2007;16:387–393. [PubMed] [Google Scholar]
- 12#.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. Mendelian randomization analysis within the CGPS and CCHS cohorts (n = 77,680) demonstrating an Lp(a) dose dependent risk for AS, with a hazard ratio of 1.6 for each 10-fold increase in genetically determined Lp(a) levels associated with the LPA SNPs rs10455872 and rs3798220 or KVI-2 repeats. [DOI] [PubMed] [Google Scholar]
- 13.Langsted A, Varbo A, Kamstrup PR, Nordestgaard BG. Elevated Lipoprotein(a) Does Not Cause Low-Grade Inflammation Despite Causal Association With Aortic Valve Stenosis and Myocardial Infarction: A Study of 100,578 Individuals from the General Population. J Clin Endocrinol Metab. 2015;100:2690–2699. doi: 10.1210/jc.2015-1096. [DOI] [PubMed] [Google Scholar]
- 14.Arsenault BJ, Boekholdt SM, Dube MP, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400. [DOI] [PubMed] [Google Scholar]
- 15.Vongpromek R, Bos S, Ten Kate GJ, et al. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J Intern Med. 2015;278:166–173. doi: 10.1111/joim.12335. [DOI] [PubMed] [Google Scholar]
- 16##.Capoulade R, Chan KL, Yeang C, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66:1236–1246. doi: 10.1016/j.jacc.2015.07.020. This study demonstrated that elevated Lp(a) [>58.5 mg/dl] as well as OxPL-apoB (>5.50 nM) are each associated with 1.5 times faster progression of AS and a 2 fold increased risk of AVR and death, in a secondary analysis of 220 patients from the ASTRONOMER trial followed for an average of 3.5 years. [DOI] [PubMed] [Google Scholar]
- 17.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JG, Luk K, Schulz CA, et al. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA. 2014;312:1764–1771. doi: 10.1001/jama.2014.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeang C, Witztum JL, Tsimikas S. ‘LDL-C’ = LDL-C + Lp(a)-C: implications of achieved ultra-low LDL-C levels in the proprotein convertase subtilisin/kexin type 9 era of potent LDL-C lowering. Curr Opin Lipidol. 2015;26:169–178. doi: 10.1097/MOL.0000000000000171. [DOI] [PubMed] [Google Scholar]
- 20.Moura LM, Ramos SF, Zamorano JL, et al. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–561. doi: 10.1016/j.jacc.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teo KK, Corsi DJ, Tam JW, et al. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol. 2011;27:800–808. doi: 10.1016/j.cjca.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Antonini-Canterin F, Hirsu M, Popescu BA, et al. Stage-related effect of statin treatment on the progression of aortic valve sclerosis and stenosis. Am J Cardiol. 2008;102:738–742. doi: 10.1016/j.amjcard.2008.04.056. [DOI] [PubMed] [Google Scholar]
- 23.Postmus I, Trompet S, Deshmukh HA, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5:5068. doi: 10.1038/ncomms6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donnelly LA, van Zuydam NR, Zhou K, et al. Robust association of the LPA locus with low-density lipoprotein cholesterol lowering response to statin treatment in a meta-analysis of 30 467 individuals from both randomized control trials and observational studies and association with coronary artery disease outcome during statin treatment. Pharmacogenet Genomics. 2013;23:518–525. doi: 10.1097/FPC.0b013e3283642fd6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deshmukh HA, Colhoun HM, Johnson T, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: importance of Lp(a) J Lipid Res. 2012;53:1000–1011. doi: 10.1194/jlr.P021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tziomalos K, Athyros VG, Wierzbicki AS, Mikhailidis DP. Lipoprotein a: where are we now? Curr Opin Cardiol. 2009;24:351–357. doi: 10.1097/HCO.0b013e32832ac21a. [DOI] [PubMed] [Google Scholar]
- 28.Yeang C, Hung MY, Byun YS, Clopton P, Yang X, Witztum JL, Tsimikas S. Effect of Therapeutic Interventions on Oxidized Phospholipids on Apolipoprotein B-100 and Lipoprotein(a) In Press. J Clin Lipidol. 2015 doi: 10.1016/j.jacl.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–1863. doi: 10.1016/j.jacc.2012.02.093. [DOI] [PubMed] [Google Scholar]
- 30.Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol. 1999;19:1218–1222. doi: 10.1161/01.atv.19.5.1218. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien KD, Reichenbach DD, Marcovina SM, et al. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol. 1996;16:523–532. doi: 10.1161/01.atv.16.4.523. [DOI] [PubMed] [Google Scholar]
- 32.Otto CM, Kuusisto J, Reichenbach DD, et al. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 33.Lou XJ, Boonmark NW, Horrigan FT, et al. Fibrinogen deficiency reduces vascular accumulation of apolipoprotein(a) and development of atherosclerosis in apolipoprotein(a) transgenic mice. Proc Natl Acad Sci U S A. 1998;95:12591–12595. doi: 10.1073/pnas.95.21.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes SD, Lou XJ, Ighani S, et al. Lipoprotein(a) vascular accumulation in mice. In vivo analysis of the role of lysine binding sites using recombinant adenovirus. J Clin Invest. 1997;100:1493–1500. doi: 10.1172/JCI119671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loscalzo J, Weinfeld M, Fless GM, Scanu AM. Lipoprotein(a), fibrin binding, and plasminogen activation. Arterioscler Thromb Vasc Biol. 1990;10:240–245. doi: 10.1161/01.atv.10.2.240. [DOI] [PubMed] [Google Scholar]
- 36.Leibundgut G, Scipione C, Yin H, et al. Determinants of binding of oxidized phospholipids on apolipoprotein(a) and lipoprotein(a) J Lipid Res. 2013;54:2815–2830. doi: 10.1194/jlr.M040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arai K, Orsoni A, Mallat Z, et al. Acute impact of apheresis on oxidized phospholipids in patients with familial hypercholesterolemia. J Lipid Res. 2012;53:1670–1678. doi: 10.1194/jlr.P027235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida H, Shoda T, Yanai H, et al. Effects of pitavastatin and atorvastatin on lipoprotein oxidation biomarkers in patients with dyslipidemia. Atherosclerosis. 2013;226:161–164. doi: 10.1016/j.atherosclerosis.2012.10.069. [DOI] [PubMed] [Google Scholar]
- 39.Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Edelstein C, Pfaffinger D, Yang M, et al. Naturally occurring human plasminogen, like genetically related apolipoprotein(a), contains oxidized phosphatidylcholine adducts. Biochim Biophys Acta. 2010;1801:738–745. doi: 10.1016/j.bbalip.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leibundgut G, Arai K, Orsoni A, et al. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J Am Coll Cardiol. 2012;59:1426–1437. doi: 10.1016/j.jacc.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsimikas S, Clopton P, Brilakis ES, et al. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: results from the Dallas Heart Study. Circulation. 2009;119:1711–1719. doi: 10.1161/CIRCULATIONAHA.108.836940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao F, Schork AJ, Maihofer AX, et al. Heritability of Biomarkers of Oxidized Lipoproteins: Twin Pair Study. Arterioscler Thromb Vasc Biol. 2015;35:1704–1711. doi: 10.1161/ATVBAHA.115.305306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byun YS, Lee JH, Arsenault BJ, et al. Relationship of oxidized phospholipids on apolipoprotein B-100 to cardiovascular outcomes in patients treated with intensive versus moderate atorvastatin therapy: The TNT trial. J Am Coll Cardiol. 2015;65:1286–1295. doi: 10.1016/j.jacc.2015.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arai K, Luke MM, Koschinsky ML, et al. The I4399M variant of apolipoprotein(a) is associated with increased oxidized phospholipids on apolipoprotein B-100 particles. Atherosclerosis. 2010;209:498–503. doi: 10.1016/j.atherosclerosis.2009.09.077. [DOI] [PubMed] [Google Scholar]
- 46.Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol. 2008;51:1653–1662. doi: 10.1016/j.jacc.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 47.Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol. 2008;52:24–32. doi: 10.1016/j.jacc.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 48.Rodenburg J, Vissers MN, Wiegman A, et al. Oxidized low-density lipoprotein in children with familial hypercholesterolemia and unaffected siblings: effect of pravastatin. J Am Coll Cardiol. 2006;47:1803–1810. doi: 10.1016/j.jacc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 49.Tsimikas S, Witztum JL, Miller ER, et al. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the MIRACL trial. Circulation. 2004;110:1406–1412. doi: 10.1161/01.CIR.0000141728.23033.B5. [DOI] [PubMed] [Google Scholar]
- 50.Faghihnia N, Tsimikas S, Miller ER, et al. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J Lipid Res. 2010;51:3324–3330. doi: 10.1194/jlr.M005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Budoff MJ, Ahmadi N, Gul KM, et al. Aged garlic extract supplemented with B vitamins, folic acid and L-arginine retards the progression of subclinical atherosclerosis: a randomized clinical trial. Prev Med. 2009;49:101–107. doi: 10.1016/j.ypmed.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Silaste ML, Rantala M, Alfthan G, et al. Changes in dietary fat intake alter plasma levels of oxidized low-density lipoprotein and lipoprotein(a) Arterioscler Thromb Vasc Biol. 2004;24:498–503. doi: 10.1161/01.ATV.0000118012.64932.f4. [DOI] [PubMed] [Google Scholar]
- 53.Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–1483. doi: 10.1016/S0140-6736(15)61252-1. [DOI] [PubMed] [Google Scholar]
- 54.Miller YI, Choi S-H, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circulation research. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horkko S, Bird DA, Miller E, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palinski W, Horkko S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–814. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedman P, Horkko S, Steinberg D, et al. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem. 2002;277:7010–7020. doi: 10.1074/jbc.M108860200. [DOI] [PubMed] [Google Scholar]
- 59.Wu R, de Faire U, Lemne C, et al. Autoantibodies to OxLDL Are Decreased in Individuals With Borderline Hypertension. Hypertension. 1999;33:53–59. doi: 10.1161/01.hyp.33.1.53. [DOI] [PubMed] [Google Scholar]
- 60.Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 61.Kiechl S, Willeit J, Mayr M, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27:1788–1795. doi: 10.1161/ATVBAHA.107.145805. [DOI] [PubMed] [Google Scholar]
- 62.Ahmadi N, Tsimikas S, Hajsadeghi F, et al. Relation of oxidative biomarkers, vascular dysfunction, and progression of coronary artery calcium. Am J Cardiol. 2010;105:459–466. doi: 10.1016/j.amjcard.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 63.Tsimikas S, Kiechl S, Willeit J, et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the Bruneck study. J Am Coll Cardiol. 2006;47:2219–2228. doi: 10.1016/j.jacc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 64.Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 65.Tsimikas S, Bergmark C, Beyer RW, et al. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J Am Coll Cardiol. 2003;41:360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- 66.Tsimikas S, Lau HK, Han KR, et al. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 2004;109:3164–3170. doi: 10.1161/01.CIR.0000130844.01174.55. [DOI] [PubMed] [Google Scholar]
- 67.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 68.Maziere C, Louvet L, Gomila C, et al. Oxidized low density lipoprotein decreases Rankl-induced differentiation of osteoclasts by inhibition of Rankl signaling. J Cell Physiol. 2009;221:572–578. doi: 10.1002/jcp.21886. [DOI] [PubMed] [Google Scholar]
- 69.Derwall M, Malhotra R, Lai CS, et al. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:613–622. doi: 10.1161/ATVBAHA.111.242594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsimikas S, Miller YI. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr Pharm Des. 2011;17:27–37. doi: 10.2174/138161211795049831. [DOI] [PubMed] [Google Scholar]
- 71.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Afonyushkin T, Oskolkova OV, Philippova M, et al. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol. 2010;30:1007–1013. doi: 10.1161/ATVBAHA.110.204354. [DOI] [PubMed] [Google Scholar]
- 73.Bochkov VN. Inflammatory profile of oxidized phospholipids. Thromb Haemost. 2007;97:348–354. [PubMed] [Google Scholar]
- 74#.Scipione CA, Sayegh SE, Romagnuolo R, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a) J Lipid Res. 2015;56:2273–2285. doi: 10.1194/jlr.M060210. The authors show that apo(a) containing OxPL induces pro-inflammatory IL-8 expression, wheras mutant apo(a) and apo(a) treated with Lp-PLA2, both lacking OxPL, do not upregulate this cytokine. This is one example of how OxPL reflects the biological activity of Lp(a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cola C, Almeida M, Li D, et al. Regulatory role of endothelium in the expression of genes affecting arterial calcification. Biochem Biophys Res Commun. 2004;320:424–427. doi: 10.1016/j.bbrc.2004.05.181. [DOI] [PubMed] [Google Scholar]
- 76.Su X, Ao L, Shi Y, et al. Oxidized low density lipoprotein induces bone morphogenetic protein-2 in coronary artery endothelial cells via Toll-like receptors 2 and 4. J Biol Chem. 2011;286:12213–12220. doi: 10.1074/jbc.M110.214619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nadlonek NA, Lee JH, Weyant MJ, et al. ox-LDL induces PiT-1 expression in human aortic valve interstitial cells. J Surg Res. 2013;184:6–9. doi: 10.1016/j.jss.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goettsch C, Rauner M, Hamann C, et al. Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells. Diabetologia. 2011;54:2690–2701. doi: 10.1007/s00125-011-2219-0. [DOI] [PubMed] [Google Scholar]
- 79.Liao L, Zhou Q, Song Y, et al. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS One. 2013;8:e82379. doi: 10.1371/journal.pone.0082379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Syvaranta S, Alanne-Kinnunen M, Oorni K, et al. Potential pathological roles for oxidized low-density lipoprotein and scavenger receptors SR-AI, CD36, and LOX-1 in aortic valve stenosis. Atherosclerosis. 2014;235:398–407. doi: 10.1016/j.atherosclerosis.2014.05.933. [DOI] [PubMed] [Google Scholar]
- 81.Bennett BJ, Scatena M, Kirk EA, et al. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2006;26:2117–2124. doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- 82.Morony S, Tintut Y, Zhang Z, et al. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice. Circulation. 2008;117:411–420. doi: 10.1161/CIRCULATIONAHA.107.707380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parhami F, Morrow AD, Balucan J, et al. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 84.Yan J, Stringer SE, Hamilton A, et al. Decorin GAG synthesis and TGF-beta signaling mediate Ox-LDL-induced mineralization of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2011;31:608–615. doi: 10.1161/ATVBAHA.110.220749. [DOI] [PubMed] [Google Scholar]
- 85.Palinski W, Yla-Herttuala S, Rosenfeld ME, et al. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10:325–335. doi: 10.1161/01.atv.10.3.325. [DOI] [PubMed] [Google Scholar]
- 86.Tintut Y, Patel J, Territo M, et al. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- 87.Blencowe C, Hermetter A, Kostner GM, Deigner HP. Enhanced association of platelet-activating factor acetylhydrolase with lipoprotein (a) in comparison with low density lipoprotein. J Biol Chem. 1995;270:31151–31157. doi: 10.1074/jbc.270.52.31151. [DOI] [PubMed] [Google Scholar]
- 88.Mahmut A, Boulanger MC, El Husseini D, et al. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: implications for valve mineralization. J Am Coll Cardiol. 2014;63:460–469. doi: 10.1016/j.jacc.2013.05.105. [DOI] [PubMed] [Google Scholar]
- 89##.Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation. 2015;132:677–690. doi: 10.1161/CIRCULATIONAHA.115.016757. This study demonstrated that Lp(a) is present in stenotic human AVs and co-localized with OxPL, Lp-PLA2 and autotaxin (an enzyme that hydrolizes lysophosphatidylcholine to lysophosphatidic acid (LPA)). LPA induces calcification by vascular interstitial cells in vitro and promotes AV calcification in a mouse model of AS. These findings add to the understanding of how Lp(a) and its lipid cargo mediate calcific AS. [DOI] [PubMed] [Google Scholar]
- 90.Miller JD, Weiss RM, Heistad DD. Calcific Aortic Valve Stenosis: Methods, Models, and Mechanisms. Circ Res. 2011;108:1392–1412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miller JD, Weiss RM, Serrano KM, et al. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schneider M, Witztum JL, Young SG, et al. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low density lipoproteins. J Lipid Res. 2005;46:769–778. doi: 10.1194/jlr.M400467-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes) J Am Coll Cardiol. 2013;62:1575–1579. doi: 10.1016/j.jacc.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 95.ClinicalTrials.gov. Thanassoulis G. Early Aortic Valve Lipoprotein(a) Lowering Trial (EAVaLL): A Pilot, Randomized Controlled-trial of Lipoprotein(a) Lowering for the Prevention of Aortic Valve Disease-translating Genomic Knowledge for Cardiovascular Prevention. Updated April 10, 2015. https://clinicaltrials.gov/ct2/show/study/NCT02109614. Accessed February 11, 2016.