

Abstract
As clinical nanomedicine has emerged over the past two decades, phototherapeutic advancements using nanotechnology have also evolved and impacted disease management. Because of unique features attributable to the light activation process of molecules, photonanomedicine (PNM) holds significant promise as a personalized, image-guided therapeutic approach for cancer and non-cancer pathologies. The convergence of advanced photochemical therapies such as photodynamic therapy (PDT) and imaging modalities with sophisticated nanotechnologies is enabling the ongoing evolution of fundamental PNM formulations, such as Visudyne®, into progressive forward-looking platforms that integrate theranostics (therapeutics and diagnostics), molecular selectivity, the spatiotemporally controlled release of synergistic therapeutics, along with regulated, sustained drug dosing. Considering that the envisioned goal of these integrated platforms is proving to be realistic, this review will discuss how PNM has evolved over the years as a preclinical and clinical amalgamation of nanotechnology with PDT. The encouraging investigations that emphasize the potent synergy between photochemistry and nanotherapeutics, in addition to the growing realization of the value of these multi-faceted theranostic nanoplatforms, will assist in driving PNM formulations into mainstream oncological clinical practice as a necessary tool in the medical armamentarium.
Introduction - The evolution of photonanomedicine
Over the past two decades, nanotechnology has experienced a rapid diversification in its potential applications. Starting out as a pure physical and materials science study of nanoscale crystalline and composite materials, the realization that nanotechnology could spearhead new approaches to overcome hurdles in pharmaceutics and in clinical disease management, paved the way for a new era of research into medical nanotechnology: nanomedicine. In many ways, the eagerly-anticipated clinical impact of nanomedicine has been somewhat delayed by the increasing evidence that some materials, as attractive as their utility might be, exert complex effects on the body. Research into improving the clinical translatability of nanomedicines has branched into multiple new avenues of exploration, where a selection of the vast existing pool of nanomaterials are fine-tuned to improve biocompatibility, tolerance and physiological efficacy. This capacity for precise alterations of the nanomaterial composition and the respective surface properties is one of the main attractions of exploring their use as novel medicines. From a chemical standpoint, the macromolecular and supramolecular modification of nanomaterials and their precursors is evolving as a new complementary perspective to more traditional synthetic chemistry approaches for small molecule therapeutics development. The explosion in peer-reviewed publications and patents reporting novel nanomaterials proposed for specific niches within the clinic serves as a direct illustration of the collective opinion held by many researchers that nanomedicine can address multiple longstanding unmet clinical needs.
According to the European Patent Office, there are 55,830 patents filed globally to date that relate to nanoparticle technologies, in addition to the 16,908 patents filed covering liposomal technologies.1 Globally, there are a total of 209 ongoing and completed clinical trials using nanoparticles for various applications in oncology, ophthalmology, dental medicine and infectious disease, amongst numerous others.2 Of those, 82 are ongoing and only 3% have been withdrawn prior to enrollment due to funding limitations and poor patient recruitment. Moreover, there are 1,641 ongoing and completed clinical trials globally using liposomes, a clinically-accepted class of nanosized drug delivery vehicles that simultaneously encapsulate hydrophilic and lipophilic agents.3, 4 Figure 1 shows the global demographic of the five largest contributors to clinical trials of both nanoparticles and liposomes to date.
Figure 1.

Distribution of the total number of clinical trials using nanoparticles and liposomes in the five highest contributing regions of the world.5
Into this mix of innovative approaches to nanomedicine has emerged photonanomedicine (PNM), which incorporates light-activatable entities in nanomaterials with potential for exquisite spatiotemporal control. This review will discuss the progress, hurdles and prospects of PNM formulations in clinical cancer diagnostics and medicine. A specific emphasis will be placed on one of the most widely explored, photochemistry-driven process in PNM, photodynamic therapy (PDT).6, 7 PDT is based on the energy-specific activation of visible and near-infrared (NIR) absorbing molecules, photosensitizers (PSs), to generate reactive molecular species (RMS) that are phototoxic to the target disease tissues, as shown in Figure 2 (A).
Figure 2.

A) A schematic representation of the Jablonski diagram showing how PDT and fluorescence induced is by the irradiation of a PS. The PS in the ground state (S0) becomes excited by incident light (hνA or hνB) to the S1 or S2 singlet excited states. The excited PS can relax to S0 by the radiative fluorescence emission of photons (hνF), which can be used for imaging and diagnostics, or can undergo a spin forbidden process termed intersystem crossing. Through the spin flip of the excited PS, the molecule occupies a long-lived triplet excited state (T1), from which photochemical reactions occur that result in the production of cytotoxic RMS, including 1O2, that is used for PDT. B) A diagrammatic representation of the use of a PS for PDT and imaging techniques. Through type I and type II photochemical reactions, the excited sensitizer generates cytotoxic 1O2 and RMS from ground state triplet oxygen (3O2) and various biochemical substrates. The concomitant fluorescence of the PSs allows for imaging and diagnostic uses, emphasizing their inherent theranostic capacity.
The generation of various therapeutic RMS molecules proceeds through two distinct, yet interchangeable photochemical processes: type I reactions that produce radicals and radical ions, and oxygen-dependent type II reactions that produce singlet oxygen (1O2) and oxygen radicals. Both photochemical reactions are initiated from the triplet excited state of the PS; however, radiative relaxation processes of photoexcited PSs give rise to fluorescent (singlet excited to singlet ground state relaxation) and phosphorescent (triplet excited state to singlet ground state relaxation) emission of light. Owing to their fluorescent properties, PSs are inherently therapeutic and diagnostic (theranostic) agents. Figure 2 (B) is a visual representation of the theranostic nature of PS molecules, where photoexcitation results in type I and type II reactions for PDT, whilst concurrently resulting in fluorescence emission that is used for a variety of imaging and diagnostic applications. Although still largely investigational, PNM was an early player in the field of nanomedicine. Visudyne®, a non-PEGylated liposomal formulation of the PS benzoporphyrin derivative (BPD), was one of the early nanotherapeutics to be approved by the U.S. Food and Drug Administration (FDA) as a first line treatment of age-related macular degeneration (AMD) and is considered an instrumental paradigm of photoactive nanotherapeutics and nanodiagnostics.8 Clinically, PDT is performed through the administration of a PS or its respective formulation, followed by the timely irradiation of the disease tissue using NIR light that optimally penetrates tissue in wavelength range know as the ‘optical window’ ranging between 600 nm and 1300 nm.9 The most effective PSs are those that have been tuned to maximally absorb NIR light within that optical window. Figure 3 is a graphical summary of clinical PDT and describes the different PS formulations that will be discussed in this review in addition to the functional and structural imaging modalities used in conjunction.
Figure 3.

A representation of the steps taken during a clinical PDT procedure using various PS formulations and the structural and functional information obtained using optical imaging techniques that enables treatment prediction and guidance. Following intravenous administration of the PS, an appropriate PS-light interval is required prior to irradiation using localized NIR light delivery. Through spatially confined PDT action on the tumor, the disease tissue is destroyed. Figure adapted with permission from Mallidi et al.10 Optical Imaging, Photodynamic Therapy and Optically Triggered Combination Treatments, The Cancer Journal, 21 (3), p194-205. (Copyright © 2015 Wolters Kluwer Health, Inc.)
The importance of the photoactivity of nanotherapeutics is not only restricted to their use in phototherapies, but also encompasses multiple parameters that distinguish such PNM formulations from the emerging pool of therapeutics. PNM formulations provide spatiotemporal control in the induction of phototherapy, spatial confinement in the phototriggered release of secondary encapsulated complementary therapeutics, temporal regulation of their respective activity and the combination of multiple theranostic agents that can be hyperspectrally and spatiotemporally resolved. This review will outline recent advancements in PNM formulations developed with respect to imaging-assisted, and rationally designed anti-cancer combination regimens. Efforts from our laboratories and others are universally driven towards achieving a unifying therapeutic and diagnostic (theranostic) nanoparticle represented in Figure 4.
Figure 4.

A schematic diagram of the visionary theranostic nanoconstruct that combines a fluorescence-based theranostic or imaging agent and a therapeutic drug encapsulated within. The surface is grafted with a targeting ligand to enable the molecular selectivity of the theranostic PNM formulation (1). Light activation can be used for image-guided therapy (2), photochemical generation of cytotoxic RMS for PDT of the disease tissue (4) and sequential, controlled release of synergistic agents for combinatorial cancer therapy (5).
The envisioned nanoconstruct realizes 1) the capacity to molecularly target tumors 2) the ability to mediate optical diagnostics and dosimetry 3) the selective induction of photodynamic action and 4) the spatiotemporal control of phototriggered release of synergistic anti-neoplastic agents. Progression driving technology closer to this visionary nanoconstruct will be highlighted in this review.
The fluorescence properties of PSs are highly effective for a variety of applications that distinguish them from conventional anti-cancer drugs that are formulated as nanomedicines. These include the potential for fluorescence pharmacokinetic (PK) quantitation of tumor PS concentrations,11 the fluorescence guidance of surgical tumor resection12 and the online monitoring of PDT dosimetry parameters through rates of PS photobleaching.13-15 The preclinical and clinical applications of multi-faceted theranostic PSs and the successive forward-looking PNM formulations will be discussed in greater detail in the Theranostics section.
Phototherapeutics are by no means restricted to the photochemistry-based PDT. Other noteworthy modalities exist and are emerging in clinical trials. Though not focused on in this review, photothermal therapy (PTT) plays a role in nanomedicine, such as plasmonic gold nanostructures.16, 17 There are currently two active clinical trials using plasmonic gold nanoshells, AuroShell®, for anti-cancer PTT specifically termed AuroLase® Therapy.18, 19 Requiring significantly higher power densities (W/cm2) than PDT (mW/cm2), the therapeutic response times for PTT ranging in the 10-3 – 1 s timeframe are significantly shorter than those of PDT (1 - 103 s).20 Power densities for PDT and PTT can oftentimes overlap when photothermal agents are utilized as mediators for PPT. However, considerable higher concentrations of photothermal agents are required for tissue thermalization with lower power densities, as compared to PDT. PDT is an attractive option when prolonged agent interaction times and a precise spatiotemporal controlled drug release is required. Unlike PTT, an attribute of PDT is the excellent healing process of normal tissue, thus scarring and collateral damage is minimized. The threshold nature of the PDT process also allows for the PS to be present in non-target sites as long as the overall PDT dose can be below the threshold for damage.21 This enables the illumination of larger volumes of tissue to ensure tumor margin sterilization. Ultimately, a selection of phototherapeutic strategies with distinct modes of action can be employed to address specific clinical needs.
Clinical nanomedicine
Past, present and future
The improved outcomes from many of the now-approved nanotechnology-based therapeutics have encouraged a sustained clinical effort, with several trials ongoing or recently completed. While the clinical advancements discussed here are by no means comprehensive, they will touch on some of the most recent results that highlight the varied capabilities offered by nanotechnology. A specific focus will be made on the nanomedicines requiring additional activation or triggered release, which can be selectively localized to the disease site. Figure 5 is a relatively comprehensive timeline outlining some of these liposomes, polymer and protein-based nanomedicines that have been approved by the FDA and other regional official medical regulatory bodies. The major driving force for the initial nanoformulations of the listed drugs was the improvement in their PK profiles and tolerability upon encapsulation. The developments of advanced nanomedicines to follow were motivated by a number of needs that were unmet by conventional nanoformulations. These include the need for increased control over activity and spatiotemporal drug release, the necessity for selectivity in targeted delivery of agents and the significance of multi-agent co-encapsulation. Highlights and technological advances in the global clinical progression of these multi-faceted ‘soft’ nanoplatorms will be described in this section.
Figure 5.

A timeline spanning the late 1990’s to date listing the chronological approval and clinical trial status of some lipid, polymer, and protein-based anti-cancer nanomedicines that are being leveraged to improve the PKs, safety profiles, and therapeutic indices of anti-neoplastic agents. The nanomedicines are classified under subgroups that describe the nature and primary utility of the clinical nanomedicines listed. These include nanomedicines that serve to improve drug PKs and safety profiles, to enable the controlled activation and release of therapeutics, to actively target and selectively deliver agents and those that act as a platform for multi-agent co-encapsulation. Visudyne®, which gained approval in 2000, is currently the only soft PNM formulation which enables controlled light activation. Controlled photoactivation of Visudyne® using 690 nm NIR light was approved for PDT of AMD, and Visudyne®-PDT is now showing significant promising in clinical trails for locally advanced pancreatic cancers.22
In the last quarter of a century, approximately 100 nanomedicine products have been introduced to the market for therapeutic or medical device applications.23 One of the most noticeable advancements has been in the field of oncology using soft organic nanoconstructs, such as liposomes, micelles, and polymeric and protein-based nanoparticles.24 The majority of these commercialized nanomedicine products are developed for intravenous injection, taking advantage of the size- and shape-dependent enhanced permeability and retention (EPR) effect in tumor vasculature for ‘passive targeting’ of solid tumors. More recently, significant clinical efforts have been made to develop targeted nanomedicine that can be activated by external stimuli, such as light, or even administered orally, which is preferred by most patients.25 Nevertheless, all these nanotechnologies have provided a solution to many hurdles in drug delivery, such as poor drug solubility and stability, suboptimal PK profiles and adverse systemic side-effects, all of which will be discussed in further detail within this review.
In the context of PNM, in 2000, the FDA approved the non-PEGylated liposomal BPD formulation, Visudyne®, for the PDT management of the wet form of AMD. To date, PDT using Visudyne® has saved the vision of millions of patients with retinal pathologies, and has been shown to work even more effectively using combination strategies.26 As discussed earlier, the liposomal PNM formulation Visudyne® pushes the boundaries of typical nanotherapeutics in that it enables the regulation of spatiotemporal control over treatment induction, and thus bears huge clinical potential with regards to the impact this regulated approach can have on combination cancer therapies.8, 27 The immediate clinical impact of Visudyne® is further evidenced in the ongoing clinical trials for pancreatic cancers.22 In our VERTPAC-01 Phase I/II trial, PDT was conducted using Visudyne® in patients with locally advanced pancreatic cancer.22 Laser light at 690 nm can be delivered via single or multiple fibers positioned percutaneously under computed tomography (CT) guidance for photochemical activation of Visudyne® within the tumor interstitium. At 40J, PDT consistently induced 12 mm of necrosis in the tumors with a low incidence of mild adverse events (e.g. abdominal pain and inflammation), thus meeting all endpoints of the study. In the same trial, CT scans prior to and following treatment showed a strong positive correlation between contrast-derived venous blood content and necrotic volume in the tumor.28 These results suggested that contrast CT could provide key surrogate dosimetry information to assess treatment response and will be discussed further in the Theranostics section. In an earlier clinical study on patients with locally advanced pancreatic cancer, Bown et al. performed PDT using the PS meso-tetrahydroxyphenyl chlorin (mTHPC), that is reconstituted in a mixture of water and ethanol containing polyethylene glycol (PEG).29 In this study, up to six optical fibers were used to deliver light percutaneously under CT guidance for effective mTHPC- PDT, which had a 100% response rate and a median overall survival of 9.5 months. More recently, Moore et al. demonstrated that the PS WST-11, a Cremophor® emulsion of the palladium-bacteriopheophorbide TOOKAD®, can be used for ultrasonography-guided vascular-targeted PDT of low-risk prostate cancer in patients that have limited therapeutic intervention options.30 It should be noted that the delivery of light can be achieved in many ways and, in the case of pancreatic cancer discussed above, ultrasound guided endoscopes and fiber optics also provide a conduit for direct light delivery into deep situated tumors. Placement of multiple fibers is also possible for treating large tumors such as glioblastoma.22, 31 To manage tumors with poorly defined margins or those of disseminated nature, light delivery over large areas is made possible clinically with diffusing tip fibers, balloons and scattering media.32
To put the growing excitement of the emerging oncological indications of PNM formulations into perspective, the current clinical status of soft nanotechnology that are often used in the building blocks of advanced PNM formulations will be reviewed. Selective tumoral activation of nanotechnology is also being clinically evaluated with Celsion™’s ThermoDox® in the Phase III HEAT trial for hepatocellular carcinoma.33 ThermoDox® is a PEGylated liposomal formulation designed to thermally release doxorubicin upon radiofrequency ablation (RFA). Results made available from Celsion™’s press releases indicate that an extended duration of RFA may be critical to improving outcomes, leading to the initiation of a new Phase III trial, OPTIMA, for hepatocellular carcinoma.34-36 Although not part of PNM, Doxil®, a liposomal formulation of doxorubicin must be mentioned in any discussion of nanomedicine, as it was the first liposomal drug to be approved by the FDA in 1995.3 Doxil® is approved for the treatment of various cancers including AIDS-related Kaposi’s sarcoma, recurrent OvCa and multiple myeloma.3 A major feature of Doxil® is the presence of surface PEG molecules that provide the liposomes with a stealth quality. ‘PEGylation’ protects the inner encapsulating doxorubicin from the external environment, prolongs circulation time to enhance passive tumor drug accumulation, and reduces cardiotoxicity associated with doxorubicin therapy. A more recent advancement in clinical nanomedicine is the FDA approval of Onivyde® (MM-398, nal-IRI), a liposomal formulation of irinotecan.37, 38 Approval for gemcitabine-refractory pancreatic cancer in 2015 was granted after the Phase III NAPOLI-1 study demonstrated an improved median overall survival using Onivyde® in combination with 5-fluorouracil and leucovorin.37, 38 Besides adding new functionality to existing FDA-approved small molecule chemotherapeutics, liposomal nanotechnology also extends to the delivery of vaccines and of PSs for cancer treatments.39
Analogous to liposomes, polymer and protein-based nanotherapeutics have also been used to improve the PK and toxicity profiles of common potent chemotherapeutics. NKTR-102 is another nanoformulation of irirnotecan made by Nektar Therapeutics™, which consists of a polymeric nanoparticle conjugated to the drug. NKTR-102 is currently in Phase I to III trials for a variety of indications.40-43 Data from the Phase III BEACON trial with NKTR-102 thus far indicates that only breast cancer patient subgroups with brain and liver metastases show a significant overall survival benefit.44, 45 Genexol ®, a PEG-poly (lactic acid) micellar formulation of paclitaxel with a diameter of ~20-50 nm, was approved for use in South Korea in patients with metastatic breast and pancreatic cancers in 2001.46 Eligard® is a poly(DL-lactide/glycolide) (PLG) nanoparticle incorporating leuprolide acetate, which was approved in 2002 for advanced prostate cancer.47 Abraxane® is an albumin-bound paclitaxel nanoparticle, ~130 nm in diameter, which was approved by the FDA in 2005 for passive targeting of metastatic breast cancer and was approved by the European Medicines Agency (EMA) for the same disease in 2008. Abraxane® received FDA approval for the treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) in 2012, which was shortly followed by approval for the treatment of metastatic adenocarcinoma of the pancreas by the FDA and EMA in 2013.48
Beyond these capabilities, nanoscale complexes, such as liposomes and/or nanoparticles, can also be leveraged for the co-encapsulation of multiple therapeutic agents, which may be mechanistically desirable. These advances in nanoformulations allow the delivery of fixed-ratio drug combinations with unified PKs, along with the option for drug compartmentalization to regulate sequential release kinetics. Furthermore, the potential for cancer targeting and light-triggered drug release may further improve treatment outcomes in cancer. Co-encapsulated nanoformulations of dual agents are already showing promise in human trials. For example, liposomally co-encapsulated cytarabine and daunorubicin (CPX-351, Celator Pharmaceuticals™), recently completed testing in Phase II studies for acute myeloid leukemia (AML).49 CPX-351 showed a significantly improved response rate and overall survival from 4.2 to 6.6 months in patients with secondary AML, when compared to patients treated with the standard combination of free cytarabine and daunorubicin.50 Preclinically, our group recently develop a photoactivatable multi-compartmental nanoconstruct that contains a ‘membrane-like’ unilamellar liposomal shell for the loading of the PS BPD and a PEG-poly (lactic-co-glycolic acid) (PLGA) nanoparticle core containing an anti-angiogenic agent.51 The anti-angiogenic agent is photo-released within the tumor to synergistically reduce both local tumor burden and distant metastases of pancreatic cancer in vivo.51 Leveraging the hydrophobic and hydrophilic compartments of folate-targeted liposomes, Morton et al. effectively co-encapsulated both hydrophobic (erlotinib) and hydrophilic (doxorubicin) therapeutics to enhance A459 tumor control in vivo via the dynamic rewiring of apoptotic signaling pathways.52
Clinical advances have also been made in actively targeted, bioconjugated nanoformulations. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes encapsulating doxorubicin were the earliest targeted chemotherapeutic immunoliposome in clinical trials, demonstrating potent antitumor activity in Phase I studies.53, 54 Merrimack Pharmaceuticals™ has also developed a human epidermal growth factor receptor-2 (HER-2) targeted doxorubicin immunoliposome, MM-302, for the treatment of breast cancer, which is currently in Phase II trials.55 BIND Therapeutics™ has also developed BIND-014, a docetaxel PEG-PLGA polymeric nanoparticle targeting prostate specific membrane antigen, which is currently in Phase II trials for prostate cancer and non-small cell lung carcinoma.56 This nanoconstruct is currently being initiated for an even broader range of indications. These examples of nanotherapeutics, amongst many others emerging for a large variety of oncological and non-oncological indications, demonstrate not only the clinical enthusiasm of nanomedicine and PNM but also pave the way for using light-activated, targeted, and combinatorial approaches that improve current drugs previously limited in their efficacy and safety.
Challenges to the clinical translation of photonanomedicine
As discussed in this review, incorporating the PDT approach with nanotechnology allows for the encapsulation of high PS payloads, improved photoactivity and appropriately-timed release and activation of therapeutic agents. As with all parenteral non-PDT nanoconstructs, understanding pharmacokinetics (PKs) of advanced nanoformulations and their respective constituents is fundamental in maximizing their therapeutic efficacy and expediting clinical translation. Each constituent exhibits individually unique PK and toxicology profiles, in addition to different metabolic and physiological clearance mechanisms. Furthermore, their cohesive behavior as an individual entity is distinctly different, as the resultant nanoformulation is essentially a new material. In vivo destabilization and degradation, therefore, results in complex multi-parametric physiological behaviors for each constituent of the nanoformulation. In addition to chemical composition, physical properties of nanoformulations govern the degree of efficacy and toxicity. For example, a 75 nm variant of Doxil®, was compared to the standard 100 nm formulation and was found to be more efficacious yet more toxic in tumor-bearing mice.57 By incorporating PS into nanoformulations, PDT using PNM formulations has its own challenges. Whilst one of the advantages that we have mentioned in the body of the manuscript is that light can act as a drug release mechanism, with that comes another challenge in the need for studying new PK parameters of the released drugs. However, despite this additional layer of complexity, preclinical evidence of the efficacy of anti-cancer PNM formulations has been very promising.
Unlike nanoformulations of small molecule therapeutics, encapsulated into a nanoconstruct, the physiological effects of advanced PNM formulations are not only restricted to the agent and the nanocarrier properties. PDT with PNM formulations involves the paired use of an administered agent or multiple agents formulated into one or more nanoconstructs, that is activated by an applied optical irradiation. Thus, the clinical approval of PNM formulations is dependent on the regulation of the PS, the secondary therapeutic agents when applicable, the nanomaterials used for their formulation and the light sources for photoactivation. As a result of complex tissue light scattering and attenuation events, NIR light penetration through vascularized soft tissue is limited to approximately 1 cm, thereby necessitating the use of secondary medical devices for the procedure, namely optical fibers.22, 58, 59 Optical fibers deliver light to deeply situated tumors, overcoming the limitation of light penetration, yet providing an additional regulatory obstacle in the approval of clinical PDT procedures. Clinically, the advances in fiber optic light conduits and image-guided approaches have empowered the applications of PDT that use an interstitial fiber placed directly within deep tumors. Placement of multiple fibers is also possible when treating large tumors, such as pancreatic tumors.22 For diffuse carcinomas with poorly defined margins, such as disseminated ovarian cancer metastases, light delivery over the large peritoneal cavity surface areas is made possible clinically with diffusing tip fibers, balloons and scattering media.60 In principle, the approval of novel PNM formulations can be expedited by the use of pre-approved and clinically approved PSs,61, 62 biocompatible nanomaterials,63, 64 targeted biologics,65, 66 light sources,62, 67 optical fibers68 and imaging modalities to guide therapy.69 Nonetheless, clinical approval of complex PNM formulations is still a big challenge because each individual formulation will become a different material and full screening becomes critical following any modification.
For the most part, downstream manufacture and quality control processes of the constituents comprising the evolving pre-clinical soft PNM formulations discussed in this review are well established in the biopharmaceutical industry. These constituents include liposomes, soft nanoparticles, photoactive nanoformulations and antibody-drug conjugates. Although amalgamations and modifications of the existing PNM formulation constituents will inevitably impact PK/pharmacodynamics (PD), toxicology and efficacy, the existing industrial infrastructure for Good Manufacturing Practice (GMP) scale-up synthesis of these advanced PNM formulations will facilitate and support their Good Laboratory Practice (GLP) toxicology studies and clinical trials. As mentioned in the previous section, one of the early PNM formulations to be approved in the clinic is Visudyne® and, therefore, there is much optimism for more complex constructs to enter into the clinic.
Forward-looking preclinical advances in inorganic PNM formulations, such as nanoscintillators70 and upconversion nanoparticles71 have enabled the conversion of deep-penetrating radiation, such as X-rays and NIR light, to visible wavelengths for deep-tissue PS excitation. However, these ambitious nanomaterials are hindered by the inherent toxicity of their components and dopants, such as heavy metal ions and rare-earth lanthanide ions. Before being considered for clinical human use, rigorous in vivo toxicology, chemical stability, physiological integrity and clearance studies are crucial to confirm their safety. Furthermore, there must be a clear clinical advantage of using such nanomaterials over more conventional direct visible and NIR-mediated PDT agents.
Despite the fact that PDT has been shown to induce distant antitumor immunity in various animal models, clinical PDT is primarily used to manage local tumors. Numerous preclinical studies have shown that PDT sensitizes tumors to inhibition by systemic cytotoxic therapies, thus reducing the spread of disease to other sites indirectly when used in combination.51, 72 Therefore, it is believed that PDT is best combined with systemic treatment modalities to maximize both local and distant tumor control. The combination of localized PDT and systemically cytotoxic therapies is also attractive for cancer treatment, which avoids overlapping toxicities and reduces the doses required to achieve the same therapeutic effect. Infrequent undesirable secondary reactions to PDT comprise skin burning, itching, and injection site reactions that include pain, edema, inflammation, extravasation, rashes, hemorrhage, and discoloration.22, 73 Although most adverse events reported with PDT were of short duration and easily manageable, it is often critical for patients to avoid direct sunlight for 24-48 hours after treatment, depending on the skin clearance rates of the PS and its respective formulation. In contrast to PDT, most chemotherapies and biologics are associated with significant and long-term side effects, such as neutropenia, diarrhea and hair-loss, and patients often require dose reduction or preemptive management. The genotoxicity of DNA-damaging cancer therapies like alkylating agents and radiation therapy is not observed with PDT, as the lack of nuclear localization of most PS molecules confines oxidative damage to the target perinuclear organelles. Unlike external beam radiation, PDT can also be repeated several times at the same site if needed, given that PS selectivity to the tumor is optimal. Moreover, studies have suggested that PDT-induced damage is generally more efficient in the tumor than tissue, thus reducing normal tissue toxicity, regardless of PS selectivity.29 Reasons for this are unclear, but elevated levels of reactive oxygen species scavengers in normal tissue are suggested.29, 74
It is becoming increasingly evident that conventional, uninformed PDT using free PSs may not be the most effective choice for cancer management. More sophisticated formulations, combination therapies, irradiation procedures and image-guidance needed to improve the clinical outcome of PDT will also impact the financial costs of the therapy. As with all other treatment modalities, additional pathological factors, such as completeness of response and the need for multiple rounds of therapy, can also influence the total costs. In general, a comprehensive treatment for pre-skin cancers may cost up to $3,500 USD for a series of treatments, and can often be covered by health insurance in the United States.75 Clinical applications of imaging-assisted PDT can be more precise, effective and less invasive, yet will lead to higher overall treatment costs. For example, clinical endoscopic PDT for early esophageal cancer can cost up to ~$5,000 (USD), which is somewhat higher than the costs of chemoradiotherapy, radiofrequency ablation and cryoablation therapy, which ranges from $1,500 to $3,700 USD. However, compared to standard esophagectomy that costs more than $25,000 USD, endoscopic PDT is considerably more cost-effective, and has a lower mortality rate with fewer major side effects.75
Theranostics
The concept of combining therapeutics with diagnostics is encompassed in the fairly well-known term, theranostics. PDT is inherently a theranostic modality as the molecules involved are both photochemically active (to achieve the therapeutic effect) and fluorescent (to achieve the diagnostic component). Because of the differences in treatment response observed between in vitro and in vivo systems and because of the high variability among patients that receive the same treatment, the ‘one size fits all’ approach for PDT is not appropriate.76 Thus, there is growing interest for developing personalized medicine and adapting the treatment in real time. This personalized medicine is a common goal of all these theranostic applications. Particular to PDT, several strategies may be employed to reach this objective: 1) individualize PDT dosimetry by monitoring PS fluorescence, 2) use the PS fluorescence properties to guide surgery, and 3) monitor the treatment efficiency by involving other imaging modalities, such as CT, magnetic resonance imaging (MRI) or photoacoustic imaging (PAI).
Optically- guided theranostics
As mentioned in the introduction, once a PS is photoexcited, it can relax either through intersystem crossing transitions eventually leading to the generation of cytotoxic RMS molecules, or by fluorescence emission that is concurrently exploited for optical imaging. Both pathways are exploited to assess PDT efficiency and perform dosimetry measurements: the singlet oxygen (1O2) generation and the photobleaching of fluorescence of the PS.
a. PS fluorescence and singlet oxygen measurements as dosimetry parameters
Both photodestruction of the PS (photobleaching) during the PDT process and the quantitation of 1O2 have been proposed as means for patient-customization of PDT dosimetry. Using in vitro experiments performed on AML5 leukemic cells undergoing PDT using 5-aminlevulinic acid (ALA), Niedre et al. demonstrated in 2003 that cell survival was directly correlated with the phosphorescence intensity of 1O2 emitted at 1270 nm.77 As a consequence, measuring the intensity of the infrared (IR) emission of 1O2 in real-time is a direct way of monitoring PDT efficiency and may even aid in predicting treatment outcomes. The 1O2 IR emission is weak and requires sophisticated imaging infrastructures; therefore, dynamic longitudinal monitoring of 1O2 generation is particularly challenging in the clinic. A more realistic method of clinically assessing the degree of 1O2 generation, and thus treatment response, is the dynamic monitoring of PS photobleaching. All PS molecules are subject to varying degrees of photobleaching directly through self-oxidation following irradiation. Jarvi et al. proposed in 2012 an in vitro comparative study investigating two dose metrics: the 1O2 emission metric, then considered as a gold-standard metric, and the metric for photobleaching of the PS.14 They demonstrated that a positive linear correlation exists between the administered 1O2 dose calculated from the phosphorescence counts of 1O2 relaxation and PS photobleaching measurements. However, they also established that the survival rate is only correlated with the degree of photobleaching as long as the oxygen partial pressure remains above 5 μM. Thus, photobleaching is not a reliable parameter for PDT under hypoxia. In 2014, Mallidi et al. published a clinical comparison between the two metrics on 26 healthy patients subjected to ALA-PDT.78 The study showed a stronger positive correlation between evolution of erythema (PDT tissue sensitization) and the PS photobleaching induced by PDT than with the 1O2 phosphorescence emission. With the evolution of better detection systems and imaging modalities, the possibility of monitoring both direct (photobleaching metrics and 1O2 phosphorescence) and indirect (structural and functional properties of the tumor) treatment prediction parameters will drive the theranostics arena towards patient personalization. Towards this goal, the following sections discuss the current developments in imaging techniques and nanotechnology geared towards personalized therapies using integrated PNM formulations.
b. Advances in imaging and spectroscopy
Outstanding progress has been made from the optical and software development perspective that allow fluorescence based optical-imaging for tumor detection, as well as video-rate intraoperative image-guided surgery.12 Reaching this level of complexity requires an improvement in fluorescent imaging modalities that face several challenges that include high tissue autofluorescence background levels and the complexity of deconvoluting chromophores with considerable spectral overlap. NIR excitation of PSs could be used for imaging, as it is less likely to induce autofluorescence, whilst narrow filters may also be employed to improve spectral separation. However, none of these solutions fully resolve the problem of PS spectral overlap with autofluorescence.79 Hyperspectral imaging has thus been introduced in an effort to overcome this hurdle in PS imaging. The principle is based on the unmixing of several spectra following the acquisition of emitted light in each pixel to resolve and identify the fluorophores contributing to the resultant combined emission profile. The systems that have been developed allow the spectral deconvolution of chromophores that are separated by just a few nanometers.80 Mansfield et al. illustrated the basis of hyperspectral imaging by successfully unmixing the spectra emitted by five quantum dots, all characterized by a different emission profile. Each distinct quantum dot was conjugated to different markers that bind to different cellular components including the mitochondria, microtubules, proliferation marker Ki-67, nucleus, and actin. The five colors were successfully unmixed and a map representing the spatial distribution of each quantum dot marker, and thus each cellular component, was compiled.
Developing cutting edge-technologies such as hyperspectral imaging leads to the improvement of fluorescence imaging techniques that can be used for several purposes including fluorescence imaging for theranostics and image-guided resection. In fact, surgery remains the primary treatment for the majority of solid tumors, which requires complete resection to ensure a positive prognosis. Although the peripheries of some tumor types are well-defined and do not require the precautionary extraction of additional seemingly healthy tissue, other diffuse tumor types, including metastases, are more difficult to differentiate from normal tissues, or reside adjacently to crucial structures. Therefore, the accurate delineation of precise tumor margins is required to achieve more complete resections. Sensitive optical imaging techniques, such as fluorescence imaging can thus guide surgery. The clinically-approved extrinsic fluorophores indocyanine green (ICG) and fluorescein sodium are examples of florescence contrast agents that have been frequently used to assist intraoperative dissection.81 More specifically, the endogenous PS protoporphyrin IX (PpIX) that accumulates in tumor tissue upon administration of the clinically-approved precursor, ALA, is currently in multiple clinical trials for assisting the resection of gliomas.82 As shown in Figure 6, the white light images and PpIX fluorescence images acquired intraoperatively from a patient with glioma are contrasted to emphasize the powerful advantage of fluorescence imaging for increasing the accurate tumor detection and boundary delineation for surgical guidance.
Figure 6.

A) Intraoperative white light image of the brain of a glioma patient with the respective fluorescence image following administration of ALA. The tumor is not visible to the naked eye, however, the endogenous PS PpIX preferentially accumulates in the tumor and enables its fluorescent detection and surgical guidance, which appears pink under blue light excitation in B) Figure reprinted from Widhalm et al. 83
The field of fluorescence-guided surgery is less well-established, as compared to other imaging modalities. However, it has recently made outstanding strides in the clinical arena, mainly to precisely delineate tumor margins and for advancing real-time NIR PSs image processing techniques during surgical procedures, as comprehensively reviewed by Elliott et al.12 During a Phase III clinical trial, image-guided tumor resection was performed following ALA administration on 322 patients with malignant glioma.84 It was observed that the resection was complete for 65% of the patients operated on under fluorescence image-guided surgery, as compared to 36% for patients operated on with white light surgery.84 Utilizing the inherent photosensitizing capacity of PSs used for fluorescence-guided resection, intraoperative PDT treatment can be used to eliminate the residual disease following surgery.32, 85 In a single-center Phase III trial on 27 glioblastoma patients, fluorescence-guided resection and post-operative PDT using a secondary administered PS doubled survival of patients, as compared to surgery performed under white light followed by radiotherapy (52.8 vs. 24.6 weeks, respectively).32 Fluorescence-guided resection is also widely investigated for bladder cancer. It has been shown in several Phase III clinical trials that PS fluorescence-guided resection is significantly more complete than following white light cystoscopy. These different results were reviewed by Jocham et al. 86
In addition to being used to guide resection, the fluorescence may also be used to diagnose or detect small nodules and metastases diffusely disseminated. For example, Zhong et al illustrated the ability to image the in vivo fluorescence signal of Visudyne® injected into the peritoneum of a mouse with disseminated micrometastases of human ovarian carcinoma (OvCa) using a high-resolution fiber optic microendoscopy modality.87 They demonstrated that this method could be quantitative and that the signal intensity is proportional to the area of the nodules disseminated throughout the peritoneal cavities, thus enabling the longitudinal fluorescence-guided monitoring of treatment response.
Some PS molecules require strategies to enhance their selectivity by directing them to biomolecules or membrane-associated enzymes that are over-expressed on tumor cells or by manipulating cancer-activated metabolism pathways.12, 81 In 2011, van Dam et al. reported the first clinical utility of targeted tumor-specific intraoperative fluorescence imaging of OvCa to improve disease staging in vivo and to maximize the extent of radical cytoreductive surgery.88 OvCa, 90-95% of which overexpress folate receptor-α, was targeted using a folate conjugate of fluorescein isothiocyanate, that could be expanded to the use of fluorescent PS conjugates. Selective theranostics using PSs have also been demonstrated preclinically using benzoporphyrin derivative (BPD) PS molecules conjugated to the anti-EGFR antibody, Cetuximab. The Cetuximab-BPD conjugates, termed photoimmunoconjugates (PICs), have been employed for the targeted imaging and PDT of non-resectable micrometastases of OvCa disseminated throughout the peritoneal cavity.11 As presented in Figure 7 (A), the PICs also enabled longitudinal fluorescence microendoscopic imaging of the remaining disseminated OvCa tumor burden following cycles of tumor-targeted activatable photoimmunotherapy (taPIT). The modality also provided robust in vivo quantitation of the imaged tumor burden (Figure 7 (B)). The details and advancements of targeted PDT using PICs and the taPIT approach will be further described in the Acquiring selectivity section.
Figure 7.

A) Images obtained by longitudinal in vivo fluorescence microendoscopy of tumor burden in a disseminated mouse model of OvCa treated with the Cet-BPD PIC without and with (taPIT) light activation (scale bar 100μm). B) Quantitative analyses of representative tumor fluorescence during the treatment. Solid lines indicate significant changes (P < 0.05, two-tailed unpaired t test). Figure adapted from Spring et al.11
Ultimately, advanced tumor-selective PNM formulations will be utilized in the same manner, where the fluorescence properties will provide the accurate delineation of tumor margins to aid surgical resection, and for the detection and photodynamic elimination of microscopic disease. Substantially prolonged circulation times of PNM formulations, as compared to free PSs or biological conjugates of PSs, will inevitably result in longer times required to attain maximal tumor-to-normal ratios. Thus, careful investigations in PKs and tumor selectivity will be critical in identifying the optimal times required to utilize the full potential of the theranostic PNM formulations.
Other imaging modalities
Theranostics also has a broader application and in this context, therapies such as chemotherapy, radiation therapy (RT), PDT or PTT can be combined with different clinical imaging techniques that include MRI, CT, positron emission tomography (PET) and NIR fluorescence.76 As a platform for the combination of therapeutic agents with imaging agents, organic carriers such as liposomes, polymers or micelles90 have been heavily investigated. In addition, inorganic nanoparticles, such as mesoporous silica nanocomposites, have also been utilized because of their biocompatibility and their high capacity for agent loading.91-93 Two types of nanodots have also been explored in the context of theranostics: quantum dots94-96 and carbon nanotubes,97 both which are utilized as photoactive nanocarriers and as visible and NIR fluorescence imaging agents. Inorganic nanoconstructs such as iron oxide nanoparticles have also be functionalized with drugs to combined MRI with a therapeutic agent.98-100 Significant interest has developed over the prospects of nanomaterials that intrinsically behave as both image contrast agents and as nanotherapeutics. One of the most prevalent examples is gold nanoparticles that, in addition to being physiologically inert, can be used as imaging agents that enhance the contrast of CT101 and surface enhance Raman spectroscopy (SERS) imaging modalities.102 Furthermore, gold nanostructures have also been shown to behave as efficacious therapeutics, such as radiosensitizers for RT,103, 104 nanoparticulate carriers of PSs for PDT105, 106 and as photothermal agents for PTT.17, 106 Gadolinium-based nanoparticles have similarly drawn such attention for their ability to improve the efficacy of RT, whilst simultaneously enhancing MRI contrast, and thus allowing for the development of MRI-guided RT theranostic nanoconstructs.107
a. Photoacoustic imaging to predict PDT efficiency
In addition to being fluorescence contrast agents, PSs are also effective photoacoustic contrast agents. PAI, a ‘light-in, sound-out’ modality provides 3D optical absorption properties of the tissue being imaged at penetration depths that are significantly deeper than fluorescence imaging. Recent work by Mallidi et al.108 and Ho et al.109 showcases the ability of PAI to monitor PS tumor uptake and directs towards the ability to personalize dosimetry based on the PS concentration at the target site. As an alternative method to assess PDT efficacy without using PS photobleaching or 1O2 luminescence, PAI can also quantify changes in blood oxygen saturation levels induced during PDT by 1O2 generation. A recent preclinical study utilized PAI to highlight the critical role of tumoral oxygen saturation levels for monitoring responsiveness to PDT.111 It was demonstrated that oxygen saturation evolution matched the tumor volume evolution and, as a consequence, was a reliable marker for PDT efficacy that could be studied non-invasively. For a PS-light-interval of 1 hour, oxygen saturation reduced to approximately 94% of the pre-PDT levels at 6 hours following treatment and increased by approximately 9% 18 hours later. In this same study, a threshold for oxygen saturation measured 6 hours and 24 hours post-PDT was established to predict tumor recurrence. Therefore, PAI could provide critical image guidance parameters when used in synchrony with PNM formulations to quantitatively monitor tumor uptake, assess the extent of the tumors response to PDT, and thus inform further cycles of therapy.
b. Computed tomography to monitor PDT
It has recently been demonstrated by preclinical and clinical studies that CT, a standard-of-care imaging method, may be used to predict the PDT dose required to treat patients with non-resectable pancreatic cancer.28 CT remains the most widely used modality to diagnose and follow the evolution of pancreatic tumors. Aside from being a reliable method to predict PDT efficiency, this protocol has the unique advantage of potentially limiting the number of procedures that patients undergo. It is an X-ray based imaging technique based on the difference of X-ray absorption coefficients of body constituents, such as bone, tissues, and vasculature. CT contrast is usually enhanced using iodine solutions, a high Z-number element that locally increase X-ray absorption.112 Elliott et al. published a preclinical study performed on rabbit orthotopic pancreatic cancer that compares several tumor controls such as blood volume, blood flow and vascular permeability surface area obtained from CT images.113 They emphasized that blood volume may be a determinant of treatment response because the lower it is, the lower the PS dose delivered. This pre-clinical study highlighted the potential of CT imaging to map the PDT dose, driven by the light attenuation in tissues. Jermyn et al. recently proposed a clinical study performed on 15 patients with locally-advanced adenocarcinoma that aimed to prove the potential of using CT imaging to predict PDT efficiency and thus adapt the treatment protocol.28 In this trial, pre-treatment and post-treatment contrast-enhanced CT scans were used. Venous blood volumes were calculated for each patient using the pre-clinical CT and then compared to the necrotic volume induced by the PDT measured using the post-treatment CT scan. The CT-derived venous blood content highly correlated with the necrotic volume normalized to the PS dose. The findings indicated that a pre-treatment CT scan may allow the prediction of the necrotic volume after PDT and, as a consequence, may inform the total PDT dose required, individualized to each patient. This clinical study corroborates the previous assumption that blood volume is a limiting factor to the effective PDT dose administered. The utility of this standard-of-care procedure could be expanded to predict patient response to PDT using parenteral PNM formulations and subsequently guide the personalization of the treatment.
c. Cerenkov emission imaging to monitor RT and PDT
Cerenkov luminescence imaging is a less common, state-of-the-art bioimaging technique currently being developed to monitor treatments such as RT, and can potentiate and guide PDT using forward-looking PNM formulations. Cerenkov imaging has recently gained substantial clinical interest with particularly strong implications in quantifying the effective dose of ionizing radiation deposited during RT.114, 115 Cerenkov optical emission spanning the UV-Visible spectrum is generated by charged particles in a dielectric medium travelling faster than the phase velocity of light in the same medium and can activate nearby PSs localized at the site of RT treatment.115, 116 Recently, the proof-of-concept of video rate Cerenkov imaging was presented, providing real time insight to the effective three-dimensional radiation dose deposited during RT, as illustrated in Figure 8.117 Although the proof-of-concept was proposed for RT of breast cancer, this dosimetry protocol may be expanded to combination treatment regimens that combine RT with PDT, providing more precise, complete and tolerable irradiation procedures. Axelsson et al. showed that Cerenkov emission is sufficient to activate the fluorescence of a PS, PpIX, although studies investigating the PDT efficacy of PSs activated using Cerenkov emission from external radiation are pending.
Figure 8.

A) The beam’s-eye-views of the applied RT fields. The blue lines refer to the multi-leaf collimator that blocks the radiation from these areas. B) The surface projection on the skin from the posterior oblique portion to be irradiated. C) Cerenkov luminescence images during RT of the posterior oblique field. The areas with the highest intensity correspond to the tissue receiving the highest effective radiation dose. Reprinted from International Journal of Radiation Oncology, 89(3), Jarvis et al.117 Cerenkov video imaging allows for the first visualization of radiation therapy in real time, 615-622. Copyright (2015), with permission from Elsevier.
β-emitting radionuclides used for PET imaging have also been known to exhibit high yields of Cerenkov emission, without the need for external activation using ionizing radiation.118 Katogiri et al. recently proposed the use of Cerenkov radiation from a β-emitting radionuclide to activate type I photochemical reactions for PDT.119 In this study, the authors reported the use of 64Cu, a radionuclide clinically used for PET imaging, to excite photocatalytic titanium dioxide (TiO2) nanoparticles through its simultaneous emission of Cerenkov radiation. This pre-clinical study demonstrated the potency of the PDT combination treatment in that tumor volumes were reduced below the detectable range when TiO2 NPs were used in combination with 64Cu. However, tumor volumes remained approximately 400 mm3 when TiO2 nanoparticles were used in conjunction with non-radioactive Cu, approximately 550 mm3 when 64Cu was used alone, and remained above 650 mm3 in the no treatment controls. Co-encapsulation of a β-emitting radionuclide with a PS in a combined novel PNM construct may promote spatiotemporally-controlled, Cerenkov-induced photodynamic activation of the PDT agent held its immediate vicinity. Furthermore, PET imaging using β-emitting radionuclides typically contrasts tumors by providing functional information, such as glucose metabolism and oxygen consumption, both critical parameters that can simultaneously monitor the extent of PDT treatment response in real time.
Although in the early stages of discovery, deep tissue PDT activation using localized Cerenkov emission provides clinically valuable advantages of either exogenous induction by ionizing radiation during RT or endogenous activation using PET probes, which can be used in concert with common clinically-accepted imaging modalities. Although PDT activation using Cerenkov radiation from β-emitting radionuclides simplifies the clinical procedure required with externally activation RT and the critical irradiation parameters associated with it, the spatial selectivity for PDT activation is entirely lost. Cerenkov-mediated PDT using an integrated PNM formulation containing a β-emitting radionuclide and a PS becomes a systemically active treatment modality, where the PK of the nanoconstruct governs induction of toxicity. Toxicity of the liver, spleen and other organs where nanoparticles tend to accumulate is likely to be observed, and therefore, tailoring size and surface properties of the PNM formulation becomes even more critical.
Additional modalities utilizing the unique inherent photoactivity of novel nanoconstructs to overcome the limited penetration of light through tissue rely on their direct interaction with deeply penetrating ionizing radiation or NIR light. Photoactive inorganic nanocrystals, such as upconverting nanoparticles71 or nanoscintillators,70, 121 respectively, are emerging as advanced deep-tissue PNM formulations, when combined with PSs to locally produce RMS at anatomical sites typically inaccessible to external photoirradiation. Because of the clinical implications of X-rays of NIR light,122 currently used for imaging and RT, these nanocrystals exhibit theranostic properties when combined with therapeutic PSs. When excited with ionizing radiation, scintillation nanoparticles emit light spanning the UV/visible/NIR spectrum and can thus internally activate associated PS with wavelengths of light typically inaccessible at such tissue depths.70 Similarly, upconverting nanoparticles absorb NIR light that penetrates tissue deeper than visible wavelengths and locally upconvert it to shorter wavelengths that efficiently excite associated PS molecules.71 Moreover, inorganic metal nanoparticles, such as gold nanoparticles, may elicit an inherent therapeutic effect including PTT or radiosensitization during RT, and their contrast-enhancing imaging properties can potentiate image guidance using CT. The diverse selection of nanomaterials provides a highly attractive pool of potential theranostic platforms; however, each nanomaterial can carry its own health risks and its clinical progression depends on the stringent investigations required to rule out immediate hazards or adverse health effects due to prolonged exposure. These concerns are more prominent when investigating inorganic nanomaterials composed of heavy metal atoms (noble metal nanoparticles), iron compounds (magnetic fluids), rare-earth metal compounds (up-converting and scintillating nanoparticles), inorganic oxides (silica, titania and zinc oxide nanoparticles) and transition metal composites (quantum dots). This concern is directly evident by the fact that currently, Feraheme®, a polyglucose sorbitol carboxymethylether-coated iron oxide nanoparticle formulation, is the only remaining approved inorganic nanotherapeutic following the withdrawal of four previously-approved nanoformulations.123 Although the reasons for their withdrawal were ambiguous, a warning emphasizing potential adverse immune reactions to Feraheme® was released by the FDA in 2015. As a consequence of the delay in the clinical translation of inorgnainc nanomaterials, this review will mainly focus on the advancement of nanoscale therapeutics and diagnostics composed of organic, biodegradable materials that are currently in clinical use.
Photochemistry and nanomedicine: meeting unmet clinical needs
Barriers to drug delivery
a. Tailoring nanomedicines to tumor physiology
Nanomedicine has been primarily introduced to reduce the inherent toxicity of cancer therapeutics by improving their selectivity towards tumor tissue. It is important to emphasize that as a cancer therapy, PDT is largely non-toxic in the absence of light. However, many PSs for PDT are hydrophobic, rendering their systemic administration impossible without the use of organic solvent mixtures. An example of such PSs is Foscan®, an EMA approved PDT agent (mTHPC/temoporfin) for the treatment of head and neck cancers, which is administered systemically in ethanol:propylene glycol (40:60 v/v). To reduce the need for such solvents and their associated toxicities, the application of nanoparticle encapsulation was primarily employed for PDT to render PSs compatible with physiological media, such as blood plasma. Efforts towards the nanoparticle encapsulation of photosensitizing agents have led to the development of a multitude of ‘second generation’ PSs, of which Visudyne® and Foslip®, a liposomal derivative of Foscan®, are notable examples.
An important unanticipated feature of nanoparticle encapsulation of PSs for their improved water solubility is the significantly increased selectivity for tumor tissue. For instance, Reddi et al. were among the first to explore liposomally encapsulate zinc phthalocyanine (ZnPC) and its drug delivery in vivo. Following the injection of 0.5 mg/kg-1 ZnPC in 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) liposomes, tumor tissue accumulated relatively high levels of ZnPC (0.6 μg/g), compared to <0. 1 ug/g in the muscle tissue surrounding the tumor 24 hfollowing intravenous administration.124 Given that PNM formulations can also improve the PS selectivity for tumor tissue, a variety of different types of nanocarriers have been investigated for the encapsulation of hydrophobic and hydrophilic PSs. Among the most studied are liposomes, (polymeric) micelles, and polymeric solid-lipid nanoparticles.125, 126 Many other investigators have reported that these PNM formulations also improve the tumor-to-normal ratio of PS in vivo, as summarized in Table 1.127-129
Table 1.
Drug selectivity for the target site, displayed as tumor:normal tissue ratio as a function of respective PNM formulation, type of PS, and time interval (hours post-injection).
| PNM formulation | Photosensitizer | Tumor:normal ratio | Time post-injection | Reference |
|---|---|---|---|---|
|
| ||||
| DPPC | ZnPC | 7.5* | 24 h | Reddi et al. (1987)130 |
|
| ||||
| DPPC:DPPG (9:1) | mTHPC | 1.5, 3.8, 9 * | 3 h, 6 h, 24 h | Reshetov et al. (2013)131 |
| 1.5, 1, 1 ** | ||||
|
| ||||
| DPPC:DPPG:DSPE-PEG (9:1:1) | mTHPC | 6, 10, 10 * | 3 h, 6 h, 24 h | |
| 1.5, 2.2, 2 ** | ||||
|
| ||||
| DPPC:DPPG:DSPE-PEG (9:1:1) | mTHPC | 10.8 ** | 7.3 h | Bucholz et al. (2005)132 |
|
| ||||
| (DMSO) | Hypocrellin A | 1.9, 2.6, 1.5 * | 3 h, 6 h, 24 h | Wang et al. (1999)133 |
| 1.2, 1.2, 1.5 ** | ||||
|
| ||||
| EPC | Hypocrellin A | 3.3, 3, 2.5 * | 3 h, 6 h, 24 h | |
| 2.1, 2.1, 1.1 ** | ||||
|
| ||||
| Visudyne® | BPD | 1.7, 5 #* | 3 h, 24 h | O’Hara et al. (2009)134 |
| 1.4, 3.5 #** | ||||
|
| ||||
| 1.2, 1.1 ##* | 3 h, 24 h | |||
| 1.9, 3 ##** | ||||
Single asterisks (*) indicate tumor:muscle ratio, double asterisks (**) indicate tumor:skin ratios. Hashtags refer to specific types of pancreatic tumor xenografts, where a single hashtag (#) refers to AsPC-1 pancreatic tumors and double hashtags (##) refer to PANC-1 pancreatic tumors. The PSs formulated in the studies summarized within the table are zinc phthalocyanine (ZnPC), meta-tetra (hydroxyphenyl)chlorin (mTHPC), Hypocrellin A and benzoporphyrin derivative (BPD). Ingredients of the PNM formulations include 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DPPG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol) (DSPE-PEG), dimethylsulfoxide (DMSO) and egg phosphatidylchloine (EPC)
The increased propensity of nanoparticles to accumulate at the tumor site is primarily effectuated by the EPR effect. The EPR effect is the result of the high metabolic demand of the proliferating tumor mass and the resultant aberrant fenestrated angiogenic vasculature that rapidly form, in addition to poor lymphatic drainage of the tumor interstitial fluid.135 The EPR effect allows for the extravasation of high-molecular weight plasma components, and can be exploited by nanocarriers of anti-cancer drugs.136 In order for nanocarriers to fully utilize the EPR effect, a steric barrier that reduces non-specific nanoparticle-protein interactions is crucial to achieve prolonged systemic circulation times. Steric stabilization of nanocarriers with hydrophilic barrier molecules, such as PEG that grants stealth-like properties to nanocarriers can significantly reduce absorption by the liver, kidney, spleen, and the reticuloendothelial system (RES).137 Thus, PEGylation enhances the extent to which the nanoparticles extravasate at the tumor site. For PDT specifically, BPD-containing liposomes that were sterically stabilized with palmityl-D-glucoronide exhibited prolonged circulation times and increased the therapeutic efficacy of the PS, in comparison to treatment with either free BPD or non-sterically stabilized liposomal BPD.129 PNM formulations utilized for non-photochemical PTT applications have also been formulated in a similar manner using identical steric stabilization approaches. An interesting advancement in the field of thermal PNM formulations is the development of the porphysome, a self-assembled nanovesicle consisting of porphyrin-conjugated phospholipid bilayers that was pioneered by Lovell et al. and was PEGylated to improve steric stability.138 Due to the high proximity of the individual lipidated porphyrin molecules within the porphysome bilayer, the chromophores exhibited up to 1500-fold fluorescence quenching and were capable of inducing phototheramlization at levels comparable to gold nanorods. The photoacoustic effect that is concurrent with photothermalization was also leveraged for imaging using PAI. Furthermore, the unique structures were expanded to pyropheophorbide, zinc pyropheophorbide and bacteriochlorophyll porphysome variants. Doxorubicin was also encapsulated within the porphysome to yield a photothermal iteration of Doxil®.
Aside from steric stability and stealth-like properties, appropriate sizing of nanocarriers is essential for the efficacy of a nanosized drug delivery platform. Sterically stabilized liposomes smaller than 70 nm were shown to be retained by the liver,139 whereas liposomes larger than 220 nm were effectively taken up by the spleen.140 As such, it was demonstrated that PEGylated liposomes with sizes between 160-220 nm exhibited the longest circulation times in vivo.140 With respect to exploiting the EPR effect for tumor-specific delivery, the vascular fenestrations of tumor-associated vessels are approximately 100-600 nm in diameter.141 In comparison, healthy vasculature typically displays fenestrations smaller than 10 nm in diameter.142 These physiological characteristics of tumor vasculature can inform the design on nanocarriers, although many factors will influence the success of nanotherapeutics, which include tumor penetration and cellular internalization. These factors include nanoparticle size, formulation material, PEG density, geomentry and electrostatics, all of which must be independently tuned in concert for a given newly-developed PNM formulation.
b. Tailoring tumor physiology to nanomedicines using photochemistry
Given that the abnormal and leaky tumor vasculature contributes to tumor growth and disease progression, there are currently ongoing investigations geared towards the normalization of the vasculature with the aim of inhibiting disease progression.143 For instance, a recent study by Chauhan et al. reports on the normalization of the abnormal tumor vessels by blocking vascular endothelial growth factor receptor-2 (VEGFR-2) signaling in angiogenic vascular endothelial cells.144 With respect to nanomedicine, it was shown that vessel normalization reduced the size of the vascular fenestrations and hindered the delivery of 125 nm quantum dots, whilst improving tumor penetration of 12 nm quantum dots.144 Vascular normalization is an example of how modulating the tumor physiology can enhance the homogenous intratumoral distribution and, subsequently, the therapeutic index of small molecular weight drugs and nanoparticles.145
Conversely, the expansion of tumor vascular fenestrae and vascular permeablization has been achieved by the controlled photochemical activation of a PS when confined to the intravascular space; a phenomenon reported early on by Henderson et al.146 and many others.6, 147-150 At sufficiently low PDT doses, vascular PDT (vPDT) has been shown to further enhance the EPR effect and to improve the extravasation of nanomedicines into the tumor. Low-dose vPDT using the PS BPD was specifically shown to enhance the EPR effect in an orthotropic rat prostate cancer model.147 The tumor uptake of 2,000-kDa FITC labeled dextran particles was enhanced following BPD-induced vPDT with a short 15 min PS-light interval.
More recently, ferritin protein nanocages loaded with the ZnF16Pc PS and surface functionalized with RGD peptide were developed by Zhen et al. (Figure 9).151 These actively targeted nanoparticles, referred to as P-RFRT, selectively bound to αvβ3 integrin receptors on endothelial cells in the tumor vasculature. The irradiance was optimized to 14 mW/cm2 (50-300 mW/cm2 is used for conventional PDT) for the maximal EPR enhancement, with minimal collateral toxicity to the tumor vasculature. Following P-RFRT-mediated vPDT, enhanced tumor accumulation of varying sized cargoes, such as albumin, quantum dots and iron oxide nanoparticles, was demonstrated in multiple xenograft tumor models. Efficacy studies were carried out under the same vPDT conditions followed by administration of Doxil® in a 4T1 tumor model. Importantly, controls studies in animals treated with P-RFRT-mediated vPDT alone showed tumor growth rates similar to saline treated animals, confirming the absence of PDT-related tumor damage. In contrast, animals treated with P-RFRT-mediated vPDT followed by Doxil® treatment experienced a potent tumor-growth inhibition of 85.9%, a 75.3% improvement in efficacy over treatment with Doxil® alone.
Figure 9.
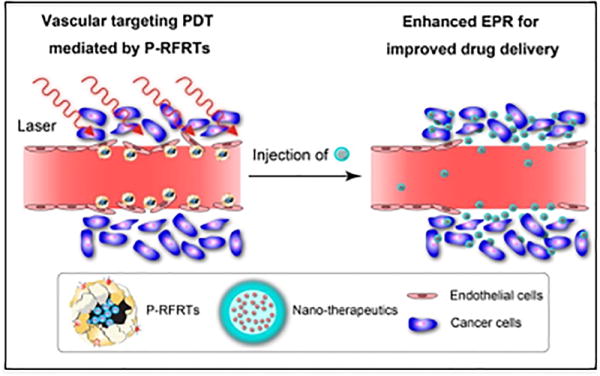
vPDT assisted EPR effect for higher tumor extravasation of nanoparticles. Proposed mechanism of action of ferritin nanoparticles loaded with ZnF16Pc PS and actively targeted with RGD-peptide (P-RFRT).151 P-RFRT mediated vPDT action first increases gaps in the tumor vasculature, thereby facilitating deeper penetration of subsequently administered therapeutic nanoparticles. Figure adapted with permission from Zhen et al.151 copyright 2014 American Chemical Society.
The tumor vasculature varies considerably between different cancer types and can influence the outcome of vPDT.150 Araki et al. employed a porphyrin-based PS loaded within PEG-poly(lactic acid) nanoparticle (PN-Por), to increase vascular permeabilization, which was subsequently followed by chemotherapy using liposomal paclitaxel (PL-PTX).152 The study was performed in murine models of a colon-26 adenocarcinoma (C26) and a B16BL6 melanoma, which vary considerably in their vascular composition and permeability. In the C26 tumor model, PN-Por-mediated vPDT itself caused considerable tumor damage via endothelial cell apoptosis; however, the subsequent PL-PTX regimen did not improve outcome. Conversely, in the B16BL6 model, vPDT enhanced diffusion of PL-PTX into the tumor core and enhanced the efficacy of PL-PTX treatment. The reason for this differential vascular response between the two tumor models was associated with pericyte coverage and vessel diameter. Thus, the overall applicability of vPDT to enhance EPR based nanoparticle accumulation depends on the precise control over light dosage, a better understanding of the different innate microvasculature of tumors and on the continued development of tailored vasculature targeted nanoparticles.
An additional physical barrier in the tumor microenvironment is the stroma. The role of the tumor stroma in cancers, such as pancreatic cancer, has become increasingly regarded as a significant hindrance to drug delivery.153 It has been found that a dense avascular tumor stroma prohibits drug diffusion throughout the tumor tissue.154 In this respect, photochemical approaches may be utilized to improve the delivery and diffusion of cancer therapeutics by disrupting the rigid stromal barriers. With the use of 3D culture models, it is known that pancreatic and ovarian tumor spheroids that develop in an extracellular matrix become increasingly insensitive to chemotherapeutic agents.155 However, neoadjuvant PDT was shown to resensitize the spheroids to chemotherapy by disrupting the nodule’s three-dimensional architecture.155, 156 As such, integrating photochemistry into nanomedicines may ultimately enable the use of forward-looking PNM formulations that can enhance their own delivery into tumors using the assistance of vPDT and even throughout the stroma.
Limiting Toxicity
Even though PDT is largely non-toxic in the absence of light, there are some toxicological issues associated with the therapy. PSs that are systemically administered in free form typically accumulate at high concentrations in the liver, spleen, lungs, and kidneys.157-159 The effective absorption of the PS by these organs potentially reduces the overall amount of PS that can accumulate at the tumor site, ultimately requiring higher drug concentrations to achieve the desired effect. Although the near lack of toxicity in the absence of light constitutes a major benefit of PSs in relation to other cancer therapeutics, PS accumulation in the skin has been a prominent adverse event associated with early PDT agents that lead to skin toxicity and ethical considerations regarding the quality of life of terminally ill patients.160 However, the later ‘second generation’ PSs have minimized these initial problems with skin phototoxicity. In contrast to conventional drugs, where the need for formulation has been driven by non-specific toxicity, the motivation of formulating PS is often a lack of solubility. Nanoformulations that enhance the solubility and molecular monomerization of PS in physiological environments can thus maximize their photoactivity in vivo.
With respect to PDT, several liposomal formulations containing the PS mTHPC, also known as temoporfin or Foscan®, have been developed by Biolitec Research GmbH™. These include a conventional liposomal formulation (Foslip®) and a PEGylated liposomal formulation (Fospeg®). The PDT efficacy of both formulations was superior to that of the free PS.161, 162 With respect to tumor selectivity, Fospeg® had significantly faster tumor accumulation kinetics and a higher tumor selectivity in comparison to Foslip®.163, 164 These results illustrate the significance of steric stabilization with respect to the efficiency of drug delivery and to the potential for reducing off-target phototoxicity with PS nanocarriers.
Reducing the toxicity has been employed extensively for cancer therapeutics so as to reduce the morbidity of these treatments. A fundamental paradigm for reducing toxicity using nanoparticule encapsulated drugs is Doxil®. This formulation contains high intraliposomal doses of doxorubicin, and was primarily developed to address the need for reducing the drug’s cardiac toxicity. Liposomes with high concentrations of doxorubicin, and their EPR-mediated accumulation in tumor tissue following systemic injection led to the slow release of doxorubicin at the tumor site. This nanomedicine showed an improvement in drug delivery and therapeutic efficacy, while significantly reducing the adverse toxicity.3
Photochemistry can play a unique role in providing an additional method for reducing toxicity of diagnostic and therapeutic agents by facilitating the controlled drug release from nanocarriers specifically at the site of the tumor. By using a PNM formulation co-encapsulating both photosensitizing and therapeutic agents, which preferentially accumulates at the tumor site, subsequent irradiation of the lesions results in the oxidative release of the adjuvant therapeutics from the nanocarrier. While the PNM formulation encapsulation shields the drug from systemically exerting its effects, the spatiotemporal controlled phototriggered release upon irradiation confines the toxicity of the drug to the irradiated site. This technique has been successfully applied by Spring et al., who showed that XL184 (cabozantinib, Cometriq®, a c-MET and VEGFR-2 inhibitor) entrapped in a polymeric nanoparticle, further encapsulated in photoactive lipid layers, was effective at a dose that was 1000-fold lower than the total dose of free XL184 administered. 51, 165
Limitations in cellular uptake
Once agents reach the target tumor tissue, another barrier to achieving their anti-neoplastic effect is the hurdles they encounter upon attempting to enter the cell. Cancer cells have evolved multiple mechanisms to evade chemical insult, the first of which are transmembrane efflux transporters, such as P-glycoprotein and ATP-binding cassette transporters (ABCs). In this context, some nanoparticles have drawn particular interest as they can evade transporter efflux of their therapeutic payload by delivering the drugs intracellularly through alternative endocytic pathways. Xu et al. presented a comprehensive and focused review on the mechanisms through which drug-encapsulating nanoparticles, termed nanotransporters when used for this specific role, evade common drug efflux transporters that give rise to multi-drug resistance in a variety of cancers.166 However, although nanoparticles can increase intracellular delivery of cytotoxic and cytotstatic agents, endosomal escape of these agents into the cytosol to perform their function can oftentimes be a second barrier that has been addressed by a photochemistry-based intervention termed photochemical internalization (PCI). Pioneered by the team of Dr. Kristian Berg in 1995, PCI utilizes low PS and light doses that are only sufficient for the selective disturbance of endosomal membranes, resulting in cytosolic release of the endosomal therapeutic contents.167, 168 PCI using the PS aluminium phthalocyanine disulfonate and bleomycin as the therapeutic agent, has been reported to improve the destruction of the peripheral margins of a human TAX-1 xenograft model, as compared to PDT when applied before external beam radiotherapy.169 The technique has rapidly progressed into the clinic and the first clinical trial using the PS tetraphenylchlorin disulfonate (Amphinex®) and bleomycin as the active chemotherapeutic for cutaneous cancers was completed in 2009.170 Currently, a Phase I/II trial using the PS Amphinex® and gemcitabine as the chemotherapeutic for the treatment of inoperable cholangiocarcinoma is ongoing.171 Recently, an elegant study reported the efficacy of haematoporphyrin-mediated PCI for the cytosolic delivery of camptothecin encapsulated within PEG-polyacetal block copolymer nanoparticles that were labile to both acid hydrolysis and photolysis. Photoirradiaton mediated the release of camptothecin from the nanoparticle as well as the endosomal compartment, and enhanced the phototoxicity of HeLa cells by up to 40%, as compared to the irradiation of cells incubated with a combination of free PS and camptothecin. 172 Understandably, PCI requires a delicate balance in the PS and light dosimitery parameters, which if they exceed a certain PS and light threshold, induce PDT tissue damage. Furthermore, it is probable that varying degrees of PDT and PCI can simultaneously occur during the irradiation of the same tumor as a result of the complex light scattering profiles throughout tissue. Thus, a definitive distinction between the two photochemical modalities can often be challenging in vivo.
Acquiring Selectivity
The concept of tumor-targeted therapeutics has been pursued since Paul Ehrlich’s proposal of the ‘magic bullet’, which comprises of a targeting moiety conjugated to a cytotoxic agent.173, 174 Antibody conjugates therefore represent an early version of nanomedicine agents. This vision is realized with antibody-based platforms such as immuno-toxins175, 176 and antibody-drug conjugates (ADCs).177-181 Lately, the field of ADCs has rapidly expanded following the FDA approval of Brentuximab vedotin and Trastuzumab emtansine, as well as the continued clinical development of several candidates.182 Parallel to ADCs, PSs have been conjugated to antibodies to form PICs.183, 184 Following seminal work by Levy et al.185-187 many examples of PICs have been reported by us11, 188-198, 199 , 200 and others,184, 201-205 where biological conjugates of PSs have been deployed as tumor-targeted agents for photoimmunotherapy (PIT) over the past three decades. While conceptually similar to ADCs, PICs present several additional advantages. In addition to antibody targeting, PICs afford a second level of selectivity by focused delivery of light to activate PDT mediated cytotoxicity only in the tumor microenvironment. Unlike ADCs, the inherent fluorescence property of the PS allows optical imaging, thus enabling various diagnostic modalities as discussed in the Theranostics section.11, 201, 206 ADCs and immuno-toxins, antibody conjugates of toxins, require cellular internalization followed by cytosolic release of the cytotoxic cargo, often requiring elaborate endosomal escape strategies. In contrast, PIT does not necessarily require internalization of the PIC and can kill cells even whilst being localized to the cell surface.89, 207 PICs are able to realize mechanistically distinct features of PDT such as its ability to sensitize chemo-resistant cancer cells156, 208 and elicit the bystander effect to extend toxicity to neighboring supporting cells beyond the primary cancer cell target.209-211 Despite these promising features and tremendous efforts there are currently no FDA approved PICs.
Goff et al. employed primary cells obtained directly from patient tissues, spanning multiple cancer types and stages of resistance, to develop various PICs.196 The authors reported the use of a murine antibody, OC-125, which recognizes the cell surface antigen CA 125 expressed by 80% of non-mucinous human OvCa. The PS chlorin e6-monoethylenediamine monoamide (CMA), linked to a poly-glutamic acid polymer, was site-specifically conjugated to oxidized carbohydrate residues of OC-125 to form an anionic PIC. This anionic PIC was also tested ex vivo in ascites or pleural fluid cells from 15 patients with ovarian and non-ovarian cancers. Viability studies in these mixed-cellular models exerted a selective PDT efficacy in OvCa cells over non-ovarian cancers. The killing of non-cancer cells was attributed in part to the bystander effect of PDT. Importantly, OvCa cells from end-stage drug-resistant patients also showed a significant response to the PDT. These chemoresistant cells often lack cross-resistance to PDT. In fact, sublethal doses of PDT can be used to resenstize drug resistant cancer cells to chemotherapy. These encouraging results prompted explorations on the combination of PICs with traditional chemotherapy agents, which otherwise would be sub-optimal in drug-resistant cancer cells. Such mechanistically non-overlapping combinations of PDT with chemotherapy could overcome chemoresistance and potentially exert a synergistic effect in stubbornly refractive tumors.156, 193, 212 Duska et al. used the F(ab′)2 fragment of OC-125 conjugated to a cationic poly-lysine polymer that was modified with multiple chlorin e6 (Ce6) PS molecules.194, 208 This PIC was assessed in 5 human cancer cell lines as well as 19 primary cultures obtained from solid tumors and ascites samples derived from platinum-responsive and platinum-resistant OvCa patients. Ex vivo viability studies using a combination of PICs with cisplatin demonstrated a potent 12.9-fold higher synergistic effect against platinum-resistant cells.208
PICs have also been designed to target the tumor neovasculature to improve the selectivity of vascular PDT (vPDT), which is discussed in greater detail in the Manipulation at the macrophysoiologcial level section. Palumbo et al. employed a PIC called SIP(L19)-PS, which was composed of a porphyrin PS conjugated to an anti-L19 antibody. The PIC targeted the spliced extra-domain B of fibronectin in the small immune protein format, a marker of tumor angiogenesis (Figure 10 (A)).89, 207
Figure 10.

A) Schematic representation of tumor vasculature targeted PIC composed of an antibody, in the small immune protein format (SIP), and PS molecules coupled to the antibody’s lysine residues.89 B) Demonstration of SIP(L19)-PS efficacy in a subcutaneous xenograft model of squamous-cell carcinoma at different time points following targeted PDT.89 Reprinted by permission of Macmillan Publishers Ltd. on behalf of Cancer Research UK: Paulmbo et al.89 British Journal of Cancer, copyright 2011.
This PIC selectively localized to tumor vessels in vivo and, upon irradiation, led to the complete eradication of a subcutaneous squamous cell carcinoma tumor at relatively low administered PS dosages. Figure 10 (B) shows the regression of the subcutaneous tumor up to 16 days following PDT. Interestingly, no tumor relapse was observed for up to 100 days after treatment. The authors confirmed the role of natural killer cells in the observed long-term response, as inhibition of the natural killer cells resulted in the poor retardation of tumor growth following PDT, eventually leading to disease progression. Finally the authors demonstrated the selective staining of vascular structures by the L19 antibody, amongst other vascular targeting antibodies, in normal skin and squamous cell carcinoma lesions extracted from patients. Occult, non-resectable drug-resistant micrometastases spread throughout the peritoneal cavity are widely responsible for treatment failure and disease relapse in OvCa.11, 88 To tackle this challenge, Spring et al. enabled a further level of selectivity in the PIC platform, namely tumor cell activation.11 Specifically this next generation PIC (Cet-BPD) was composed of the EGFR-specific chimeric antibody Cetuximab and the FDA-approved PS BPD (Figure 11).
Figure 11.

A) Pictorial representation of Cetuximab-BPD (Cet-BPD) structure and switch mechanism based on lysosomal degredation and quenching of BPD molecules for tumor-targeted activatable photoimmunotherapy (taPIT). B) An illustrative comparison of the tumor-focused phototoxicity of taPIT vs non-specific action of perennially activated agents. Adapted from Spring et al.11
Overexpression of the EGFR has been observed in up to 70% of OvCa cases and is a biomarker for poor patient prognosis. This tumor-targeted and cell-activatable PIC relied on the static quenching of the BPD molecules whilst the PIC remained intact (OFF-state).11 Following tumor cell binding, receptor-mediated endocytosis and subsequent intracellular endolysosomal degradation of the antibody, the BPD molecules were dequenched and activated (ON-state). This approach greatly simplifies dosimetry and helps reduce bowel toxicity, a major limitation to peritoneal PDT in the clinic. As the ultimate test for the PIC’s tumor selectivity, a disseminated micrometastatic OvCa in vivo model was used. Confocal microscopy demonstrated the PIC’s capacity for EGFR selectivity through an 81% preferential localization of the PIC in epithelial OvCa cells over normal endothelial cells. Furthermore, a clinically used cisplatin/taxol combination produced only a modest 50% reduction in tumor burden after two rounds of chemotherapy, likely due to the chemoresistance of OvCa cells. In contrast, PDT using the Cet-BPD PIC resulted in 89% reduction in tumor burden. The combination of PIC-mediated PDT (1 cycle) with chemotherapy (1 cycle) resulted in an exceptional 97% reduction of peritoneal micrometastatic burden.
In addition to full-length antibody PICs, PSs have been conjugated to various smaller antibody fragments.194, 213-215 Single-domain antibodies (SDAs), are approximately 10-times smaller than full-length antibodies and thus, faster physiological clearance rates mean that SDA- PICs can reach maximal tumor-to-normal ratios faster than larger PICs. Heuker et al. functionalized two EGFR-targeting SDAs with a silicon phthalocynanine PS derivative (IRDye® 700DX).216 Both monovalent (non-internalizing) and biparatopic (internalizing) SDAs were derivatized with the PS to investigate the dependence of cellular internalization on efficacy. Selectivity of cellular uptake and phototoxicity was determined in four cell lines with varying EGFR expression levels. In phototoxicity studies, the biparatopic SDA-PIC was more potent, which is consistent with the general observation that intracellular PIC-mediated PDT is more effective. A phototoxicity study in a co-culture of 14C cells (high EGFR) and HeLa cells (low EGFR) using both SDA-PICs showed an EGFR-selective reduction in viability in the 14C cells. However, the rate of the phototoxicity of the biparatopic internalizing SDA-PIC was significantly higher than that of the monovalent non-internalizing SDA-PICs.
The field of PICs has shown tremendous promise in cancer therapy and diagnostics and there has been a marked rise in the number of PIC studies reporting their effectiveness as targeted molecular imaging probes and PS conjugates that are selective to the tumor and components of the tumor stroma. 184, 201, 205, 217, 218 However, the clinical translation of PICs has lagged behind their ADC counterparts. Other challenges in PIC such as stability, clearance, tumor penetration and toxicity are common to ADCs and biological therapeutics. Specifically for PDT, complex light dosimetry parameters need to be further tackled with novel PIC and agent delivery strategies. None-the-less, an active clinical trial supported by Aspyrian Therapeutics, Inc.™ is currently recruiting for EGFR-selective PIT in patients with recurrent head and neck cancers.219 Such exciting initiatives motivate a further drive in expanding PICs as precursors to targeted PNM formulations, whereby the long-standing preclinical efficacy of PIT is further advanced through nanotechnology.
Targeted nanotherapeutics as mediators for selectivity
As discussed previously, nanocarriers that are passively targeted to tumor tissue hold significant benefits over the use of free pharmaceutical agents, particularly when the drug freely escape from the carrier or can exert its effecty whilst still encapsulated. However, many agents with intracellular targets are unable to diffuse across membranes freely. Conjugation of small molecules, peptides, or antibodies onto the carrier surface may potentiate cell-particle interactions, leading to enhanced uptake and intracellular accumulation of the drug. There is a wide variety of ligands used to target nanocarriers to cancer tissue, which includes folate,220 RGD peptides,221 full-length antibodies,53 Fab’ fragments,222 and single domain antibodies.223 Depending on the PKs of the nanoconstruct, it should be emphasized that ligand-targeted nanotherapeutics does not always improve drug delivery to the target site in comparison to untargeted nanocarriers, but has the benefit of facilitating selective cellular uptake to improve drug efficacy and treatment response.53 However, nanoparticle targeting may also be achieved without the use of conjugated targeting ligands. For instance, liposomes carrying a positive surface charge are taken up specifically by angiogenic endothelial cells, and can thus be used to specifically target the tumor vasculature.224-226 As cationic liposomes can interact with the cell membrane, it is thought that the distorted blood flow at the tumor site leads to shedding of the barrier-forming glycocalyx on endothelial cells and thus, increases the potential for liposome-cell membrane interaction.225-227 Although a plethora of different cationic nanocarriers have shown potential as drug delivery systems, it is currently unclear whether all cationic nanoparticles exert selectivity for the tumor vasculature or whether this phenomenon is specific for liposomes.227
In the field of PDT, targeted PNM formulations may be instrumental in achieving higher vascular selectivity of the treatment. As an advancement to PICs with the additional capacity to selectively deliver high payloads of co-encapsulated secondary agents, targeted PNM formulations hold significant potential in future clinical applications of PDT. However, targeted nanocarriers for PDT have not been explored to a great extent. Targeted nanocarriers developed for PDT applications include gold, silica and polymeric nanoparticles functionalized with anti-HER2 antibodies, lectins (targeted to oncofetal glycans), anti-EGFR antibodies, folate molecules, a tumor vascular targeting F3 peptide and mannose.228-233 All these targeted PNM platforms have enabled selective cancer cell uptake and have enhanced the PDT efficacies (Figure 12).
Figure 12.

A diagrammatic representation of the different targeting ligands used to functionalize nanotherapeutics carrying PSs, to mediate the molecular selectivity of PDT damage. These include antibodies and antibody fragments, glycans targeting endogenous cell surface lectins, exogenous lectins targeting cell surface glycans, and folate molecules targeting the folate receptor.
With regards to liposomes, targeting of both hydrophobic and hydrophilic PSs has been explored with liposomal PSs conjugated to transferrin (sulfonated aluminum phthalocyanine), Uro-10 IgG antibodies (pyropheophorbide) and Cetuximab (BPD).234-236 However, selective in vivo PDT using these targeted PNM formulations have yet to be fully investigated in vivo. The application of a BPD-containing liposomal formulation targeted to the neoangiogenic tumor vasculature by the conjugation of a custom APRPG peptide has been investigated in vivo. The results did not show enhanced tumor localization of BPD in comparison to non-targeted BPD-liposomes, yet targeted PDT yielded a 3-fold increase in efficacy.237 The findings emphasize the importance of spatial control of damage and the necessity for targeted PNM formulations.
With respect to theranostic applications of targeted PNM formulations, silica nanoparticles containing both gadolinium and the PS Ce6 that were targeted with mannose were successfully developed and tested in a rat glioma model. 238 Similarly, polyacrylamide nanoparticles with entrapped porfimer sodium and the MRI contrast agent iron oxide that were functionalized with the nucleolin-binding peptide F3.239 The targeted nanoparticles were shown to substantially enhance the selectivity of PDT on human MBA-MB-435 breast cancer cells in vitro by more than 80%, comparted to therapy using non-targeted nanoparticles. Another interesting development in targeted PNM formulations is the progression of the aforementioned porphysome structure that was used primarily for PTT.138 Jin el al. used folate-derivatized porphysomes, where the lipidated pyropheophorbide PS molecules that were 99.1% quenched became destabilized and dequenched following in vivo targeting. The targeted porphysomes bound to the folate-overexpressing KB tumors and subsequent cellular internalization resulted in the recovery of the lipidated PS’s photoactivity.233 Thus, targeting lead to effective PDT-mediated tumor photosensitization and a 6-fold inhibition in tumor grown, as compared to the non-targeted counterpart. Such exciting studies utilizing advanced PNM formulations for the targeted, tumor-specific induction of PDT are paving the way for further investigations in multiplexed PNM formulation-based combinatorial cancer therapy that will be discussed in the Enhancing combination therapies using photonanomedicine section.
Approaches to manipulate cancer tissue using photochemistry and nanotechnology
Effects and manipulation at the molecular and cellular level
In recent years, the biological responses of PDT have increasingly gained more attention. This information has led to a better understanding on the cell death mechanisms induced by various forms of PDT, as well the compensatory molecular phenomena that occur in the target tissue in its attempt to escape the treatment. It is becoming more evident that the most effective therapies for complicated cancers will involve multi-faceted combination treatments. Figure 13 is a conceptual diagram representing the mechanisms of RMS-mediated tumor damage induced by PDT and how the knowledge of pro-survival molecular pathways induced by an initial therapy can inform synergistic secondary treatment regimens to enhance the efficacy of PDT.
Figure 13.

A conceptual depiction of the molecular responses to PDT and the rationale for mechanism-based combinations. PDT, like other mono-therapies, successfully kills a proportion of cancer cells while others survive. During the process of dying or injury, the cells mount a variety of pro-survival molecular responses that help the injured cells survive and the surviving cells proliferate. The figure gives only some examples of these pro-survival pathways/molecules that are secreted, upregulated or activated as a process of the “shock” of the PDT process. The pro-survival molecular responses elicited by the residual cancer cells following therapy require additional interventions, which can be intelligently co-delivered to the tumors using technologies such as theranostic PNM formulations. A strong, fundamental mechanistic justification for the selected combination maximizes the synergistic therapeutic effect, whist minimizing toxicities. Examples of pro-survival molecular responses targeted in combination therapy with PDT include c-MET/VEGFR-2 signaling using the inhibitor XL184,51 EGFR signaling using the inhibitor erlotinib,240 VEGF induction using a p38 MAPK inhibitor241 and cyclooxygenase-2 (COX-2) induction using the inhibitor NS-389.242
Numerous preclinical studies have already shown that PDT can synergize with chemotherapies and biological inhibitors, as well as re-sensitizing cancer cells to other therapeutic modalities. We strongly believe that designing PDT-based combination regimens that target multiple non-overlapping pathways, such that each component enhances the others, will most effectively improve the outcomes while minimizing systemic toxicities. In this section, we will shortly review the cell death mechanisms induced by PDT, discuss the survival and growth strategies tumors engage to survive PDT, and how these mechanisms can be exploited to enhance the efficacy of anti-cancer treatments. This information will be of vital importance in the design and application of novel PNM formulations that can deliver mechanism-based combination therapies to synergistically improve treatment outcome. PS excitation leads to the production of highly oxidative RMS with short diffusion ranges. Specifically, 1O2 has an area of action that is limited to a spherical radius of diffusion of 100 nm.243 Thus, the PDT effect is highly dependent on the intracellular localization of the PS.160
Hydrophobic PSs that reside within the lipid bilayer of the plasma membrane, for example porfimer sodium and phthalocyanines,61 are capable of oxidizing unsaturated fatty acids within the membrane, causing lipid packing defects that may culminate in necrosis. PSs that localize at the mitochondria, such as BPD,61 may do the same for the mitochondrial membranes, resulting in mitochondrial membrane permeabilization and the release of apoptogenic factors.244 Moreover, the mitochondrial membranes are replete with anti-apoptotic BCL2-family proteins, which are oxidized by the PDT-produced RMS, resulting in their dysfunction and prolongation of the apoptotic pathway.245 Other prominent intracellular PS accumulation sites are the endoplasmic reticulum (ER) and the Golgi-apparatus.61 Following PDT, the damage to these organelles induces ER-stress, protein aggregation, uncoupling of the intracellular Ca2+ homeostasis, and apoptosis via both mitochondrial-dependent and independent pathways.246, 247 Lysosomal PSs exert their effects through lysosomal rupture, of which the released cathepsins are capable of relaying pro-apoptotic signaling, culminating in mitochondrial membrane permeabilization and apoptosis.248, 249 However, when tumors are exposed to sublethal oxidative damage as a result of insufficient photosensitization or the inability of the excitation light to encompass the entire lesion, tumor cells activate coping mechanisms in their attempt to survive therapy. These survival mechanisms involve an antioxidant response, an inflammatory response, a hypoxic stress response, an immediate early response, and the ER-stress response, which have been elaborately reviewed.247
With the available knowledge on the cellular biological and molecular mechanisms by which PDT exerts its therapeutic effects, attempts have been made to enhance the therapeutic efficacy by using biomodulatory approaches to increase intracellular PS levels and to use adjuvant agents that block the PDT-related survival mechanisms. As these approaches entail multiple agents that may all have variable toxicities and individual PK profiles, the co-encapsulation of these agents into single PNM formulations may bear significant therapeutic potential.
For superficial tumors, PDT with the pro-PS ALA that is enzymatically converted into PpIX, is an effective treatment modality. Several biomodulatory approaches to increase the effect of ALA-PDT have focused on increasing PS accumulation in the tumor tissue. Vitamin D analogues calcitriol, cholecalciferol and calcipotriene have been shown to increase intracellular PpIX accumulation following ALA incubation,250, 251 as a result of upregulated CCAAT enhancer binding protein β and coproporphyrinogen oxidase.252 Similarly, methotraxate facilitates PpIX buildup in tumor cells by upregulating coproporphyrinogen oxidase and can exert additional chemotherapeutic effects by inhibiting DNA synthesis.253 With these biomodulatory approaches, the intrinsic aberrant metabolic activity of tumor cells can be exploited using agents that are used clinically for a variety of pathologies. As both ALA and Vitamin D analogues are typically hydrophobic, their combined incorporation into a nanoparticle may be beneficial with respect to their plasma concentrations, PK behavior, cellular uptake, and synergistic effects on tumor photosensitization. With respect to survival pathway inhibition strategies, adjuvant therapies that prevent these survival pathways have yielded promising results. PDT’s oxidative stress activates oxidation-sensitive activators of the NRF2 pathway that subsequently induces the transcription of genes that promote production of antioxidants such as glutathione. Hampering this antioxidant response using inhibitors of glutathione synthesis has been shown to be beneficial for the efficacy of PDT.254, 255 With respect to hypoxia, the intratumoral depletion of oxygen is caused by its photochemical conversion into RMS, and is exacerbated by the vascular shutdown that ensues from PDT. The hypoxic tumor microenvironment activates hypoxia inducible factor 1, which stimulates anaerobic glycolysis, promotes angiogenesis and reduces sensitivity to cell death.256 Interfering with these survival responses in combination with PDT has resulted in an enhancement of PDT efficacy in both in vitro and in vivo models of cancer.257-261 In addition to the antioxidant and hypoxic stress responses, the ER-stress response is engaged as a result of massive protein oxidation and the consequential misfolding, unfolding and aggregation of the oxidized proteins.247, 262, 263 Heat shock proteins and other chaperone proteins aid in the refolding or degradation of the oxidized polypeptides. Therefore, combining PDT with agents that prevent the execution of these chaperone-mediated survival mechanism have yielded promising results with respect to cell killing efficiency.263-265 Studies during which PDT was combined with inhibitors of the inflammatory response and immediate early stress responses have yielded non-coherent results, potentially owing to the dichotomous functions of these signaling pathways with respect to cell survival and cell death.247, 266-268
At a molecular biological level, it is important to emphasize that PDT and more conventional cancer therapeutics, such as chemotherapy and RTs, do not have overlapping mechanisms of cytotoxicity. Classic chemotherapeutic agents and radiotherapy rely on inflicting DNA damage that translates into an apoptotic pathway via the nucleus 269, 270. In comparison, PDT may either cause direct by directly permeabilizing the plasma membrane of tumor cells, or by inducing apoptotic signaling via organelle damage.244, 271, 272 Rizvi et al. have shown that ovarian spheroids treated with BPD-PDT were hypersensitized to carboplatin.156 Similar results have been obtained with different PSs and chemotherapeutic agents,155, 273, 274 underscoring that PDT and chemotherapy may act synergistically to eradicate tumor tissue more effectively. Several approved chemotherapeutic agents encompass molecular inhibitors of pro-survival pathways that have been implicated in cellular resistance to PDT, such as bortezomib, an inhibitor of the ER-stress response. The findings summarized herein underline the potential of combination therapies to synergistically enhance cancer treatment outcomes, which can be potentiated by the co-encapsulation of both phototherapeutic and chemotherapeutic compounds into singular nanoparticulate formulations. However, it must be noted that PNM formulations not only alter PK and biodistribution profiles of PSs, but also govern their intracellular fate. Thus, manipulating tumor tissue as informed by the cellular effects of PDT using a particular PS may not be as effective when the biomodulatory agent and the same PDT agent are co-encapsulated into a single PNM formulation.
Enhancing combination therapies using photonanomedicine
Combination chemotherapy is the front line anti-cancer treatment regimen currently used in the clinic.275 However, this approach based on the co-administration of two or more drugs, is far from ideal and suffers from varying bioavailability, PKs, bio-distribution and systemic toxicity.276 These challenges have spurred the development of both organic and inorganic nanoparticles capable of delivering multiple chemotherapeutics and diagnostic agents.276-278 These multi-agent delivery vehicles enable stoichiometric cargo loading, controlled delivery, prolonged circulation times and tumor targeting via the EPR effect. While these constructs have been reviewed elsewhere, here we focus on nanoparticle platforms that leverage light to employ mechanistically-informed therapeutic modalities at the ‘right place and the right time’.
As discussed in the Effects and manipulation at the molecular and cellular level section, drug resistance mechanisms and escape pathways are a major hurdle to maximizing the therapeutic effects of drugs. In an effort to tackle these challenges, Spring et al. recently developed a multi-agent platform termed a photoactivatable multi-inhibitor nanoliposome (PMIL).51 The PMIL is based on a core-shell architecture where a lipid layer embedded with hydrophobic BPD PS molecules encases a PEG-PLGA nanoparticle that entraps a secondary hydrophobic multi-kinase inhibitor, XL184 (cabozantinib, Cometriq®). The water soluble block copolymer nanoparticles were physically entrapped in BPD-containing photoactive lipid bilayers, and possibly monolayers that are known to self-assemble onto PLGA nanoparticles.279 The PMIL platform combines three complimentary treatment modalities: PDT, VEGFR-2 receptor inhibition and c-MET receptor inhibition to elicit the anti-vascular and anti-tumoral properties of PDT with the anti-angiogenic and anti-invasive effects of XL184. Upon NIR light illumination, PDT damages tumor cells and induces thrombotic cascades that ultimately lead to vascular occlusion. It is speculated that a macrophysiological effect, such as vascular occlusion, can lead to prolonged intratumoral retention of nanotherapeutics. The complex vascular effects of PDT and its specific role on the delivery of nanomedicines will be described in further detail in the Manipulation at the macrophysoiologcial level section. The PMIL was designed to shield XL184-containing nanoparticles by the photoactive lipid encapsulation, until photoirradiation of the BPD PS with 690 nm light disrupts the lipid layer and triggers controlled release of the XL184. Subsequent slow-release biodegradable nanoparticles, such as the PEG-PLGA particle encapsulating the XL184, can be chemically tuned to modulate the drug release kinetics and sustain tumor exposure to the agent, thus presenting an additional layer of complexity to combinatorial PNM formulations such as the PMIL. XL184 inhibits VEGFR-2 and c-MET signaling, thus suppressing several escape pathways that govern tumor angiogenesis, vascular regrowth, tumor cell invasion and metastatic escape. This synergistic three-way mode of action elicited by PMIL-based combinatorial PDT is summarized in Figure 14.
Figure 14.

Schematic representation of the three-way mode of action of photoactivatable multi-inhibitor nanoliposomes (PMIL).51
In a subcutaneous mouse model of pancreatic cancer, combinatorial PDT using the PMIL reduced the mean tumor volume by 92% compared to the no-treatment control group, whilst the monotherapies and the co-administered same nanoformulations of BPD and XL184 packaged individually had no significant effect. Moreover, in an orthotopic metastatic pancreatic model, PMIL showed a reduction in microvascularity of the tumors and a 99% mean reduction in liver and retroperitoneal lymph node metastasis, with respect to the no-treatment control group. Interestingly the dosage of XL184 in the PMIL was 1000-fold lower than that used in standard oral preclinical regimens needed to achieve the same therapeutic efficacy. This increased drug potency underscores a substantial advantage of PNM formulations, considering the systemic toxicity of multi-kinase inhibitors like XL184. The light-assisted, controlled release of secondary agents such as XL184 from PNM formulations is critical to their improved tolerance. The improved efficacy of the PMIL is multi-factorial, owing to the fact that such PNM formulations improve bioavailability of therapeutics, increase tumor accumulation, prime and resensitize tumors to inhibitors through PDT and most importantly, provide a sophisticated platform for the spatiotemporal induction of multiple combined treatment regimes.
In a complimentary approach to overcome PDT escape pathways mediated by VEGF, Tangutoori et al. aimed to neutralize intracellular VEGF prior to its secretion.280 To realize this goal, a nanophotoactivatable liposome (nanoPAL) platform was developed.280 This multi-agent vehicle was loaded with BPD embedded in the liposomal bilayer and the clinically-approved anti-VEGF monoclonal antibody, bevacizumab (Avastin®), encapsulated in the aqueous core, for the photo-triggered intracellular delivery. The synthesis parameters were rigorously optimized to preserve the antibody’s biological activity, to maintain BPD in its monomeric photoactive state, to offer favorable payload release and to promote cellular uptake whilst limiting systemic toxicity though a moderate cationic surface charge. Figure 15 (A) shows the TEM images of the nanoPAL before and after PDT using 1 J/cm2 of 690 nm light, which results in the destabilization of the spherical structure. Loss of liposomal integrity translates to the therapeutic phototriggered release of bevacizumab. In vitro studies using human pancreatic ductal adenocarcinoma cells (AsPC1) demonstrated that the integrated nanoPAL platform enhanced the cellular co-delivery of fluorescently labeled bevacizumab and BPD, as compared to the co-incubation of the individual liposomal BPD formulation, Visudyne®, and free bevacizumab. This trend was also observed in cytotoxicity studies in the same cell line, with nanoPAL eliciting 82% cell death relative to only 54% by the co-incubated independent agents upon light irradiation. Finally, in a subcutaneous AsPC1 xenograft model of pancreatic cancer, treatment with a co-administered regimen of independent Visudyne® and bevacizumab agents only stabilized tumor growth. In contrast, co-administration through the nanoPAL platform induced extensive necrotic tumor damage and arrested tumor growth. Minimal regrowth of the tumor was observed through completion of the study at 35 days.
Figure 15.

Multi-agent nanoconstructs for mechanistically informed combination therapy A) TEM images of nanophotoactivatable liposome (nanoPAL) before and after PDT. (Reprinted from Tangutoori et al.280 with permission from Elsevier.) B) Pictorial representation of the structure and composition of the NCP@pyrolipid (Adapted with permission from He et al.281 copyright 2015 American Chemical Society.) and C) the nanoporphyrin platform (NP) (Reprinted by permission of Macmillan Publishers Ltd. Li et al.282 Nature Communications, copyright 2014).
In another rendition of the core-shell design, He et al. developed a multi-agent nanoparticle to deliver cisplatin and the PS pyropheophorbide (Figure 15 (B)).281 The core was based on their previously established nanoscale coordination polymer (NCP) platform within which cisplatin was coordinated as a prodrug with Pt in the +IV oxidation state. The shell was composed of a lipid bilayer doped with a phospholipid-porphyrin amphiphile (pyrolipid), originally developed by Zheng and Lovell for the porphysome.138, 283, 284 The self-assembled ~108 nm nanoparticle, termed NCP@pyrolipid, resulted in quenching of the lipidated porphyrin pyropheophorbide PS molecules. In vitro studies demonstrated that efficient uptake of NCP@pyrolipid within SQ20B head and neck cancer cells occurred within 1 hour and gradually destabilized after 2 hours, as indicated by an increase in PS fluorescence. The cytotoxicity of NCP@pyrolipid was tested in cisplatin-sensitive and cisplatin-resistant cell lines. The particle showed a potent synergistic action between PDT and cisplatin, reducing the IC50 values in the JSQ3 cisplatin-resistant head and neck cancer line from 13.33 μM using free cisplatin to 1.21 μM using NCP@pyrolipid PDT when irradiated at 690 nm. Encouraged by these in vitro studies, the authors investigated the NCP@pyrolipid efficacy in a cisplatin-resistant SQ20B subcutaneous xenograft mouse model. Combinatorial NCP@pyrolipid-PDT resulted in approximately 83% reduction in tumor volume, whilst controls including chemotherapy alone, PDT alone and NCP@pyrolipid without irradiation did not result in any tumor regression at the administered dose. An elegant micellar construct called Nanoporphyrin (NP) was introduced by Li et al. as an ‘all-in-one’ theranostic platform (Figure 15 (C)).282 NP were based on linear PEG chains and a dendritic oligomer composed of the PS pyropheophorbide a and cholic acid. To improve stability in the blood, NPs were further stabilized by disulfide cross-linking. The incorporation of the PS within the delivery vehicle itself lies at the core of the NP design. The PS is quenched in the NP and activated upon NP dissociation via light activation and disulfide reduction in the tumor microenvironment, thus reducing non-specific off-target phototoxicity. Irradiation of the PS with NIR light generated RMS and localized thermalization to induce both PDT and PTT therapeutic modalities, respectively. Further, doxorubicin was also loaded into the hydrophobic core of the cross-linked NPs. The therapeutic efficacy of combined PDT, PTT and doxorubicin was successfully demonstrated in transgenic breast cancer and OvCa xenograft mouse models. The innate metal-chelating capability of NP enabled loading of Gd(III) and 64Cu(II) for to provide theranostic PNM formulations that are capable of acting as contrast agents for MRI and PET imaging respectively, in addition to the NIR fluorescence imaging capabilities. NP’s ability to detect tumors by NIR fluorescence imaging, MRI, PET as well as dual MRI-PET were demonstrated in multiple mouse models.
Wang et al. engineered a dual-switch mesoporus silica nanoparticle (MSN) platform capable of selectively delivering drugs to either the extracellular or intracellular environment.285 This was achieved by using two gates that trapped the cargo in the MSN pores, and opened upon either UV-light exposure or a decrease in pH levels. The switch mechanism was based on binding between α-cyclodextrin with phenyl groups (pH responsive) and β-cyclodextrin with azobenzene groups (UV-responsive). The controlled dual-delivery was demonstrated with fluorescent dyes and the anti-cancer drugs octreotide acetate and epirubicin. Octreotide acetate elicits its cytotoxicity by binding to somatostatin receptors on the cell surface while epirubicin blocks DNA/RNA machinery in the nucleus. Cell viability assays using A549 cells demonstrated maximal efficacy of the dual-drug loaded MSNs with both UV and pH-activation, compared to either trigger alone. While this proof of concept study is encouraging, the applicability of UV light for deep tissue photoactivation is limited and potential long-term toxicity issues related to MSN administration need to be fully investigated.
Manipulation at the macrophysoiologcial level
As discussed earlier in the Tailoring tumor physiology to nanomedicines using photochemistry section, low-dose vascular PDT (vPDT) can be used to enhance the delivery of nanomedicines by expanding the tumor vascular fenestrae, and thus further enhance the EPR effect. Within the context of therapeutic PDT tissue damage, the anti-tumor mechanisms of photochemistry include damaging the pathological vasculature, in addition to the aforementioned direct tumor cell insults.286 vPDT relies on the preferential accumulation of the PS in the neovasculature over the tumor and is characterized by a short PS-light interval in its application, as shown in Figure 16.147, 286-289
Figure 16.

A Schematic representation of passive and active vascular PDT (vPDT).286 Passive vPDT relies on the PS’s inherent pharmacological ability to preferntially accumulate in the tumor vasculature while active vPDT leverages a tumor vascular homing ligand chemically conjugated to the PS or PS loaded nanoparticle. (Reproduced with permission of Begell House Inc. Publishers Chen et al.286 Critical reviews in eukaryotic gene expression via the Copyright Clearance Center.)
High-dose vPDT causes thrombus formation, vasoconstriction, occlusion and decreased blood perfusion, whilst low-dose vPDT can be used to enhance vascular permeability. The most well known clinical application of high-dose vPDT has been for treating AMD with Visudyne®.290, 291 In the anti-cancer arena TOOKAD has been widely studied as an intravascular PS for recurrent prostate cancer.147, 292 vPDT can be achieved by either passive or active targeting. The former relies on the PK properties of the PS or the PNM formulation to preferentially accumulate in tumor vasculature within a specific time-period following intravenous injection, thus providing a therapeutic window.287, 293, 294 Active targeting involves the conjugation of vasculature-specific ligands that selectively bind to proteins such as VEGFRs and integrins.286, 295 In this section, nanoparticle-assisted vPDT by both passive and active targeting strategies will be the focus.
Different nanoparticle formulations used to deliver the same PS can result in cellular PDT, vPDT or a combination of both. In a study by Garcia-Diaz et al., the PS temocene was delivered in three formats: micellar, liposomal and free form.296 In vivo studies were performed as either vascular or cellular PDT using a 15 minute or 24 hour PS-light interval, respectively. Interestingly, the most effective treatment was observed in the case of temocene delivered in micelles with a short PS-light interval through vPDT, whilst liposmal temocene was more potent with the longer PS-light interval inducing cellular PDT. The Russell group developed surface functionalized gold nanoparticles with a zinc phthalocyanine-based PS.297 PDT efficacy studies in a murine model of amelanotic melanoma were performed with a PS-light interval of 3 hours and 24 hours following injection of the nanoparticles, which correspond to the highest accumulation time points in the serum and the tumor, respectively. A delay in tumor regrowth was observed in the shorter interval treatment regimen, implying a more vPDT-based mechanism of action. This was conclusively demonstrated with electron microscopy images of tumor tissues that showed extensive capillary damage and leakage of erythrocytes.
Active targeting of nanoparticles to the tumor vasculature with peptide ligands is a promising complimentary strategy.232, 295 A landmark study by Reddy et al. used Photofrin® encapsulated in polyacrylamide particles functionalized with the vascular homing, 31 residue F3 peptide.232 The F3 peptide binds to the cell surface protein nucleolin, which is a marker for angiogenic endothelial cells in the tumor vasculature. Guided by MRI imaging through the integration of iron oxide within the PNM formulation, PDT treatment demonstrated higher survival rates in glioma-bearing rats treated with the targeted nanoparticles, as compared to non-targeted particles or un-encapsulated Photofrin®.
Recently, Bechet et al. developed a hybrid silica-based nanoparticle with encapsulated gadolinium oxide for MRI imaging and a chlorin PS for PDT.298, 299 These ultra-small particles, approximately 2.8 nm in diameter, were surface-functionalized with a short peptide sequence (ATWLPPR) that targets tumor vasculature by binding to the VEGF receptor neuropilin-1 (NRP-1).300, 301 Following in vitro validation of NRP-1 targeting and phototoxicity, the multi-functional particles were used to treat glioblastoma multiforme in an orthotopic rat model. MRI imaging of the tumor tissue allowed real-time stereotactic interstitial PDT. Interestingly, the particles localized to the proliferating angiogenic endothelial cells lining the neovessels in the tumor periphery. Following interstitial PDT, multiple non-invasive imaging modalities such as perfusion MRI, proton magnetic resonance spectroscopy and PET-CT were employed to monitor the tumor status. Reduced blood perfusion rates, increased fatty acid production, as well as histological demonstration of vascular disruption and edema, proved a potent in vivo PDT effect.
As with most treatment modalities, more than one mode of action that unite and complement each other can provide significantly improved therapeutic outcomes. A unique advantage of PDT, and subsequent advanced PNM formulations developed thereof, that distinguish them from single modality treatment regimens, is their capacity for high spatiotemporal control over treatment induction, and therefore, mechanism of action. The combined effect of vascular and cellular PDT damage has thus far been achieved through the optimized PS-light interval; however, PNM formulations targeted towards both cellular and vascular tumor components further promise to enhance selectivity and control over PDT action.
Summary and Prospective
Nanomedicine has progressed significantly in recent years because of the high surface area-to-volume ratio of nanoconstructs, their capacity for enhanced site-specific targeting, their capability for cell membrane internalization and their significant biocompatibility following breakthroughs in appropriate surface modifications. Numerous soft nanoconstructs have shown great promise for improving drug stability, altering PK and PD profiles, reducing systemic toxicity, and increasing drug concentrations at tumor site, while many are already approved for human clinical use. These soft nanoconstructs can be relatively easily modified with small photosensitizing molecules adding new functionality to existing FDA-approved nanomedicines. Exciting progress of inorganic ‘hard’ nanomedicines is promising in preclinical studies and clinical trials as therapeutics, whilst few are granted for medical imaging applications in humans. Many of these hard nanomedicines may already possess unique photoresponsive properties (e.g. the surface plasmon resonance properties of gold nanoconstructs) and have be intelligently combined with photosensitizing molecules. In addition to their photochemical therapeutic effects, these hard nanoparticles can image disease tissues via multiple non-invasive and minimally-invasive imaging methods, such as photoacoustic imaging, CT imaging and florescence imaging. These emerging imaging techniques have the potential to aid early diagnostics and treatment regimens with potential applications in detecting early-stage cancer, as well as in guiding drug delivery, light dosimetry, and even surgery. The ability to use photoactive nanomedicines as theranostic contrast agents has attracted increased attention, as they can be overcome common drawbacks such as weak photostability, low sensitivity and physiological compatibility that are associated with traditional contrast agents.
In summary, the substantial chemical and optical advances in nanotechnology has led to a paradigm shift in the field of photomedicine. Although one of the early-approved nanotherapeutics was a PNM formulation (Visudyne®), there are relatively few PNM formulations for cancer in clinical studies compared to the number of PNM-related publications. In principle, these promising preclinical PNM formulations possess the ability to image malignant tissues and spatiotemporally control the release of chemical and biological therapeutic payloads, resensitizing tumor to the agents which they ultimately synergize with. Furthermore, the pre-clinical PNM formulations can also modulate tumor microenvironments to enhanced tumor delivery of drugs and nanoparticles. Much of this potential remains to be realized in complex systems prior to clinical translation but does offer exciting opportunities to further advance cancer theranostics. The true clinical impact of these advanced optical nanoplatforms is expected in the coming decade as the convergence in the scientific and medical understanding of photomedicine and nanotechnology ultimately realizes the niche potential for such state-of-the-art PNM formulations.
Acknowledgments
We thank Dr. Bryan Spring, Dr. Srivalleesha Mallidi and Dr. Akilan Palanisami for insightful discussions. The support of the National Institutes of Health Grants P01CA084203, R01CA156177, and R01CA160998 to T.H. is gratefully acknowledged. Financial support by the Bullock-Wellman Postdoctoral Fellowship # 223794 for G.O. and the MGH-Tosteson-Funds for Medical Discovery (FMD) Fellowship #224889 for H-C.H. is also gratefully acknowledged.
Abbreviation
- 1O2
Singlet oxygen
- 3O2
Triplet oxygen
- ABC
ATP-binding cassette (transporters)
- ALA
5-aminolevulinic acid
- AMD
Age-related macular degeneration
- AML
Acute myeloid leukemia
- BPD
Benzoporphyrin derivative
- Ce6
Chlorin e6
- CELSI
Cerenkov-excited luminescence-scanned imaging
- Cet
Cetuximab
- CMA
Chlorin e6-monoethylenediamine monoamide
- CT
Computed Tomography
- DMSO
Dimethylsulfoxide
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DPPG
1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol)
- DPSE-PEG
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)
- EGFR
Epidermal growth factor receptor
- EMA
European Medicines Agency
- EPC
Egg phosphatidylcholine
- EPR
Enhanced permeability and retention (effect)
- ER
Endoplasmic reticulum
- Fab
Fragment antigen binding
- FDA
Food and Drug Administration
- GLP
Good Laboratory Practice
- GMP
Good Manufacturing Practice
- iCCD
Intensified charge-coupled device
- ICG
Indocyanine green
- IR
Infrared
- MRI
Magnetic resonance imaging
- MSN
Mesoporous silica nanoparticle
- mTHPC
Meta-tetra (hydroxyphenyl)chlorin
- nanoPAL
Nanophotoactivatable liposome
- NCP
Nanoscale coordination polymer
- NIR
Near-infrared
- NP
Nanoporphyrin
- NSCLC
Non-small cell lung cancer
- OvCa
Ovarian carcinoma
- PAI
Photoacoustic imaging
- PD
Pharmacodynamics
- PCI
Photochemical internalization
- PDT
Photodynamic therapy
- PEG
Polyethylene glycol
- PET
Positron emission tomography
- PIC
Photoimmunoconjugate
- PIT
Photoimmunotherapy
- PLG
poly(DL-lactide/glycolide)
- PLGA
poly(lactide-co-glycolic acid)
- PMIL
Photoactivatable multi-inhibitor nanoliposome
- PNM
Photonanomedicine
- PpIX
Protoporphyrin IX
- PS
Photosensitizer
- PTT
Photothermal therapy
- RES
Reticuloendothelial system
- RFA
Radiofrequency ablation
- RMS
Reactive molecular species
- RT
Radiation therapy
- SDA
Single domain antibody
- SERS
Surface-enhance Raman spectroscopy
- taPIT
Tumor-targeted activatable photoimmunotherapy
- TiO2
Titanium dioxide
- UV
Ultraviolet
- VEGF
Vascular endothelial growth factor
- vPDT
Vascular photodynamic therapy
- XL184
Cabozantinib, Cometriq®
- ZnPC
Zinc phthalocyanine
References
- 1.http://worldwide.espacenet.com.
- 2.https://clinicaltrials.gov, search ‘nanoparticles’.
- 3.Barenholz Y. J Controlled Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 4.https://clinicaltrials.gov, search ‘liposomes’.
- 5.https://clinicaltrials.gov.
- 6.Henderson BW, Dougherty TJ. Photochem Photobiol. 1992;55:145–157. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- 7.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Participants VR. Retina (Philadelphia, Pa) 2005;25:119–134. doi: 10.1097/00006982-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Anderson RR, Parrish JA. J Invest Dermatol. 1981;77:13–19. doi: 10.1111/1523-1747.ep12479191. [DOI] [PubMed] [Google Scholar]
- 10.Mallidi S, Spring BQ, Chang S, Vakoc B, Hasan T. Cancer J. 2015;21:194–205. doi: 10.1097/PPO.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai Z, Anbil S, Sears RB, Mensah LB, Goldschmidt R, Erdem SS, Oliva E, Hasan T. Proc Natl Acad Sci U S A. 2014;111:E933–942. doi: 10.1073/pnas.1319493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott JT, Dsouza AV, Davis SC, Olson JD, Paulsen KD, Roberts DW, Pogue BW. Biomed Opt Express. 2015;6:3765–3782. doi: 10.1364/BOE.6.003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pogue BW, Sheng C, Benevides J, Forcione D, Puricelli B, Nishioka N, Hasan T. J Biomed Opt. 2008;13:034009. doi: 10.1117/1.2937476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarvi MT, Patterson MS, Wilson BC. Biophys J. 2012;102:661–671. doi: 10.1016/j.bpj.2011.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentine RM, Ibbotson SH, Brown CT, Wood K, Moseley H. Photochem Photobiol. 2011;87:242–249. doi: 10.1111/j.1751-1097.2010.00829.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Wallace M, Melancon MP. Nanomedicine (London, U K) 2014;9:2041–2057. doi: 10.2217/nnm.14.136. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, El-Sayed IH, Qian W, El-Sayed MA. J Am Chem Soc. 2006;128:2115–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 18.https://clinicaltrials.gov, NCT01679470.
- 19.https://clinicaltrials.gov, NCT02680535.
- 20.Boulnois J-L. Lasers Med Sci. 1986;1:47–66. [Google Scholar]
- 21.Patterson MS, Wilson BC, Graff R. Photochem Photobiol. 1990;51:343–349. doi: 10.1111/j.1751-1097.1990.tb01720.x. [DOI] [PubMed] [Google Scholar]
- 22.Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, Kent E, Bown SG, Hasan T, Pogue BW, Pereira SP. Br J Cancer. 2014;110:1698–1704. doi: 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. Nanomedicine. 2013;9:1–14. doi: 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissig V, Pettinger TK, Murdock N. Int J Nanomed. 2014;9:4357–4373. doi: 10.2147/IJN.S46900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC. Sci Transl Med. 2013;5:213ra167. doi: 10.1126/scitranslmed.3007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barak Y, Heroman WJ, Tezel TH. Middle East Afr J Ophthalmol. 2012;19:43–51. doi: 10.4103/0974-9233.92115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichikawa K, Takeuchi Y, Yonezawa S, Hikita T, Kurohane K, Namba Y, Oku N. Cancer Lett. 2004;205:39–48. doi: 10.1016/j.canlet.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Jermyn M, Davis SC, Dehghani H, Huggett MT, Hasan T, Pereira SP, Bown SG, Pogue BW. Phys Med Biol. 2014;59:1911–1921. doi: 10.1088/0031-9155/59/8/1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bown SG, Rogowska AZ, Whitelaw DE, Lees WR, Lovat LB, Ripley P, Jones L, Wyld P, Gillams A, Hatfield AW. Gut. 2002;50:549–557. doi: 10.1136/gut.50.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore CM, Azzouzi AR, Barret E, Villers A, Muir GH, Barber NJ, Bott S, Trachtenberg J, Arumainayagam N, Gaillac B, Allen C, Schertz A, Emberton M. BJU Int. 2015;116:888–896. doi: 10.1111/bju.12816. [DOI] [PubMed] [Google Scholar]
- 31.Krishnamurthy S, Powers SK, Witmer P, Brown T. Lasers Surg Med. 2000;27:224–234. doi: 10.1002/1096-9101(2000)27:3<224::aid-lsm4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 32.Eljamel MS, Goodman C, Moseley H. Lasers Med Sci. 2008;23:361–367. doi: 10.1007/s10103-007-0494-2. [DOI] [PubMed] [Google Scholar]
- 33.https://clinicaltrials.gov, NCT00617981.
- 34.https://clinicaltrials.gov:, NCT02112656.
- 35.http://investor.celsion.com/releasedetail.cfm?ReleaseID=737033.
- 36.http://investor.celsion.com/releasedetail.cfm?ReleaseID=897613.
- 37.Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, Macarulla T, Lee KH, Cunningham D, Blanc JF, Hubner RA, Chiu CF, Schwartsmann G, Siveke JT, Braiteh F, Moyo V, Belanger B, Dhindsa N, Bayever E, Von Hoff DD, Chen LT, Group NS. Lancet. 2015 doi: 10.1016/S0140-67361500986-1. [DOI] [Google Scholar]
- 38.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm468654.htm.
- 39.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 40.https://clinicaltrials.gov, NCT00802945.
- 41.Awada A, Garcia AA, Chan S, Jerusalem GH, Coleman RE, Huizing MT, Mehdi A, O’Reilly SM, Hamm JT, Barrett-Lee PJ, Cocquyt V, Sideras K, Young DE, Zhao C, Chia YL, Hoch U, Hannah AL, Perez EA, Group NS. Lancet Oncol. 2013;14:1216–1225. doi: 10.1016/S1470-2045(13)70429-7. [DOI] [PubMed] [Google Scholar]
- 42.https://clinicaltrials.gov, NCT02312622.
- 43.https://clinicaltrials.gov, NCT00806156.
- 44.https://clinicaltrials.gov, NCT01492101.
- 45.http://ir.nektar.com/releasedetail.cfm?ReleaseID=902141.
- 46.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Pharm Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sartor O. Urology. 2003;61:25–31. doi: 10.1016/s0090-4295(02)02396-8. [DOI] [PubMed] [Google Scholar]
- 48.Saif MW. JOP. 2013;14:686–688. doi: 10.6092/1590-8577/2028. [DOI] [PubMed] [Google Scholar]
- 49.Lancet JE, Cortes JE, Hogge DE, Tallman MS, Kovacsovics TJ, Damon LE, Komrokji R, Solomon SR, Kolitz JE, Cooper M, Yeager AM, Louie AC, Feldman EJ. Blood. 2014;123:3239–3246. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.https://clinicaltrials.gov, NCT01696084.
- 51.Spring BQ, Sears RB, Zheng LK, Mai Z, Watanabe R, Sherwoord ME, Schoenfeld DA, Pogue BW, Pereira SP, Villa E, Hasan T. Nat Nanotechnol. 2016 doi: 10.1038/nnano.2015.311. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morton SW, Lee MJ, Deng ZJ, Dreaden EC, Siouve E, Shopsowitz KE, Shah NJ, Yaffe MB, Hammond PT. Sci Signaling. 2014;7:ra44. doi: 10.1126/scisignal.2005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mamot C, Ritschard R, Wicki A, Stehle G, Dieterle T, Bubendorf L, Hilker C, Deuster S, Herrmann R, Rochlitz C. Lancet Oncol. 2012;13:1234–1241. doi: 10.1016/S1470-2045(12)70476-X. [DOI] [PubMed] [Google Scholar]
- 54.Cancer Discovery - Immunoliposomes Are Active in EGFR-Overexpressing Solid Tumors. 2013;3:OF15. [Google Scholar]
- 55.https://clinicaltrials.gov, NCT02213744.
- 56.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Sci Transl Med. 2012;4:128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 57.Cui J, Li C, Guo W, Li Y, Wang C, Zhang L, Zhang L, Hao Y, Wang Y. J Controlled Release. 2007;118:204–215. doi: 10.1016/j.jconrel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 58.Steiner R. ch. 2. Laser-Tissue Interactions In: Christian Raulin SK, editor. Laser and IPL Technology in Dermatology and Aesthetic Medicine. Springer Science & Business Media; 2011. [Google Scholar]
- 59.Jacques SL. J Biomed Opt. 2010;15:051608. doi: 10.1117/1.3494561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hahn SM, Fraker DL, Mick R, Metz J, Busch TM, Smith D, Zhu T, Rodriguez C, Dimofte A, Spitz F, Putt M, Rubin SC, Menon C, Wang HW, Shin D, Yodh A, Glatstein E. Clin Cancer Res. 2006;12:2517–2525. doi: 10.1158/1078-0432.CCR-05-1625. [DOI] [PubMed] [Google Scholar]
- 61.O’Connor AE, Gallagher WM, Byrne AT. Photochem Photobiol. 2009;85:1053–1074. doi: 10.1111/j.1751-1097.2009.00585.x. [DOI] [PubMed] [Google Scholar]
- 62.http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=p990048.
- 63.http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm468728.htm.
- 64.http://www.fda.gov/safety/medwatch/safetyinformation/ucm359951.htm.
- 65.http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm278957.htm.
- 66.http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm340913.htm.
- 67.http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm089780.htm.
- 68.http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cftopic/pma/pma.cfm?num=p020021.
- 69.Pogue BW, Elliott JT, Kanick SC, Davis SC, Samkoe KS, Maytin EV, Pereira SP, Hasan T. Phys Med Biol. 2016;61:R57–89. doi: 10.1088/0031-9155/61/7/R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bulin A-L, Truillet C, Chouikrat R, Lux F, Frochot C, Amans D, Ledoux G, Tillement O, Perriat P, Barberi-Heyob M, Dujardin C. J Phys Chem C. 2013;117:21583–21589. [Google Scholar]
- 71.Chen G, Qiu H, Prasad PN, Chen X. Chem Rev. 2014;114:5161–5214. doi: 10.1021/cr400425h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kosharskyy B, Solban N, Chang SK, Rizvi I, Chang Y, Hasan T. Cancer Res. 2006;66:10953–10958. doi: 10.1158/0008-5472.CAN-06-1793. [DOI] [PubMed] [Google Scholar]
- 73.Triesscheijn M, Baas P, Schellens JH, Stewart FA. Oncologist. 2006;11:1034–1044. doi: 10.1634/theoncologist.11-9-1034. [DOI] [PubMed] [Google Scholar]
- 74.Miller AC, Henderson BW. Radiat Res. 1986;107:83–94. [PubMed] [Google Scholar]
- 75.http://www.health.alberta.ca/documents/AHTDP-PDT-EEC-UofA-STE.pdf.
- 76.Mura S, Couvreur P. Adv Drug Delivery Rev. 2012;64:1394–1416. doi: 10.1016/j.addr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 77.Niedre MJ, Secord AJ, Patterson MS, Wilson BC. Cancer Res. 2003;63:7986–7994. [PubMed] [Google Scholar]
- 78.Mallidi S, Anbil S, Lee S, Manstein D, Elrington S, Kositratna G, Schoenfeld D, Pogue B, Davis SJ, Hasan T. J Biomed Opt. 2014;19:028001. doi: 10.1117/1.JBO.19.2.028001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mansfield JR, Gossage KW, Hoyt CC, Levenson RM. J Biomed Opt. 2005;10:9. doi: 10.1117/1.2032458. [DOI] [PubMed] [Google Scholar]
- 80.Zimmermann T, Rietdorf J, Pepperkok R. FEBS Lett. 2003;546:87–92. doi: 10.1016/s0014-5793(03)00521-0. [DOI] [PubMed] [Google Scholar]
- 81.Nguyen QT, Tsien RY. Nat Rev Cancer. 2013;13:653–662. doi: 10.1038/nrc3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stummer W, Reulen HJ, Novotny A, Stepp H, Tonn JC. In: Local therapies for glioma; Present status and future developments. Westphal M, Tonn JC, Ram Z, editors. [Google Scholar]; Springer-Verlag Wien KG, Sachsenplatz 4-6, A-1200, Vienna, Austria. Vol. 88. Springer-Verlag New York Inc.; 175 Fifth Avenue, New York, NY, 10010-7858, USA: 2003. pp. 9–12. [Google Scholar]
- 83.Widhalm G, Wolfsberger S, Minchev G, Woehrer A, Krssak M, Czech T, Prayer D, Asenbaum S, Hainfellner JA, Knosp E. Cancer. 2010;116:1545–1552. doi: 10.1002/cncr.24903. [DOI] [PubMed] [Google Scholar]
- 84.Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Hans-Jurgen R, Ala-Glioma Study G. Lancet Oncol. 2006;7:392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 85.Kostron H. In: Photodynamic Therapy: Methods and Protocols. Gomer CJ, editor. Vol. 635. Humana Press Inc; Totowa: 2010. pp. 261–280. [Google Scholar]
- 86.Jocham D, Stepp H, Waidelich R. Eur Urol. 2008;53:1138–1150. doi: 10.1016/j.eururo.2007.11.048. [DOI] [PubMed] [Google Scholar]
- 87.Zhong W, Celli JP, Rizvi I, Mai Z, Spring BQ, Yun SH, Hasan T. Br J Cancer. 2009;101:2015–2022. doi: 10.1038/sj.bjc.6605436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, de Jong JS, Arts HJ, van der Zee AG, Bart J, Low PS, Ntziachristos V. Nat Med. 2011;17:1315–1319. doi: 10.1038/nm.2472. [DOI] [PubMed] [Google Scholar]
- 89.Palumbo A, Hauler F, Dziunycz P, Schwager K, Soltermann A, Pretto F, Alonso C, Hofbauer GF, Boyle RW, Neri D. Br J Cancer. 2011;104:1106–1115. doi: 10.1038/bjc.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blanco E, Kessinger CW, Sumer BD, Gao J. Exp Biol Med. 2009;234:123–131. doi: 10.3181/0808-MR-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee JE, Lee N, Kim T, Kim J, Hyeon T. Acc Chem Res. 2011;44:893–902. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 92.Mai WX, Meng H. Integr Biol. 2013;5:19–28. doi: 10.1039/c2ib20137b. [DOI] [PubMed] [Google Scholar]
- 93.Chen Y, Chen HR, Shi JL. Adv Mater. 2013;25:3144–3176. doi: 10.1002/adma.201205292. [DOI] [PubMed] [Google Scholar]
- 94.Al-Jamal WT, Kostarelos K. Acc Chem Res. 2011;44:1094–1104. doi: 10.1021/ar200105p. [DOI] [PubMed] [Google Scholar]
- 95.Savla R, Taratula O, Garbuzenko O, Minko T. J Controlled Release. 2011;153:16–22. doi: 10.1016/j.jconrel.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 96.Zdobnova TA, Stremovskiy OA, Lebedenko EN, Deyev SM. PLoS One. 2012;7:8. doi: 10.1371/journal.pone.0048248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang K, Feng LZ, Shi XZ, Liu Z. Chem Soc Rev. 2013;42:530–547. doi: 10.1039/c2cs35342c. [DOI] [PubMed] [Google Scholar]
- 98.Hadjipanayis CG, Machaidze R, Kaluzova M, Wang LY, Schuette AJ, Chen HW, Wu XY, Mao H. Cancer Res. 2010;70:6303–6312. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ho D, Sun XL, Sun SH. Acc Chem Res. 2011;44:875–882. doi: 10.1021/ar200090c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McCarthy JR, Weissleder R. Adv Drug Delivery Rev. 2008;60:1241–1251. doi: 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hainfeld JF, Slatkin DN, Focella TM, Smilowitz HM. Br J Radiol. 2006;79:248–253. doi: 10.1259/bjr/13169882. [DOI] [PubMed] [Google Scholar]
- 102.Lane LA, Qian XM, Nie SM. Chem Rev. 2015;115:10489–10529. doi: 10.1021/acs.chemrev.5b00265. [DOI] [PubMed] [Google Scholar]
- 103.Hainfeld JF, Slatkin DN, Smilowitz HM. Phys Med Biol. 2004;49:N309–N315. doi: 10.1088/0031-9155/49/18/n03. [DOI] [PubMed] [Google Scholar]
- 104.Misawa M, Takahashi J. Nanomedicine. 2011;7:604–614. doi: 10.1016/j.nano.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 105.Wieder ME, Hone DC, Cook MJ, Handsley MM, Gavrilovic J, Russell DA. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2006;5:727–734. doi: 10.1039/b602830f. [DOI] [PubMed] [Google Scholar]
- 106.Dykman L, Khlebtsov N. Chem Soc Rev. 2012;41:2256–2282. doi: 10.1039/c1cs15166e. [DOI] [PubMed] [Google Scholar]
- 107.Sancey L, Lux F, Kotb S, Roux S, Dufort S, Bianchi A, Cremillieux Y, Fries P, Coll JL, Rodriguez-Lafrasse C, Janier M, Dutreix M, Barberi-Heyob M, Boschetti F, Denat F, Louis C, Porcel E, Lacombe S, Le Duc G, Deutsch E, Perfettini JL, Detappe A, Verry C, Berbeco R, Butterworth KT, McMahon SJ, Prise KM, Perriat P, Tillement O. Br J Radiol. 2014;87:15. doi: 10.1259/bjr.20140134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mallidi S, Huang H-C, Liu JY, Mensah L, Mai Z, Hasan T. Cancer Research. 2013;73:3923. [Google Scholar]
- 109.Ho CJ, Balasundaram G, Driessen W, McLaren R, Wong CL, Dinish US, Attia AB, Ntziachristos V, Olivo M. Sci Rep. 2014;4:5342. doi: 10.1038/srep05342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen Y, Sajjad M, Wang Y, Batt C, Nabi HA, Pandey RK. ACS Med Chem Lett. 2011;2:136–141. doi: 10.1021/ml100211g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mallidi S, Watanabe K, Timerman D, Schoenfeld D, Hasan T. Theranostics. 2015;5:289–301. doi: 10.7150/thno.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosen JE, Serge Y, Ameena M, Mohit V, Frank XG. J Nanomed Nanotechnol. 2011;2:1000115. [Google Scholar]
- 113.Elliott JT, Samkoe KS, Gunn JR, Stewart EE, Gardner TB, Tichauer KM, Lee TY, Hoopes PJ, Pereira SP, Hasan T, Pogue BW. Acad Radiol. 2015;22:572–579. doi: 10.1016/j.acra.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tanha K, Pashazadeh AM, Pogue BW. Biomed Opt Express. 2015;6:3053–3065. doi: 10.1364/BOE.6.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang RX, D’Souza AV, Gunn JR, Esipova TV, Vinogradov SA, Glaser AK, Jarvis LA, Gladstone DJ, Pogue BW. Opt Lett. 2015;40:827–830. doi: 10.1364/OL.40.000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Axelsson J, Davis SC, Gladstone DJ, Pogue BW. Med Phys. 2011;38:4127–4132. doi: 10.1118/1.3592646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jarvis LA, Zhang RX, Gladstone DJ, Jiang SD, Hitchcock W, Friedman OD, Glaser AK, Jermyn M, Pogue BW. Int J Radiat Oncol, Biol, Phys. 2014;89:615–622. doi: 10.1016/j.ijrobp.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 118.Ross HH. Anal Chem. 1969;41:1260–1265. [Google Scholar]
- 119.Kotagiri N, Sudlow GP, Akers WJ, Achilefu S. Nat Nanotechnol. 2015;10:370–379. doi: 10.1038/nnano.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Silindir M, Ozer AY, Erdogan S. Drug Delivery. 2012;19:68–80. doi: 10.3109/10717544.2011.635721. [DOI] [PubMed] [Google Scholar]
- 121.Chen W, Zhang J. J Nanosci Nanotechnol. 2006;6:1159–1166. doi: 10.1166/jnn.2006.327. [DOI] [PubMed] [Google Scholar]
- 122.Rosenthal EL, Warram JM, de Boer E, Chung TK, Korb ML, Brandwein-Gensler M, Strong TV, Schmalbach CE, Morlandt AB, Agarwal G, Hartman YE, Carroll WR, Richman JS, Clemons LK, Nabell LM, Zinn KR. Clin Cancer Res. 2015;21:3658–3666. doi: 10.1158/1078-0432.CCR-14-3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anselmo AC, Mitragotri S. AAPS J. 2015;17:1041–1054. doi: 10.1208/s12248-015-9780-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reddi E, Lo Castro G, Biolo R, Jori G. Br J Cancer. 1987;56:597–600. doi: 10.1038/bjc.1987.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 126.Li L, Huh KM. Biomater Res. 2014;18:19. doi: 10.1186/2055-7124-18-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Richter AM, Waterfield E, Jain AK, Canaan AJ, Allison BA, Levy JG. Photochem Photobiol. 1993;57:1000–1006. doi: 10.1111/j.1751-1097.1993.tb02962.x. [DOI] [PubMed] [Google Scholar]
- 128.Wang Z-J, He Y-Y, Huang C-G, Huang J-S, Huang Y-C, An J-Y, Gu Y, Jiang L-J. Photochem Photobiol. 1999;70:773–780. [PubMed] [Google Scholar]
- 129.Oku N, Saito N, Namba Y, Tsukada H, Dolphin D, Okada S. Biol Pharm Bull. 1997;20:670–673. doi: 10.1248/bpb.20.670. [DOI] [PubMed] [Google Scholar]
- 130.Milanesi C, Biolo R, Reddi E, Jori G. Photochem Photobiol. 1987;46:675–681. doi: 10.1111/j.1751-1097.1987.tb04831.x. [DOI] [PubMed] [Google Scholar]
- 131.Reshetov V, Lassalle HP, Francois A, Dumas D, Hupont S, Grafe S, Filipe V, Jiskoot W, Guillemin F, Zorin V, Bezdetnaya L. Int J Nanomed. 2013;8:3817–3831. doi: 10.2147/IJN.S51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buchholz J, Kaser-Hotz B, Khan T, Rohrer Bley C, Melzer K, Schwendener RA, Roos M, Walt H. Clin Cancer Res. 2005;11:7538–7544. doi: 10.1158/1078-0432.CCR-05-0490. [DOI] [PubMed] [Google Scholar]
- 133.Wang ZJ, He YY, Huang CG, Huang JS, Huang YC, An JY, Gu Y, Jiang LJ. Photochem Photobiol. 1999;70:773–780. [PubMed] [Google Scholar]
- 134.O’Hara J, Samkoe KS, Chen A, Hoopes PJ, Rizvi I, Hasan T, Pogue BW. 2009 [Google Scholar]
- 135.Maeda H. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 136.Maeda H. Bioconjugate Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 137.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Cancer Res. 1994;54:987–992. [PubMed] [Google Scholar]
- 138.Lovell JF, Jin CS, Huynh E, Jin H, Kim C, Rubinstein JL, Chan WC, Cao W, Wang LV, Zheng G. Nat Mater. 2011;10:324–332. doi: 10.1038/nmat2986. [DOI] [PubMed] [Google Scholar]
- 139.Liu D, Mori A, Huang L. Biochim Biophys Acta, Biomembr. 1992;1104:95–101. doi: 10.1016/0005-2736(92)90136-a. [DOI] [PubMed] [Google Scholar]
- 140.Awasthi VD, Garcia D, Goins BA, Phillips WT. Int J Pharm. 2003;253:121–132. doi: 10.1016/s0378-5173(02)00703-2. [DOI] [PubMed] [Google Scholar]
- 141.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Am J Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Pharmacol Rev. 1999;51:691–744. [PubMed] [Google Scholar]
- 143.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chauhan VP, Stylianopoulos T, Martin JD, Popovi Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Nat Nanotechnol. 2012;7:383–388. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jain RK. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 146.Henderson BW, Waldow SM, Mang TS, Potter WR, Malone PB, Dougherty TJ. Cancer Res. 1985;45:572–576. [PubMed] [Google Scholar]
- 147.Chen B, Pogue BW, Luna JM, Hardman RL, Hoopes PJ, Hasan T. Clin Cancer Res. 2006;12:917–923. doi: 10.1158/1078-0432.CCR-05-1673. [DOI] [PubMed] [Google Scholar]
- 148.Kobayashi H, Watanabe R, Choyke PL. Theranostics. 2013;4:81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Snyder JW, Greco WR, Bellnier DA, Vaughan L, Henderson BW. Cancer Res. 2003;63:8126–8131. [PubMed] [Google Scholar]
- 150.Wang Y, Gonzalez M, Cheng C, Haouala A, Krueger T, Peters S, Decosterd LA, van den Bergh H, Perentes JY, Ris HB, Letovanec I, Debefve E. Lasers Surg Med. 2012;44:318–324. doi: 10.1002/lsm.22013. [DOI] [PubMed] [Google Scholar]
- 151.Zhen Z, Tang W, Chuang YJ, Todd T, Zhang W, Lin X, Niu G, Liu G, Wang L, Pan Z, Chen X, Xie J. ACS nano. 2014;8:6004–6013. doi: 10.1021/nn501134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Araki T, Ogawara K, Suzuki H, Kawai R, Watanabe T, Ono T, Higaki K. J Controlled Release. 2015;200:106–114. doi: 10.1016/j.jconrel.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 153.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, DeNicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sherman Mara H, Yu Ruth T, Engle Dannielle D, Ding N, Atkins Annette R, Tiriac H, Collisson Eric A, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng Tiffany W, Dawson David W, Donahue Timothy R, Masamune A, Shimosegawa T, Apte Minoti V, Wilson Jeremy S, Ng B, Lau Sue L, Gunton Jenny E, Wahl Geoffrey M, Hunter T, Drebin Jeffrey A, O’Dwyer Peter J, Liddle C, Tuveson David A, Downes M, Evans Ronald M. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Celli JP, Solban N, Liang A, Pereira SP, Hasan T. Lasers Surg Med. 2011;43:565–574. doi: 10.1002/lsm.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW, Finkelstein D, Hasan T. Cancer Res. 2010;70:9319–9328. doi: 10.1158/0008-5472.CAN-10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bellnier DA, Ho YK, Pandey RK, Missert JR, Dougherty TJ. Photochem Photobiol. 1989;50:221–228. doi: 10.1111/j.1751-1097.1989.tb04152.x. [DOI] [PubMed] [Google Scholar]
- 158.Gomer CJ, Ferrario A. Cancer Res. 1990;50:3985–3990. [PubMed] [Google Scholar]
- 159.Chan WS, Marshall JF, Svensen R, Bedwell J, Hart IR. Cancer Res. 1990;50:4533–4538. [PubMed] [Google Scholar]
- 160.Weijer R, Broekgaarden M, Kos M, Vught R, Rauws EAJ, van Gulik TM, Storm G, Heger M. J Photochem Photobiol, C. 2015;23:103–131. [Google Scholar]
- 161.Bovis MJ, Woodhams JH, Loizidou M, Scheglmann D, Bown SG, MacRobert AJ. J Controlled Release. 2012;157:196–205. doi: 10.1016/j.jconrel.2011.09.085. [DOI] [PubMed] [Google Scholar]
- 162.Buchholz J, Kaser-Hotz B, Khan T, Rohrer Bley C, Melzer K, Schwendener RA, Roos M, Walt H. Clin Cancer Res. 2005;11:7538–7544. doi: 10.1158/1078-0432.CCR-05-0490. [DOI] [PubMed] [Google Scholar]
- 163.Reshetov V, Lassalle HP, François A, Dumas D, Hupont S, Gräfe S, Filipe V, Jiskoot W, Guillemin F, Zorin V, Bezdetnaya L. Int J Nanomed. 2013;8:3817–3831. doi: 10.2147/IJN.S51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.de Visscher SAHJ, Ka akova S, de Bruijn HS, van den Heuvel AvdP, Amelink A, Sterenborg HJCM, Robinson DJ, Roodenburg JLN, Witjes MJH. Lasers Surg Med. 2011;43:528–536. doi: 10.1002/lsm.21082. [DOI] [PubMed] [Google Scholar]
- 165.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman HA, Christensen JG, Aftab DT, McDonald DM. Cancer Discovery. 2012;2:270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xue X, Liang XJ. Chin J Cancer. 2012;31:100–109. doi: 10.5732/cjc.011.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Berg K, Selbo PK, Prasmickaite L, Tjelle TE, Sandvig K, Moan J, Gaudernack G, Fodstad O, Kjolsrud S, Anholt H, Rodal GH, Rodal SK, Hogset A. Cancer Res. 1999;59:1180–1183. [PubMed] [Google Scholar]
- 168.Berg K, Dietze A, Kaalhus O, Hogset A. Clin Cancer Res. 2005;11:8476–8485. doi: 10.1158/1078-0432.CCR-05-1245. [DOI] [PubMed] [Google Scholar]
- 169.Norum OJ, Gaustad JV, Angell-Petersen E, Rofstad EK, Peng Q, Giercksky KE, Berg K. Photochem Photobiol. 2009;85:740–749. doi: 10.1111/j.1751-1097.2008.00477.x. [DOI] [PubMed] [Google Scholar]
- 170.https://clinicaltrials.gov, NCT00993512.
- 171. https://clinicaltrials.gov, NCT01900158.
- 172.Pasparakis G, Manouras T, Vamvakaki M, Argitis P. Nature communications. 2014;5:3623. doi: 10.1038/ncomms4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Elliott RL. Surg Oncol. 2012;21:53–55. doi: 10.1016/j.suronc.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 174.Strebhardt K, Ullrich A. Nat Rev Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 175.Pastan I, Willingham MC, FitzGerald DJ. Cell. 1986;47:641–648. doi: 10.1016/0092-8674(86)90506-4. [DOI] [PubMed] [Google Scholar]
- 176.Antignani A, Fitzgerald D. Toxins. 2013;5:1486–1502. doi: 10.3390/toxins5081486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kovtun YV, Goldmacher VS. Cancer Lett. 2007;255:232–240. doi: 10.1016/j.canlet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 178.Garnett MC. Adv Drug Delivery Rev. 2001;53:171–216. doi: 10.1016/s0169-409x(01)00227-7. [DOI] [PubMed] [Google Scholar]
- 179.Smaglo BG, Aldeghaither D, Weiner LM. Nat Rev Clin Oncol. 2014;11:637–648. doi: 10.1038/nrclinonc.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Koshkaryev A, Sawant R, Deshpande M, Torchilin V. Adv Drug Delivery Rev. 2013;65:24–35. doi: 10.1016/j.addr.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chari RV, Miller ML, Widdison WC. Angew Chem, Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 182.Sassoon I, Blanc V. Methods Mol Biol. 2013;1045:1–27. doi: 10.1007/978-1-62703-541-5_1. [DOI] [PubMed] [Google Scholar]
- 183.Bullous AJ, Alonso CM, Boyle RW. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2011;10:721–750. doi: 10.1039/c0pp00266f. [DOI] [PubMed] [Google Scholar]
- 184.van Dongen GA, Visser GW, Vrouenraets MB. Adv Drug Delivery Rev. 2004;56:31–52. doi: 10.1016/j.addr.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 185.Mew D, Wat CK, Towers GH, Levy JG. J Immunol. 1983;130:1473–1477. [PubMed] [Google Scholar]
- 186.Wat CK, Mew D, Levy JG, Towers GH. Prog Clin Biol Res. 1984;170:351–359. [PubMed] [Google Scholar]
- 187.Mew D, Lum V, Wat CK, Towers GH, Sun CH, Walter RJ, Wright W, Berns MW, Levy JG. Cancer Res. 1985;45:4380–4386. [PubMed] [Google Scholar]
- 188.Savellano MD, Hasan T. Photochem Photobiol. 2003;77:431–439. doi: 10.1562/0031-8655(2003)077<0431:tctote>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 189.Savellano MD, Hasan T. Clin Cancer Res. 2005;11:1658–1668. doi: 10.1158/1078-0432.CCR-04-1902. [DOI] [PubMed] [Google Scholar]
- 190.Abu-Yousif AO, Moor AC, Zheng X, Savellano MD, Yu W, Selbo PK, Hasan T. Cancer Lett. 2012;321:120–127. doi: 10.1016/j.canlet.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Molpus KL, Hamblin MR, Rizvi I, Hasan T. Gynecol Oncol. 2000;76:397–404. doi: 10.1006/gyno.1999.5705. [DOI] [PubMed] [Google Scholar]
- 192.Soukos NS, Hamblin MR, Keel S, Fabian RL, Deutsch TF, Hasan T. Cancer Res. 2001;61:4490–4496. [PMC free article] [PubMed] [Google Scholar]
- 193.Rizvi I, Dinh TA, Yu W, Chang Y, Sherwood ME, Hasan T. Isr J Chem. 2012;52:776–787. doi: 10.1002/ijch.201200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Duska LR, Hamblin MR, Bamberg MP, Hasan T. Br J Cancer. 1997;75:837–844. doi: 10.1038/bjc.1997.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hamblin MR, Miller JL, Hasan T. Cancer Res. 1996;56:5205–5210. [PubMed] [Google Scholar]
- 196.Goff BA, Bamberg M, Hasan T. Cancer Res. 1991;51:4762–4767. [PubMed] [Google Scholar]
- 197.Del Governatore M, Hamblin MR, Shea CR, Rizvi I, Molpus KG, Tanabe KK, Hasan T. Cancer Res. 2000;60:4200–4205. [PubMed] [Google Scholar]
- 198.Goff BA, Hermanto U, Rumbaugh J, Blake J, Bamberg M, Hasan T. Br J Cancer. 1994;70:474–480. doi: 10.1038/bjc.1994.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Oseroff AR, Ohuoha D, Hasan T, Bommer JC, Yarmush ML. Proc Natl Acad Sci U S A. 1986;83:8744–8748. doi: 10.1073/pnas.83.22.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hamblin MR, Del Governatore M, Rizvi I, Hasan T. Br J Cancer. 2000;83:1544–1551. doi: 10.1054/bjoc.2000.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Nat Med. 2011;17:1685–1691. doi: 10.1038/nm.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vrouenraets MB, Visser GW, Stigter M, Oppelaar H, Snow GB, van Dongen GA. Cancer Res. 2001;61:1970–1975. [PubMed] [Google Scholar]
- 203.Vrouenraets MB, Visser GW, Stewart FA, Stigter M, Oppelaar H, Postmus PE, Snow GB, van Dongen GA. Cancer Res. 1999;59:1505–1513. [PubMed] [Google Scholar]
- 204.Hemming AW, Davis NL, Dubois B, Quenville NF, Finley RJ. Surg Oncol. 1993;2:187–196. doi: 10.1016/0960-7404(93)90006-k. [DOI] [PubMed] [Google Scholar]
- 205.Schmidt S, Wagner U, Schultes B, Oehr P, Decleer W, Ertmer W, Lubaschowski H, Biersack HJ, Krebs D. Fortschr Med. 1992;110:298–301. [PubMed] [Google Scholar]
- 206.Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T. Chem Rev. 2010;110:2795–2838. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Casi G, Neri D. J Controlled Release. 2012;161:422–428. doi: 10.1016/j.jconrel.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 208.Duska LR, Hamblin MR, Miller JL, Hasan T. J Natl Cancer Inst. 1999;91:1557–1563. doi: 10.1093/jnci/91.18.1557. [DOI] [PubMed] [Google Scholar]
- 209.Dahle J, Kaalhus O, Moan J, Steen HB. Proc Natl Acad Sci U S A. 1997;94:1773–1778. doi: 10.1073/pnas.94.5.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dahle J, Angell-Petersen E, Steen HB, Moan J. Photochem Photobiol. 2001;73:378–387. doi: 10.1562/0031-8655(2001)073<0378:beicdi>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 211.Poyer F, Thomas CD, Garcia G, Croisy A, Carrez D, Maillard P, Lupu M, Mispelter J. Photodiagn Photodyn Ther. 2012;9:303–309. doi: 10.1016/j.pdpdt.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 212.Postiglione I, Chiaviello A, Palumbo G. Cancers. 2011;3:2597–2629. doi: 10.3390/cancers3022597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bhatti M, Yahioglu G, Milgrom LR, Garcia-Maya M, Chester KA, Deonarain MP. Int J Cancer. 2008;122:1155–1163. doi: 10.1002/ijc.23206. [DOI] [PubMed] [Google Scholar]
- 214.Staneloudi C, Smith KA, Hudson R, Malatesti N, Savoie H, Boyle RW, Greenman J. Immunology. 2007;120:512–517. doi: 10.1111/j.1365-2567.2006.02522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kuimova MK, Bhatti M, Deonarain M, Yahioglu G, Levitt JA, Stamati I, Suhling K, Phillips D. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2007;6:933–939. doi: 10.1039/b708320c. [DOI] [PubMed] [Google Scholar]
- 216.Heukers R, van Bergen en Henegouwen PM, Oliveira S. Nanomedicine. 2014;10:1441–1451. doi: 10.1016/j.nano.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 217.Sato K, Nagaya T, Choyke PL, Kobayashi H. Theranostics. 2015;5:698–709. doi: 10.7150/thno.11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sato K, Hanaoka H, Watanabe R, Nakajima T, Choyke PL, Kobayashi H. Mol Cancer Ther. 2015;14:141–150. doi: 10.1158/1535-7163.MCT-14-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.https://clinicaltrials.gov, NCT02422979.
- 220.Zhao X, Li H, Lee RJ. Expert Opin Drug Delivery. 2008;5:309–319. doi: 10.1517/17425247.5.3.309. [DOI] [PubMed] [Google Scholar]
- 221.Temming K, Schiffelers RM, Molema G, Kok RJ. Drug Resist Updates. 2005;8:381–402. doi: 10.1016/j.drup.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 222.Quarta A, Bernareggi D, Benigni F, Luison E, Nano G, Nitti S, Cesta MC, Di Ciccio L, Canevari S, Pellegrino T, Figini M. Nanoscale. 2015;7:2336–2351. doi: 10.1039/c4nr04426f. [DOI] [PubMed] [Google Scholar]
- 223.Oliveira S, Heukers R, Sornkom J, Kok RJ, van Bergen en Henegouwen PMP. J Controlled Release. 2013;172:607–617. doi: 10.1016/j.jconrel.2013.08.298. [DOI] [PubMed] [Google Scholar]
- 224.Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, Hanahan D, McDonald DM. J Clin Invest. 1998;101:1401–1413. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Campbell RB, Ying B, Kuesters GM, Hemphill R. J Pharm Sci. 2009;98:411–429. doi: 10.1002/jps.21458. [DOI] [PubMed] [Google Scholar]
- 226.Abu Lila A, Ishida T, Kiwada H. Pharm Res. 2009;27:1171–1183. doi: 10.1007/s11095-010-0110-1. [DOI] [PubMed] [Google Scholar]
- 227.Bilensoy E. Expert Opin Drug Delivery. 2010;7:795–809. doi: 10.1517/17425247.2010.485983. [DOI] [PubMed] [Google Scholar]
- 228.Stuchinskaya T, Moreno M, Cook MJ, Edwards DR, Russell DA. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2011;10:822–831. doi: 10.1039/c1pp05014a. [DOI] [PubMed] [Google Scholar]
- 229.Obaid G, Chambrier I, Cook MJ, Russell DA. Angew Chem, Int Ed. 2012;51:6158–6162. doi: 10.1002/anie.201201468. [DOI] [PubMed] [Google Scholar]
- 230.Meyers JD, Cheng Y, Broome AM, Agnes RS, Schluchter MD, Margevicius S, Wang X, Kenney ME, Burda C, Basilion JP. Particle & particle systems characterization : measurement and description of particle properties and behavior in powders and other disperse systems. 2015;32:448–457. doi: 10.1002/ppsc.201400119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gary-Bobo M, Mir Y, Rouxel C, Brevet D, Basile I, Maynadier M, Vaillant O, Mongin O, Blanchard-Desce M, Morère A, Garcia M, Durand JO, Raehm L. Angew Chem, Int Ed. 2011;50:11425–11429. doi: 10.1002/anie.201104765. [DOI] [PubMed] [Google Scholar]
- 232.Reddy GR, Bhojani MS, McConville P, Moody J, Moffat BA, Hall DE, Kim G, Koo YE, Woolliscroft MJ, Sugai JV, Johnson TD, Philbert MA, Kopelman R, Rehemtulla A, Ross BD. Clin Cancer Res. 2006;12:6677–6686. doi: 10.1158/1078-0432.CCR-06-0946. [DOI] [PubMed] [Google Scholar]
- 233.Jin CS, Cui L, Wang F, Chen J, Zheng G. Adv Healthcare Mater. 2014;3:1240–1249. doi: 10.1002/adhm.201300651. [DOI] [PubMed] [Google Scholar]
- 234.Gijsens A, Derycke A, Missiaen L, De Vos D, Huwyler J, Eberle A, de Witte P. Int J Cancer. 2002;101:78–85. doi: 10.1002/ijc.10548. [DOI] [PubMed] [Google Scholar]
- 235.Bergstrom LC, Vucenik I, Hagen IK, Chernomorsky SA, Poretz RD. J Photochem Photobiol, B. 1994;24:17–23. doi: 10.1016/1011-1344(94)07008-3. [DOI] [PubMed] [Google Scholar]
- 236.Mir Y, Elrington SA, Hasan T. Nanomedicine. 2013;9:1114–1122. doi: 10.1016/j.nano.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ichikawa K, Hikita T, Maeda N, Yonezawa S, Takeuchi Y, Asai T, Namba Y, Oku N. Biochim Biophys Acta, Biomembr. 2005;1669:69–74. doi: 10.1016/j.bbamem.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 238.Benachour H, Sève A, Bastogne T, Frochot C, Vanderesse R, Jasniewski J, Miladi I, Billotey C, Tillement O, Lux F, Barberi-Heyob M. Theranostics. 2012;2:889–904. doi: 10.7150/thno.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Reddy GR, Bhojani MS, McConville P, Moody J, Moffat BA, Hall DE, Kim G, Koo YEL, Woolliscroft MJ, Sugai JV, Johnson TD, Philbert MA, Kopelman R, Rehemtulla A, Ross BD. Clin Cancer Res. 2006;12:6677–6686. doi: 10.1158/1078-0432.CCR-06-0946. [DOI] [PubMed] [Google Scholar]
- 240.Edmonds C, Hagan S, Gallagher-Colombo SM, Busch TM, Cengel KA. Cancer Biol Ther. 2012;13:1463–1470. doi: 10.4161/cbt.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Solban N, Selbo PK, Sinha AK, Chang SK, Hasan T. Cancer Res. 2006;66:5633–5640. doi: 10.1158/0008-5472.CAN-06-0604. [DOI] [PubMed] [Google Scholar]
- 242.Ferrario A, Von Tiehl K, Wong S, Luna M, Gomer CJ. Cancer Res. 2002;62:3956–3961. [PubMed] [Google Scholar]
- 243.Hatz S, Poulsen L, Ogilby PR. Photochem Photobiol. 2008;84:1284–1290. doi: 10.1111/j.1751-1097.2008.00359.x. [DOI] [PubMed] [Google Scholar]
- 244.Garg A, Nowis D, Golab J, Agostinis P. Apoptosis. 2010;15:1050–1071. doi: 10.1007/s10495-010-0479-7. [DOI] [PubMed] [Google Scholar]
- 245.Kessel D, Castelli M. Photochem Photobiol. 2001;74:318–322. doi: 10.1562/0031-8655(2001)074<0318:etbitt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 246.Buytaert E, Dewaele M, Agostinis P. Biochim Biophys Acta. 2007;1776:86–107. doi: 10.1016/j.bbcan.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 247.Broekgaarden M, Weijer R, van Gulik TM, Hamblin MR, Heger M. Cancer Metastasis Rev. 2015;34:643–690. doi: 10.1007/s10555-015-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Moan J, Berg K, Anholt H, Madslien K. Int J Cancer. 1994;58:865–870. doi: 10.1002/ijc.2910580620. [DOI] [PubMed] [Google Scholar]
- 249.Wilson PD, Firestone RA, Lenard J. J Cell Biol. 1987;104:1223–1229. doi: 10.1083/jcb.104.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Anand S, Wilson C, Hasan T, Maytin EV. Cancer Res. 2011;71:6040–6050. doi: 10.1158/0008-5472.CAN-11-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Anand S, Rollakanti KR, Horst RL, Hasan T, Maytin EV. Photochem Photobiol. 2014;90:1126–1135. doi: 10.1111/php.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Anand S, Hasan T, Maytin EV. Mol Cancer Ther. 2013;12:1638–1650. doi: 10.1158/1535-7163.MCT-13-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sinha AK, Anand S, Ortel BJ, Chang Y, Mai Z, Hasan T, Maytin EV. Br J Cancer. 2006;95:485–495. doi: 10.1038/sj.bjc.6603273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Nowis D, Legat M, Grzela T, Niderla J, Wilczek E, Wilczynski GM, Glodkowska E, Mrowka P, Issat T, Dulak J. Oncogene. 2006;25:3365–3374. doi: 10.1038/sj.onc.1209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kimani SG, Phillips JB, Bruce JI, MacRobert AJ, Golding JP. Photochem Photobiol. 2012;88:175–187. doi: 10.1111/j.1751-1097.2011.01022.x. [DOI] [PubMed] [Google Scholar]
- 256.Semenza GL. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 257.Ferrario A, Gomer C. J Environ Pathol, Toxicol Oncol. 2006;25:251–260. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i1-2.160. [DOI] [PubMed] [Google Scholar]
- 258.Ferrario A, Lim S, Xu F, Luna M, Gaffney KJ, Petasis NA, Schönthal AH, Gomer CJ. Cancer Lett. 2011;304:33–40. doi: 10.1016/j.canlet.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 259.Broekgaarden M, Weijer R, Weijer M, van den IJssel B, Kos M, Alles LK, van Wijk AC, Bikadi Z, Hazai E, van Gulik TM, Heger M. Nano Res. 2015;16:19960–19977. [Google Scholar]
- 260.Weijer R, Broekgaarden M, Krekorian M, Alles LK, van Wijk AC, Mackaaij C, Verheij J, van der Wal AC, van Gulik TM, Storm G, Heger M. Oncotarget. 2015;7:3341–3356. doi: 10.18632/oncotarget.6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ferrario A, von Tiehl KF, Rucker N, Schwarz MA, Gill PS, Gomer CJ. Cancer Res. 2000;60:4066–4069. [PubMed] [Google Scholar]
- 262.Tsaytler PA, O’Flaherty MC, Sakharov DV, Krijgsveld J, Egmond MR. J Proteome Res. 2008;7:3868–3878. doi: 10.1021/pr800189q. [DOI] [PubMed] [Google Scholar]
- 263.Szokalska A, Makowski M, Nowis D, Wilczynski GM, Kujawa M, Wójcik C, Mlynarczuk-Bialy I, Salwa P, Bil J, Janowska S, Agostinis P, Verfaillie T, Bugajski M, Gietka J, Issat T, Glodkowska E, Mrówka P, Stoklosa T, Hamblin MR, Mróz P, Jakóbisiak M, Golab J. Cancer Res. 2009;69:4235–4243. doi: 10.1158/0008-5472.CAN-08-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ferrario A, Gomer CJ. Cancer Lett. 2010;289:188–194. doi: 10.1016/j.canlet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Nonaka M, Ikeda H, Inokuchi T. Photochem Photobiol. 2004;79:94–98. [PubMed] [Google Scholar]
- 266.Coupienne I, Bontems Sb, Dewaele M, Rubio N, Habraken Y, Fulda S, Agostinis P, Piette J. Biochem Pharmacol. 2011;81:606–616. doi: 10.1016/j.bcp.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 267.Chen HM, Liu CM, Yang H, Chou HY, Chiang CP, Kuo MY. J Oral Pathol Med. 2011;40:483–489. doi: 10.1111/j.1600-0714.2010.00973.x. [DOI] [PubMed] [Google Scholar]
- 268.Broekgaarden M, Kos M, Jurg FA, van Beek AA, van Gulik TM, Heger M. Int J Mol Sci. 2015;16:19960–19977. doi: 10.3390/ijms160819960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bartek J, Lukas J. Curr Opin Cell Biol. 2007;19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 270.Vogelstein B, Lane D, Levine AJ. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 271.Kessel D. Photochem Photobiol. 2014;90:1211–1213. doi: 10.1111/php.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Spring BQ, Rizvi I, Xu N, Hasan T. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2015;14:1476–1491. doi: 10.1039/c4pp00495g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nonaka M, Ikeda H, Inokuchi T. Cancer Lett. 2002;184:171–178. doi: 10.1016/s0304-3835(02)00208-2. [DOI] [PubMed] [Google Scholar]
- 274.Baas P, van Geel IP, Oppelaar H, Meyer M, Beynen JH, van Zandwijk N, Stewart FA. Br J Cancer. 1996;73:945–951. doi: 10.1038/bjc.1996.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.LoRusso PM, Canetta R, Wagner JA, Balogh EP, Nass SJ, Boerner SA, Hohneker J. Clin Cancer Res. 2012;18:6101–6109. doi: 10.1158/1078-0432.CCR-12-2455. [DOI] [PubMed] [Google Scholar]
- 276.Zhang H, Wang G, Yang H. Expert Opin Drug Delivery. 2011;8:171–190. doi: 10.1517/17425247.2011.547470. [DOI] [PubMed] [Google Scholar]
- 277.Iyer AK, Singh A, Ganta S, Amiji MM. Adv Drug Delivery Rev. 2013;65:1784–1802. doi: 10.1016/j.addr.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 278.Hu CM, Aryal S, Zhang L. Ther Delivery. 2010;1:323–334. doi: 10.4155/tde.10.13. [DOI] [PubMed] [Google Scholar]
- 279.Mieszawska AJ, Gianella A, Cormode DP, Zhao Y, Meijerink A, Langer R, Farokhzad OC, Fayad ZA, Mulder WJ. Chem Commun (Cambridge, U K) 2012;48:5835–5837. doi: 10.1039/c2cc32149a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Tangutoori S, Spring BQ, Mai Z, Palanisami A, Mensah L, Hasan T. Nanomedicine. 2015 doi: 10.1016/j.nano.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.He C, Liu D, Lin W. ACS nano. 2015;9:991–1003. doi: 10.1021/nn506963h. [DOI] [PubMed] [Google Scholar]
- 282.Li Y, Lin TY, Luo Y, Liu Q, Xiao W, Guo W, Lac D, Zhang H, Feng C, Wachsmann-Hogiu S, Walton JH, Cherry SR, Rowland DJ, Kukis D, Pan C, Lam KS. Nature communications. 2014;5:4712. doi: 10.1038/ncomms5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lovell JF, Jin CS, Huynh E, MacDonald TD, Cao W, Zheng G. Angew Chem, Int Ed. 2012;51:2429–2433. doi: 10.1002/anie.201108280. [DOI] [PubMed] [Google Scholar]
- 284.Carter KA, Shao S, Hoopes MI, Luo D, Ahsan B, Grigoryants VM, Song W, Huang H, Zhang G, Pandey RK, Geng J, Pfeifer BA, Scholes CP, Ortega J, Karttunen M, Lovell JF. Nature communications. 2014;5:3546. doi: 10.1038/ncomms4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wang W, Wen Y, Xu L, Du H, Zhou Y, Zhang X. Chemistry. 2014;20:7796–7802. doi: 10.1002/chem.201402334. [DOI] [PubMed] [Google Scholar]
- 286.Chen B, Pogue BW, Hoopes PJ, Hasan T. Crit Rev Eukaryotic Gene Expression. 2006;16:279–305. doi: 10.1615/critreveukargeneexpr.v16.i4.10. [DOI] [PubMed] [Google Scholar]
- 287.Chen B, Pogue BW, Hoopes PJ, Hasan T. Int J Radiat Oncol, Biol, Phys. 2005;61:1216–1226. doi: 10.1016/j.ijrobp.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 288.Chen B, Pogue BW, Goodwin IA, O’Hara JA, Wilmot CM, Hutchins JE, Hoopes PJ, Hasan T. Radiat Res. 2003;160:452–459. doi: 10.1667/RR3059. [DOI] [PubMed] [Google Scholar]
- 289.Fingar VH, Kik PK, Haydon PS, Cerrito PB, Tseng M, Abang E, Wieman TJ. Br J Cancer. 1999;79:1702–1708. doi: 10.1038/sj.bjc.6690271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Schmidt-Erfurth U, Hasan T. Surv Ophthalmol. 2000;45:195–214. doi: 10.1016/s0039-6257(00)00158-2. [DOI] [PubMed] [Google Scholar]
- 291.Kramer M, Miller JW, Michaud N, Moulton RS, Hasan T, Flotte TJ, Gragoudas ES. Ophthalmology. 1996;103:427–438. doi: 10.1016/s0161-6420(96)30675-1. [DOI] [PubMed] [Google Scholar]
- 292.Trachtenberg J, Bogaards A, Weersink RA, Haider MA, Evans A, McCluskey SA, Scherz A, Gertner MR, Yue C, Appu S, Aprikian A, Savard J, Wilson BC, Elhilali M. J Urol. 2007;178:1974–1979. doi: 10.1016/j.juro.2007.07.036. discussion 1979. [DOI] [PubMed] [Google Scholar]
- 293.Pegaz B, Debefve E, Borle F, Ballini JP, van den Bergh H, Kouakou-Konan YN. J Photochem Photobiol, B. 2005;80:19–27. doi: 10.1016/j.jphotobiol.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 294.Pegaz B, Debefve E, Ballini JP, Konan-Kouakou YN, van den Bergh H. J Photochem Photobiol, B. 2006;85:216–222. doi: 10.1016/j.jphotobiol.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 295.Neri D, Bicknell R. Nat Rev Cancer. 2005;5:436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 296.Garcia-Diaz M, Kawakubo M, Mroz P, Sagrista ML, Mora M, Nonell S, Hamblin MR. J Controlled Release. 2012;162:355–363. doi: 10.1016/j.jconrel.2012.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Camerin M, Magaraggia M, Soncin M, Jori G, Moreno M, Chambrier I, Cook MJ, Russell DA. European journal of cancer. 2010;46:1910–1918. doi: 10.1016/j.ejca.2010.02.037. [DOI] [PubMed] [Google Scholar]
- 298.Bechet D, Auger F, Couleaud P, Marty E, Ravasi L, Durieux N, Bonnet C, Plenat F, Frochot C, Mordon S, Tillement O, Vanderesse R, Lux F, Perriat P, Guillemin F, Barberi-Heyob M. Nanomedicine. 2015;11:657–670. doi: 10.1016/j.nano.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 299.Couleaud P, Bechet D, Vanderesse R, Barberi-Heyob M, Faure AC, Roux S, Tillement O, Porhel S, Guillemin F, Frochot C. Nanomedicine (London, U K) 2011;6:995–1009. doi: 10.2217/nnm.11.31. [DOI] [PubMed] [Google Scholar]
- 300.Thomas N, Bechet D, Becuwe P, Tirand L, Vanderesse R, Frochot C, Guillemin F, Barberi-Heyob M. J Photochem Photobiol, B. 2009;96:101–108. doi: 10.1016/j.jphotobiol.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 301.Thomas N, Tirand L, Chatelut E, Plenat F, Frochot C, Dodeller M, Guillemin F, Barberi-Heyob M. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2008;7:433–441. doi: 10.1039/b718259g. [DOI] [PubMed] [Google Scholar]