

Abstract
OBJETIVE
Almost all patients with Fragile X Syndrome (FXS) exhibit a CGG repeat expansion (full mutation) in the Fragile Mental Retardation 1 gene (FMR1). Here, we report five unrelated males with FXS harboring a somatic full mutation/deletion mosaicism.
METHODS
Mutational profiles were only elucidated by using a combination of molecular approaches (CGG-based PCR, Sanger sequencing, MS-MLPA, Southern blot and mPCR).
RESULT
Four patients exhibited small deletions encompassing the CGG repeats tract and flanking regions, whereas the remaining had a larger deletion comprising at least exon 1 and part of intron 1 of FMR1 gene. The presence of a 2–3 base pairs microhomology in proximal and distal non-recurrent breakpoints without scars supports the involvement of microhomology mediated induced repair (MMBIR) mechanism in three small deletions.
CONCLUSION
Our data highlights the importance of using different research methods to elucidate atypical FXS mutational profiles, which are clinically undistinguishable and may have been underestimated.
Keywords: Fragile X syndrome, FMR1 gene, copy number variation, deletion, mosaicism
Introduction
Fragile X syndrome [FXS, OMIM 300624] is the most common cause of intellectual disability (ID) and the leading monogenic cause of autism spectrum disorders, affecting approximately 1/5000 males and 1/6000 females [1,2]. In almost all FXS cases, the causative mutation is a large CGG-repeat expansion (>200 CGG repeats) in the 5′ untranslated region of the FMR1 gene [OMIM 309550], which triggers a cascade of epigenetics modifications, resulting in the deficiency or complete absence of the encoded product, FMRP [3]. FMRP is a selective mRNA-binding protein that regulates the translation of a subset of dendritic mRNAs. Its absence causes increased protein synthesis in postsynaptic dendritic and consequently synaptic dysregulation [4]. Besides moderate to severe ID, the clinical spectrum of FXS in males includes a broad spectrum of behavioral, cognitive, neurologic, and physical problems, whereas in females the phenotype can be milder, likely due to X chromosome inactivation [5].
The CGG repeats tract length is highly polymorphic and can be divided into four distinct FMR1 allelic categories: a) normal and generally stable alleles (6–44 CGG repeats); intermediate gray zone alleles slightly unstable on parental transmission (45–54 CGG repeats); c) premutation unstable alleles (CGG 55–200), which are at a high risk of maternal transgenerational expansion and (d) full mutation alleles (greater than 200 CGG repeats), which leads to FMR1 silencing and FXS phenotype (reviewed in [6]).
Molecular diagnosis of FXS relies on the size of the number of CGG repeats and methylation patterns in the FMR1, which has been historically investigated by a combination of both Southern Blotting (SB) analysis and polymerase chain reaction (PCR)-based methods [7–12]. However, point mutations, insertions or deletions at the FMR1 gene, although rare, have been documented and could increase the overall diagnostic yield and help to account for a portion of undiagnosed ID cases [13]. Here, we report on five males with FXS harboring a full mutation/deletion mosaicism, who were identified by a combination of methodologies used to analyze their FMR1 gene structure.
Materials and Methods
Patients
The five Brazilian unrelated males (patients 1033, 1234, 1337, 1513 and 1629) were referred to the Human Genetics Service at the State University of Rio de Janeiro (Rio de Janeiro, Brazil) for FXS investigation, due to an idiopathic history of ID. They took part from a cohort of 247 unrelated males aged from 4 to 22 years (x= 10.15 ± 4.77 years) referred to our laboratory during the last five years for FXS testing from different pediatric/neurologic public centers in Rio de Janeiro.
The Institutional Ethics Committee from State University of Rio de Janeiro approved the research protocols and written informed consent was obtained from legal guardians.
All five patients exhibited a normal karyotype and inconclusive results were found using conventional CGG-based polymerase chain reaction (PCR) [9] (Supplementary Figure 1). Patients 1033, 1234, 1337 and 1629 showed small deletions around the CGG repeats, represented by faint bands visualized on a 6% polyacrylamide gel apparently below the normal range, whereas a weak normal-sized fragment was found for patient 1513 (Supplementary Figure 1).
Molecular Analysis
Methylation Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA, SALSA ME029 probe mix) was performed following the manufacturer’s instructions [MRC-Holland, The Netherlands]. Each MS-MLPA reaction generates two products: one gives information about copy number variations on FMR1/AFF2 genes and the other gives information about FMR1/AFF2 methylation status. After MS-MLPA reaction, samples were submitted to capillary electrophoresis on an ABI3130 Genetic Analyzer [Thermo Fisher Scientifc Inc., USA] and data were analyzed with GeneMarker v.2.4.0 software [SoftGenetics, USA].
Southern Blot analysis (SB) with probe StB12.3 was conducted as previously described [14].
High Resolution Methylation PCR (mPCR) was performed using AmplideX FMR1 mPCR kit [Asuragen Inc., Austin, TX, USA] [12]. For segregation analysis, patients’ mothers were evaluated also by Triplet Repeat Primed-PCR (RTP-PCR) through AmplideX FMR1 PCR kit [Asuragen Inc.] [11].
Direct Sanger sequencing of standard CGG-based PCR amplicons [Fu et al, 1991] was applied to validate the small deletions and to define deletions’ breakpoints. To gain insight into the underlying mechanism(s) of the rearrangements, bioinformatic analysis of the sequences flanking the deletions’ breakpoints were performed with RepeatMasker Documentation program [101]. For patient 1513, attempt of fine mapping the deletion breakpoints was performed by iterative rounds of regular PCR and quantitative Real Time PCR (qPCR, primers sequences on Supplementary Table 1).
qPCR reactions were conducted on a 7500 Fast Real Time PCR System [Thermo Fisher Scientific Inc.] and data analysis was performed according to the ΔΔCt method, using PORCN gene (Xp11.23) as a normalizer.
FMR1 mRNA expression analysis was accomplished by RT-qPCR with primers on exons 3/4 of FMR1 (forward: 5′ GAA GTT GAG GTG TAT TCC AGA GC 3′; reverse: 5′ AAC TCA CCC TTT ATC ATC CTC AC 3′) in four patients (1033, 1234, 1337, 1513). Total RNA was extracted from peripheral blood stored in RNAlater solution [Thermo Fisher Scientific Inc.] with RiboPure blood kit [Ambion, USA]. cDNA was generated starting from 170 to 900 ng of total RNA using the Superscript III First-Strand Synthesis System, containing random hexamer primers and Superscript III reverse transcriptase [Thermo Fisher Scientific Inc.]. RT-qPCR reactions were conducted on a 7500 Fast Real Time PCR System [Thermo Fisher Scientific Inc.] and data analysis was achieved according to the ΔΔCt method, using GAPDH and GUSB genes as normalizers. FMRP expression was not accomplished due to the lack of fresh blood samples.
Results
After conventional FMR1 CGG-based PCR [9], patients 1033, 1234, 1337 and 1629 presented weak suspicious bands below the normal range, indicating the presence of possible deletions [Supplementary Figure 1]. Neither a premutation nor a normal sized allele was detected in addition to the potential deleted fragments. Thus, to uncover the nature of the suspicious CGG-based PCR fragments and to clarify the FMR1 methylation status of the patients included in this study, three additional methodologies were applied for comparative/complemental purposes. Direct Sanger sequencing of the amplicons confirmed deletions comprising the whole CGG repeats tract and flanking regions in these four patients (Table 1 and Supplemental Figures 2–5). In addition, to determine if the patients harbored a single deletion or if they were mosaic for a full mutation, and also to resolve the low intensity of the CGG repeat amplification of patient 1513, we employed MS-MLPA, SB and mPCR analyses. All five patients exhibited abnormal methylation patterns for FMR1 promoter in MS-MLPA (Supplementary Figure 6). Additionally, patient 1513 displayed a deletion of seven consecutive probes located at exon 1 and intron 1 of the FMR1 gene, comprising at least 1042 bp (ChrX:147.911.906-147.912.457; UCSC Genome Browser, hg38) (Supplementary Figure 7), concomitantly with abnormal FMR1 methylation. As expected, MS-MLPA was not suitable to detect copy number variation concerning small deletions within the CGG repeats tract due to impossibility of probe design. These results suggest that the patients could have a methylated deleted allele originated from a full mutated allele during early embryogenesis or that they harbor a full mutation allele together with the deletion (somatic mosaicism). In SB and mPCR analyses, all five patients exhibited a methylated full mutation profile, confirming that they have full mutations. However, the small deleted alleles were not observed using either methodologies in any of the patients (1033, 1234, 1337 and 1629) (Supplementary Figures 8 and 9). TRP-PCR showed that all mothers are carriers of a premutation allele, with the exception of patient 1234, whose mother additionally exhibited a full mutation allele besides the premutation one, which was undetected by SB analysis (Supplementary Figure 10).
Table 1.
Genomic locations of the deletions identified in the five mosaic patients.
| Patient | Deletion (GRCh38/hg38) | Extension (bp) |
|---|---|---|
|
| ||
| 1033 | 147912023–147912150* | 128 |
| 1234 | 147912043–147912135* | 93 |
| 1337 | 147912039–147912110* | 72 |
| 1513 | 147911695–147912736** | 1042 |
| 1629 | 147912021–147912113* | 93 |
The position of the deletions is approximated, due to the presence of microhomology of 2–3 base pairs at the breakpoints.
Precise extension of this deletion is unknown.
FMR1 mRNA expression analysis by RT-qPCR on patients 1033, 1234 and 1513 showed a very low expression of FMR1 mRNA (less/equal to 0.05), whereas patient 1337 exhibited a residual expression (0.18) (Supplementary Table 2). Patient 1629 was not analyzed due to the unavailability of the RNA sample.
To evaluate if the different mutations presented by our patients reflected in the severity of the clinical phenotype, we fulfilled a checklist developed for Brazilian patients concerning ten main clinical and behavioral FXS features [15]. Patients 1033, 1234, 1337 and 1629 obtained scores of 7, 5, 4 and 5, respectively, whereas patient 1513 had the highest score of 8 (Table 2, Supplementary Figure 11), revealing that this later patient with a family history of ID has a more severe phenotype including elongated face, large ears, hyperextensible finger joints, large testes, plantar crease, tactile defensiveness, poor eye contact, aggressive behavior and lack of words’ articulation to form sentences.
Table 2.
Main clinical and behavioral features presented by the five mosaic patients according to the checklist elaborated by Christofolini and colleagues [15].
| Patients | Age (years)* |
Family history of ID |
Elongated face |
Large ears |
Hyperextensible finger joints |
Large testes |
Plantar crease |
Hand biting |
Hand flapping |
Tactile defensiveness |
Poor eye contact |
Total score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1033 | 33 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 7 |
| 1234 | 18 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 5 |
| 1337 | 14 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 4 |
| 1513 | 12 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 8 |
| 1629 | 18 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 5 |
0 is for absence, whereas 1 is for presence.
Age (years) at the moment of the last clinical evaluation.
Bioinformatics inquiry of the sequences flanking the deletion breakpoints (300 bp for each side) showed no common repetitive elements (LINEs, SINEs and LTRs). However, a 2 or 3-bp microhomology sequence was identified at the junction in three of the four small deletions without the introduction of additional nucleotides. For patient 1513, due to the larger extension of the deletion, we applied iterative rounds of PCR and qPCR for fine mapping the deletion breakpoints. Nevertheless, all regions analyzed exhibited normal qPCR values, probably because of the interference of high abundance full mutation alleles, whose genetic structure outside the CGG repeats tract is normal, concomitantly to possible normal-sized alleles detected by the initial CGG-based PCR [9] (Supplementary Figure 1).
Discussion
In this study, we report five patients with FXS presenting a somatic full mutation/deletion mosaicism, which represents 2.02 % of our cohort versus a prevalence of 8.09 % for “pure” full mutation cases. Four cases represent small deletions encompassing the CGG repeats and flanking regions, whereas the remaining has a larger deletion comprising exon 1 and part of intron 1 of FMR1 gene. Mosaicism between a premutation and a full mutation is a common situation among males with FXS, with an estimated frequency of 12–41% [16,17]. Conversely, mosaicism cases involving deleted and full mutation alleles are less reported in FXS [18]. As corroborated by our patients, the majority of the deletions comprising the CGG repeat together with a full mutation or a premutation allele occurs during the transmission of a maternal premutation allele to the offspring (for review see [18]). After expansion into a full mutation allele during meiosis, a deletion can occur mitotically during the embryonic cell divisions, resulting in distinct subpopulations of cells carrying deletion and full mutation alleles. In most of the deletions involving the CGG repeats reported so far, the deleted alleles were unmethylated, suggesting that the deletion occurred before the 11th week of gestation [19–22]. However, SB methodology, that would be able to inform about the methylation status of the deletion, failed to detect the smallest alleles in our study likely due to the low abundance of these deleted alleles.
In patients exhibiting the small deletions, the deletion has left the transcription start site and the translation initiation codon intacts, but very low expression levels of FMR1 mRNA was detected. This is most likely due to the presence of low percentage of the deleted alleles or maybe to the loss of some regulatory element. Patient 1337 presented with residual mRNA expression of FMR1 gene (18%) due to his methylation mosaicism (26% unmethylated full mutation allele) concomitant to a possible contribution of the deleted allele. Patient 1513 presents with a large deletion, which is similar to what has been reported in two mosaic individuals with FXS: (a) a mosaicism including a full mutation, a premutation and a deleted allele (which included 8.7 kb comprising 85 bp distal to the repeats, the entire repeat tract up to part of intron 1 of the FMR1 gene) [23]; (b) a mosaicism including a full mutation and a deletion of 486 bp involving 168 bp upstream of the CGG repeats, the entire CGG repeat tract and 138 bp downstream of the repeats comprising part of intron 1 [24]. The deletions in both reported cases affected the transcription start site and the translation start codon, leading to the absence of FMR1 mRNA expression. In our case (patient 1513), 3% of FMR1 mRNA expression was observed, suggesting that the transcription of the FMR1 gene may not be completely compromised by the deletion (Supplementary Table 2). Nonetheless, it should be noted that the FMR1 mRNA profile in leucocytes may not accurately reflect the one present in other tissues, particularly in the brain [25,26].
According to the checklist applied [15], the five patients scored values over the cutoff of 4, indicating that they are clinically undistinguishable neither from patients routinely submitted to FXS screening nor from confirmed typical FXS males. However, patient 1513, presenting with the larger size of the deletion, exhibited a more significant impairment, compatible with the higher clinical score (8), whereas patient 1337 presenting a methylation mosaicism exhibited a milder phenotype with the lower score (4).
Different mechanisms leading to deletions within the FMR1 gene have been proposed so far. Firstly, the Chi-like element, a sequence located approximately 70 bp from the CGG repeats (5′ GGTGGAGG 3′), was reported as a hotspot for deletions caused by CGG instability [19]. Corroborating this fact, the deletions of patients 1033, 1234, 1337 and 1629 are located between 8–29 bp near the CGG repeats (Figure 1 and Supplementary Figures 2–5). Besides, Mononen and colleagues [27] proposed that realignment between the CGG repeats tract and dispersed non-adjacent homologous repetitive sequences, such as the sequence 5′ GGCGGCGGCGG 3′, located 37 bp upstream of the proximal end of the repeat tract may also play a role in repeat instability leading to duplications/deletions in FMR1 gene.
Figure 1.
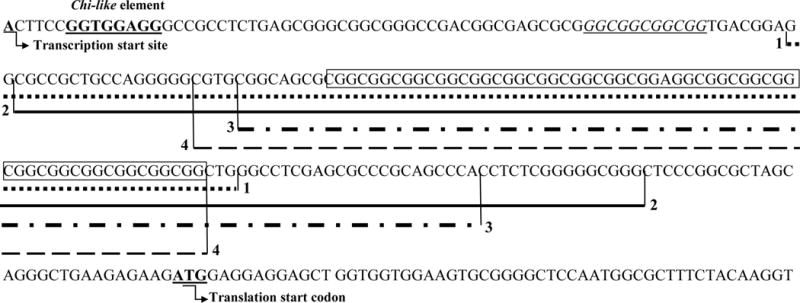
Extension of the small FMR1 deletions (5′-3′) described in patients 1629 (1), 1033 (2), 1234 (3) and 1337 (4).
Wild type sequence (NG_007529.1) with a (CGG)19 repeat tract; CGG repeat sequences are boxed; 1- Patient 1629; 2- Patient 1033; 3- Patient 1234; 4- Patient 1337; Chi-like sequence (5′ GGTGGAGG 3′) previously reported to be involved in deletions comprising CGG repeats [19] is indicated in bold underlined text; The homologous simple repeat sequence (5′ GGCGGCGGCGG 3′) proposed by Mononen and colleagues [27] to play a role in repeat instability leading to duplications/deletions in FXS is in italicized and underlined.
In light of the recent knowledge emerging from copy number variation (CNV) formation, we evaluated the deletions’ breakpoints in patients 1033, 1234, 1337 and 1629 to infer the most likely mechanism(s) [28]. The presence of a 2–3 base pairs microhomology between the proximal and distal non recurrent breakpoints of three small deletions (patients 1033, 1337 and 1629) (Supplementary Figures 7–10) supports the involvement of non-homologous end joining (NHEJ), fork stalling and template switching (FoSteS) or microhomology mediated induced repair (MMBIR) mechanisms. NHEJ usually generate small scars in breakpoints [29,30], which was not the case. FoSTeS, in turn, usually leads to more complex rearrangements, i.e., deletions concomitant with inversions, insertions concomitant with deletions, among others [28,31]. So, MMBIR is the most likely CNV mechanism involved in the small deletions cases. MMBIR repairs double strand breaks with unique ends, when sequences of DNA from single strands are available and share microhomology of 2–3 bp with the 3′ end of the single strand of the stalled fork [28]. Nevertheless, which genomic feature could predispose the full mutation allele to be reverted to a deletion during the early developmental stages in some cases but not in others remains to be elucidated. It is possible that such replication errors happen relatively frequently due to the described microhomology, but deleted alleles would only be detectable if present in high enough relative concentration.
In this study, we employed different methodologies to analyze the CGG repeats in the FMR1 gene. Interestingly, conventional PCR followed by direct Sanger sequencing was the unique methodology able to detect the small deletions, but it masked the full mutation alleles, which are refractory to amplification. MS-MLPA, in turn, had the advantage of being efficient to evaluate CNVs along FMR1 gene besides the methylation status, but the repetitive nature of the CGG repeats made the detection of deleted alleles within this region difficult. SB was incapable of detecting low abundance alleles, such as the small deletions in the probands and the full mutation in the mother of patient 1234, probably because these alleles are below the detection threshold. Finally, mPCR was not able to detect the deletions within the CGG repeats tract concomitant to the full mutation alleles. However, it has been proved to be a highly sensitive methodology, revealing profiles previously undetected by SB, such as the full mutation exhibited by the mother of patient 1234 and the methylation mosaicism for full mutation alleles in patient 1337 [12].
The panorama we described here demonstrates that evaluating FXS atypical mutations that are clinically undistinguishable is still a challenge, even in light of high throughput methodologies, like exome sequencing, which at present is not suitable for identifying trinucleotide repeat expansions having limited value in FXS detection [32,33]. Because of the technical limitations and the specific pitfalls of each method, they could induce potential misinterpretation of the genotype if used alone. In this sense, the deletions we described could be potentially missed by FXS testing using SB, MS-MLPA or TRP-PCR/mPCR. Moreover, we should take into account that the amplicon size of the FMR1 CGG-based PCR we used is relatively large (557 bp for a 27 CGG repeat allele). Most laboratories have used a first line PCR test with primers closer to the repeat region, which would not have the sensitivity to detect the much shorter product and further contributes to the likely underestimated prevalence of the deleted alleles.
Interestingly, recent advances in targeted therapies to reverse the neurobiological abnormalities in FXS have shown a predictive response based on the degree of FMR1 methylation. Improvements in hallmarks symptoms of FXS were observed for FXS individuals with a fully methylated allele, but not for those with methylation mosaicism [34], so that the therapeutic response in deletion/full mutation mosaicism should be evaluated in future research. Besides, deletions in regions of FMR1 other than those bearing CGG repeats may be missed, since these regions are not routinely interrogated during FXS testing. So, the remaining questions reside on which is the actual prevalence of full mutation/deletion mosaicism in males with FXS, how to measure the percent of cells carrying the deletion in the brain compared to those that carry the full mutation and which is the clinical meaning and therapeutics implications of this kind of mosaicism. It is likely that in the near future knowledge on the prevalence of the non-CGG repeat FMR1 mutations will be provided by the advent of rapid DNA sequencing methodologies and mainly single molecule sequencing, which might illuminate the existence of all types of mutation in FMR1.
Supplementary Material
Supplementary Figure 1: Silver-stained 6% polyacrylamide gel, showing PCR amplification of FMR1 CGG repeats [9]. The lower band of 223 bp indicated on the right constitutes the internal control for the PCR reaction. A higher band representing the amplification product containing the (CGG)n repeat lies between 488 bp (6 CGG repeats) and 632 bp (54 CGG repeats) in normal individuals. Artifactual bands, probably heteroduplexes, are seen above the normal polymorphic bands. Full mutations are refractory to upper band amplification. Lane 1: size standards from a 1-kb ladder (Thermo Fisher Scientific Inc.) with molecular sizes, from the bottom up, of 201, 220, 298, 344, 396, 506 and 517 bp. The 220- and 517-bp bands are indicated on the left; Lane 2: negative control; Lane 3: normal control male; Lane 4: full mutation control male with absence of the product containing the (CGG)n repeats; Lanes 5–8: patients 1033, 1234, 1337 and 1629, respectively presenting small bands for the (CGG)n repeats region (red circles); Lane 9: patient 1513 presenting a low abundance normal-sized band for the (CGG)n repeats region (green circle).
Supplementary Table 1: Genomic locations and sequences of qPCR primers used for mapping the deletion in patient 1513.
Supplementary Figure 2: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1033 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1033 presenting the small deletion involving the whole CGG repeats tract and 29 bp proximal and 39 bp distal flanking regions (c.-156_-29del128); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 2 bp (GG) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 3: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1234 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1234 presenting the small deletion involving the whole CGG repeats tract and 9 bp proximal and 24 bp distal flanking regions (c.-136_-44del93); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. No microhomology was seen in proximal and distal breakpoints.
Supplementary Figure 4: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1337 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1033 presenting the small deletion involving the whole CGG repeats tract in addition to 29 bp proximal and 39 bp distal flanking regions (c.-142_-70del72); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 3 bp (GGC) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 5: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1629 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1629 presenting the small deletion involving the whole CGG repeats tract and 30 bp proximal and 3 bp distal flanking regions (c.-153_-61del93); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 2 bp (GG) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 6: Electropherograms showing the results of FMR1 MS-MLPA methylation analysis in the five mosaic patients after HhaI digestion. Red arrows indicate the peaks from methylated probes located near of the CGG repeat tract on exon 1 of FMR1, blue arrows indicate the absence of signal of control probes located in other regions of FMR1 gene and green arrows indicate the expected absence of digestion signal in control probes. As expected all patients showed abnormal methylation near of the CGG repeats represented by the signal given by the methylation specific probes.
Supplementary Figure 7: FMR1 MS-MLPA analysis for copy number variation in patient 1513. Green squares located between the green lines represent FMR1 specific probes with normal results (ratios between 0.7 and 1.3), blue squares represent the control probes and the seven red squares under the green line represent the deleted probes located on exon 1 and intron 1 of FMR1 (03727-L03157, 03725-L13188, 03722-L03182, 12916-L14361, 12915-L14360, 04037-L03722, 12918-L12030) with ratios next to 0 (ChrX:147.911.906-147.912.457; UCSC Genome Browser Hg38).
Supplementary Figure 8: Southern blot analysis of the five mosaic patients and their mothers.
Supplementary Figure 9: Electropherograms showing results from the FMR1 mPCR analysis in the five mosaic patients. Blue peaks represent the CGG repeat size and green peaks represent the methylation status of each peak. The two peaks are compared and the percentage of methylation is given above each green peak. Methylation percentages between 20%–80% are considered partially methylated and above 80% are considered fully methylated and methylation percentages that are nominally calculated in excess of 100% were scaled to 100%, according to the manufacturer’s instructions [Asuragen Inc.]. All patients presented fully methylated full mutations peaks (>200 CGG repeats), with the exception of patient 1337 (c), who also presented a partially methylated full mutated peak with 26% of methylation.
Supplementary Figure 10: Electropherograms showing TRP-PCR segregation analysis in the mothers of the five mosaic patients. a) Mother of patient 1033 presenting a normal peak with 31 CGG repeats and two peaks correspondent to premutation alleles of 99 and 106 CGG repeats. b) Mother of patient 1234 exhibiting a normal peak with 36 CGG repeats, a premutated peak with 174 CGG repeats and a full mutated peak with more than 200 CGG repeats. c) Mother of patient 1337 showing a normal peak with 30 CGG repeats and a premutated peak with 79 CGG repeats. d) Mother of patient 1513 presenting a normal peak with 30 CGG repeats and two peaks with 99 and 105 CGG repeats.
Supplementary Table 2: FMR1 mRNA expression analysis through RT-qPCR from the five mosaic patients. ΔΔCt method was applied for comparative purposes with two control males, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and beta glucuronidase (GUSB) genes as normalizers. The results (mean ± standard deviation) were obtained from three consecutive experiments.
Supplementary Figure 11: Pictures of the affected males with the FMR1 full mutation. Note some facial dysmorphies as elongated face and large ears. (a) patient 1033; (b) patient 1234; (c) patient 1337; (d) patient 1513. No pictures were available for patient 1629.
Key Issues.
-
-
Fragile X syndrome (FXS) is the most common cause of intellectual disability and the leading monogenic cause of autism spectrum disorders.
-
-
The causative mutation in almost all FXS cases is a CGG-repeat expansion in the 5′ UTR region of the FMR1 gene, resulting in partial or complete epigenetic gene silencing.
-
-
Current molecular diagnosis of FXS relies on the size of the number of CGG repeats and methylation patterns in the FMR1 gene.
-
-
We describe five FXS males harboring full mutation/deletion mosaicism, which were identified only by combining different molecular approaches.
-
-
Four patients exhibited small deletions encompassing the CGG repeats tract, whereas the remaining had a larger deletion of at least 1042 bp, all of them arising during the transmission of a maternal premutation allele to the offspring.
-
-
Bioinformatics inquiry of the sequences flanking the breakpoints in the small deletions supports the involvement of microhomology mediated induced repair (MMBIR) mechanism.
-
-
Mosaicism cases involving deleted and full mutation alleles are apparently rare and clinically undistinguishable, but they could be potentially missed by routine FXS testing due to technical limitations.
-
-
Future research resides on the actual prevalence of this kind of mosaicism and its clinical meaning and therapeutics implications.
Acknowledgments
We thank the patients and their families for their kind participation in this study, Luciana Guedes for technical help in RT-qPCR experiments, CNPq and FAPERJ (Brazil) for financial support. This work was also supported by NICHD grant HD02274.
Footnotes
Website
101. RepeatMasker Documentation program – http://repeatmasker.genome.washington.edu
Conflicts of interest
Authors do not have any conflict of interest.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–14. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherman S. Epidemiology. In: Hagerman RJ, Hagerman PJ, editors. Fragile X syndrome: diagnosis, treatment and research. The Johns Hopkins University Press; 2012. pp. 136–168. [Google Scholar]
- 3*.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. One of the first publications describing the molecular mechanism involved in FMR1 silencing. [DOI] [PubMed] [Google Scholar]
- 4.Chen E, Joseph S. Fragile X mental retardation protein: A paradigm for translational control by RNA-binding proteins. Biochimie. 2015;114:147–54. doi: 10.1016/j.biochi.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saldarriaga W, Tassone F, González-Teshima LY, Forero-Forero JV, Ayala-Zapata S, Hagerman R. Fragile X syndrome. Colomb Med (Cali) 2014;45:190–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–45. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 7*.Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boué J, Bertheas MF, Mandel JL. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. One of the first publications describing the molecular mechanism involved in FMR1 silencing. [DOI] [PubMed] [Google Scholar]
- 8*.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–58. doi: 10.1016/0092-8674(91)90283-5. The first publication describing a PCR method for FXS analysis based on refractory amplification of full mutation. [DOI] [PubMed] [Google Scholar]
- 9.Haddad LA, Mingroni-Netto RC, Vianna-Morgante AM, Pena SD. A PCR-based test suitable for screening for fragile X syndrome among mentally retarded males. Hum Genet. 1996;97:808–12. doi: 10.1007/BF02346194. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, Pan R, Hagerman PJ, Stenzel TT, Tassone F, Latham GJ. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn. 2010;12:589–600. doi: 10.2353/jmoldx.2010.090227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, Hagerman PJ, Stenzel TT, Hadd AG, Latham GJ, Tassone F. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. Landmark publication describing the development of a new FMR1 PCR technology than can accurately categorize the spectrum of FMR1 alleles, including alleles previously considered too large to amplify. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Hadd AG, Sah S, Houghton JF, Filipovic-Sadic S, Zhang W, Hagerman PJ, Tassone F, Latham GJ. High-resolution methylation polymerase chain reaction for fragile X analysis: evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet Med. 2011;13:528–38. doi: 10.1097/GIM.0b013e31820a780f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wells RD. Mutation Spectra in Fragile X Syndrome Induced by Deletions of CGG·CCG Repeats. J Mol Chem. 2009;284:7407–11. doi: 10.1074/jbc.R800024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–9. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christofolini DM, Abbud EM, Lipay MV, Costa SS, Vianna-Morgante AM, Bellucco FT, Nogueira SI, Kulikowski LD, Brunoni D, Juliano Y, Ramos MA, Melaragno MI. Evaluation of clinical checklists for fragile X syndrome screening in Brazilian intellectually disabled males: proposal for a new screening tool. J Intellec Disabil. 2009;13:239–48. doi: 10.1177/1744629509348429. [DOI] [PubMed] [Google Scholar]
- 16.Nolin SL, Glicksman A, Houck GE, Jr, Brown WT, Dobkin CS. Mosaicism in fragile X affected males. Am J Med Genet. 1994;51:509–12. doi: 10.1002/ajmg.1320510444. [DOI] [PubMed] [Google Scholar]
- 17.Merenstein SA, Sobesky WE, Taylor AK, Riddle JE, Tran HX, Hagerman RJ. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet. 1996;64:388–94. doi: 10.1002/(SICI)1096-8628(19960809)64:2<388::AID-AJMG31>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18**.Coffee B, Ikeda M, Budimirovic DB, Hjelm LN, Kaufmann WE, Warren ST. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: a case report and review of the literature. Am J Med Genet A. 2008;146A:1358–67. doi: 10.1002/ajmg.a.32261. This publication reviews the literature of FMR1 deletions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Graaff E, Rouillard P, Willems PJ, Smits AP, Rousseau F, Oostra BA. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum Mol Genet. 1995;4:45–9. doi: 10.1093/hmg/4.1.45. [DOI] [PubMed] [Google Scholar]
- 20.Petek E, Kroisel PM, Schuster M, Zierler H, Wagner K. Mosaicism in a fragile X male including a de novo deletion in the FMR1 gene. Am J Med Genet. 1999;84:229–32. [PubMed] [Google Scholar]
- 21.Han XD, Powell BR, Phalin JL, Chehab FF. Mosaicism for a full mutation, premutation, and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high-functioning fragile X male. Am J Med Genet Part A. 2006;140A:1463–71. doi: 10.1002/ajmg.a.31291. [DOI] [PubMed] [Google Scholar]
- 22.Govaerts LC, Smit AE, Saris JJ, VanderWerf F, Willemsen R, Bakker CE, De Zeeuw CI, Oostra BA. Exceptional good cognitive and phenotypic profile in a male carrying a mosaic mutation in the FMR1 gene. Clin Genet. 2007;72:138–44. doi: 10.1111/j.1399-0004.2007.00829.x. [DOI] [PubMed] [Google Scholar]
- 23.Quan F, Grompe M, Jakobs P, Popovich BW. Spontaneous deletion in the FMR1 gene in a patient with fragile X syndrome and cherubism. Hum Mol Genet. 1995;4:1681–84. doi: 10.1093/hmg/4.9.1681. [DOI] [PubMed] [Google Scholar]
- 24.Schmucker B, Seidel J. Mosaicism for a full mutation and a normal size allele in two fragile X males. Am J Med Genet. 1999;84:221–5. [PubMed] [Google Scholar]
- 25.Bonarrigo FA, Russo S, Vizziello P, Menni F, Cogliati F, Giorgini V, Monti F, Milani D. Think About it: FMR1 gene mosaicism. J Child Neurol. 2014;29:NP74–7. doi: 10.1177/0883073813503187. [DOI] [PubMed] [Google Scholar]
- 26.Pretto DI, Mendoza-Morales G, Lo J, Cao R, Hadd A, Latham GJ, Durbin-Johnson B, Hagerman R, Tassone F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet. 2014;51:309–18. doi: 10.1136/jmedgenet-2013-102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mononen T, von Koskull H, Airaksinen RL, Juvonen V. A novel duplication in the FMR1 gene: implications for molecular analysis in fragile X syndrome and repeat instability. Clin Genet. 2007;72:528–31. doi: 10.1111/j.1399-0004.2007.00903.x. [DOI] [PubMed] [Google Scholar]
- 28**.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. Provides a description of microhomology-mediated break-induced replication and comparison of many different mechanisms leading to CNVs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen JM, Cooper DN, Férec C, Kehrer-Sawatzki H, Patrinos GP. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20:222–233. doi: 10.1016/j.semcancer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–81. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singleton AB. Exome sequencing: a transformative technology. Lancet Neurology. 2011;10:942–6. doi: 10.1016/S1474-4422(11)70196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keogh MJ, Chinnery PF. Next generation sequencing for neurological diseases: new hope or new hype? Clin Neurol Neurosurg. 2013;115:948–53. doi: 10.1016/j.clineuro.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K, Brun A, Bussy G, Gasparini F, Hilse T, Floesser A, Branson J, Bilbe G, Johns D, Gomez-Mancilla B. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3:64ra1. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Silver-stained 6% polyacrylamide gel, showing PCR amplification of FMR1 CGG repeats [9]. The lower band of 223 bp indicated on the right constitutes the internal control for the PCR reaction. A higher band representing the amplification product containing the (CGG)n repeat lies between 488 bp (6 CGG repeats) and 632 bp (54 CGG repeats) in normal individuals. Artifactual bands, probably heteroduplexes, are seen above the normal polymorphic bands. Full mutations are refractory to upper band amplification. Lane 1: size standards from a 1-kb ladder (Thermo Fisher Scientific Inc.) with molecular sizes, from the bottom up, of 201, 220, 298, 344, 396, 506 and 517 bp. The 220- and 517-bp bands are indicated on the left; Lane 2: negative control; Lane 3: normal control male; Lane 4: full mutation control male with absence of the product containing the (CGG)n repeats; Lanes 5–8: patients 1033, 1234, 1337 and 1629, respectively presenting small bands for the (CGG)n repeats region (red circles); Lane 9: patient 1513 presenting a low abundance normal-sized band for the (CGG)n repeats region (green circle).
Supplementary Table 1: Genomic locations and sequences of qPCR primers used for mapping the deletion in patient 1513.
Supplementary Figure 2: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1033 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1033 presenting the small deletion involving the whole CGG repeats tract and 29 bp proximal and 39 bp distal flanking regions (c.-156_-29del128); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 2 bp (GG) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 3: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1234 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1234 presenting the small deletion involving the whole CGG repeats tract and 9 bp proximal and 24 bp distal flanking regions (c.-136_-44del93); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. No microhomology was seen in proximal and distal breakpoints.
Supplementary Figure 4: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1337 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1033 presenting the small deletion involving the whole CGG repeats tract in addition to 29 bp proximal and 39 bp distal flanking regions (c.-142_-70del72); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 3 bp (GGC) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 5: Electropherogram showing the direct Sanger sequencing after FMR1 CGG-based PCR [8] from the patient 1629 compared to a normal control male. (a) normal control male exhibiting a (CGG)20 repeats tract; (b) Patient 1629 presenting the small deletion involving the whole CGG repeats tract and 30 bp proximal and 3 bp distal flanking regions (c.-153_-61del93); (c) scheme from the deleted sequence (represented by the dotted line) compared to the reference sequence. Note the microhomology of 2 bp (GG) between the distal and proximal breakpoints (underlined in blue).
Supplementary Figure 6: Electropherograms showing the results of FMR1 MS-MLPA methylation analysis in the five mosaic patients after HhaI digestion. Red arrows indicate the peaks from methylated probes located near of the CGG repeat tract on exon 1 of FMR1, blue arrows indicate the absence of signal of control probes located in other regions of FMR1 gene and green arrows indicate the expected absence of digestion signal in control probes. As expected all patients showed abnormal methylation near of the CGG repeats represented by the signal given by the methylation specific probes.
Supplementary Figure 7: FMR1 MS-MLPA analysis for copy number variation in patient 1513. Green squares located between the green lines represent FMR1 specific probes with normal results (ratios between 0.7 and 1.3), blue squares represent the control probes and the seven red squares under the green line represent the deleted probes located on exon 1 and intron 1 of FMR1 (03727-L03157, 03725-L13188, 03722-L03182, 12916-L14361, 12915-L14360, 04037-L03722, 12918-L12030) with ratios next to 0 (ChrX:147.911.906-147.912.457; UCSC Genome Browser Hg38).
Supplementary Figure 8: Southern blot analysis of the five mosaic patients and their mothers.
Supplementary Figure 9: Electropherograms showing results from the FMR1 mPCR analysis in the five mosaic patients. Blue peaks represent the CGG repeat size and green peaks represent the methylation status of each peak. The two peaks are compared and the percentage of methylation is given above each green peak. Methylation percentages between 20%–80% are considered partially methylated and above 80% are considered fully methylated and methylation percentages that are nominally calculated in excess of 100% were scaled to 100%, according to the manufacturer’s instructions [Asuragen Inc.]. All patients presented fully methylated full mutations peaks (>200 CGG repeats), with the exception of patient 1337 (c), who also presented a partially methylated full mutated peak with 26% of methylation.
Supplementary Figure 10: Electropherograms showing TRP-PCR segregation analysis in the mothers of the five mosaic patients. a) Mother of patient 1033 presenting a normal peak with 31 CGG repeats and two peaks correspondent to premutation alleles of 99 and 106 CGG repeats. b) Mother of patient 1234 exhibiting a normal peak with 36 CGG repeats, a premutated peak with 174 CGG repeats and a full mutated peak with more than 200 CGG repeats. c) Mother of patient 1337 showing a normal peak with 30 CGG repeats and a premutated peak with 79 CGG repeats. d) Mother of patient 1513 presenting a normal peak with 30 CGG repeats and two peaks with 99 and 105 CGG repeats.
Supplementary Table 2: FMR1 mRNA expression analysis through RT-qPCR from the five mosaic patients. ΔΔCt method was applied for comparative purposes with two control males, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and beta glucuronidase (GUSB) genes as normalizers. The results (mean ± standard deviation) were obtained from three consecutive experiments.
Supplementary Figure 11: Pictures of the affected males with the FMR1 full mutation. Note some facial dysmorphies as elongated face and large ears. (a) patient 1033; (b) patient 1234; (c) patient 1337; (d) patient 1513. No pictures were available for patient 1629.