

Summary
FGF21 contributes to the metabolic response to dietary protein restriction, and prior data implicate GCN2 as the amino acid sensor linking protein restriction to FGF21 induction. Here we demonstrate the persistent and essential role of FGF21 in the metabolic response to protein restriction. We show that FGF21-KO mice are fully resistant to low protein (LP)-induced changes in food intake, energy expenditure (EE), body weight gain and metabolic gene expression for 6 months. GCN2-KO mice recapitulate this phenotype, but LP-induced effects on food intake, EE, and body weight subsequently begin to appear after 14 days on diet. We show that this delayed emergence of LP-induced metabolic effects in GCN2-KO mice coincides with a delayed but progressive increase of hepatic FGF21 expression and blood FGF21 concentrations over time. These data indicate that FGF21 is essential for the metabolic response to protein restriction, but that GCN2 is only transiently required for LP-induced FGF21.
eTOC Blurb
Laeger et al demonstrate that FGF21 is required for adaptive metabolic responses to protein restriction. The amino acid sensor GCN2 initially contributes to the induction of FGF21, but additional mechanisms compensate for its absence over longer periods.
Introduction
Fibroblast growth factor 21 (FGF21) is a circulating hormone associated with metabolic responses to nutrient restriction, as initial work demonstrated that FGF21 is increased during fasting, starvation, and a ketogenic diet (Badman et al., 2007; Inagaki et al., 2007; Potthoff et al., 2009; Potthoff et al., 2012). However, our recent data suggests that reduced protein intake is the primary regulator of FGF21 during these interventions, and that FGF21-deficient mice fail to exhibit the increases in food intake, increases in energy expenditure (EE) and reductions in growth observed in wild-type mice consuming low protein diet (Laeger et al., 2014a). The mechanisms linking reduced protein intake to increased hepatic FGF21 expression and secretion are currently unclear, but previous work implicates the amino acid sensor GCN2 (De Sousa-Coelho et al., 2012; Laeger et al., 2014a). GCN2 phosphorylates eIF2α in response to depletion of cellular amino acids (Wek et al., 1995), leading to the inhibition of general protein synthesis while increasing the translation of specific transcription factors, such as ATF4. GCN2-dependent phosphorylation of eIF2α and resultant activation of ATF4 links amino acid availability to metabolism, particularly in the liver (Anthony et al., 2004; Dudek and Semenkovich, 1995; Guo and Cavener, 2007; Hamanaka et al., 2005; Xiao et al., 2011; Zhang et al., 2002). The FGF21 promoter contains amino acid response elements (AARE), and both depletion of amino acids and activation of this eIF2α/ATF4 pathway increases FGF21 (De Sousa-Coelho et al., 2012; Kim et al., 2013; Schaap et al., 2013; Wilson et al., 2015). Consistent with these reports, it was demonstrated that hepatic eIF2α-phosphorylation is induced in multiple settings of dietary protein restriction, and that LP-induced increases in FGF21 and eIF2α phosphorylation are blunted in GCN2-deficient mice (Laeger et al., 2014a).
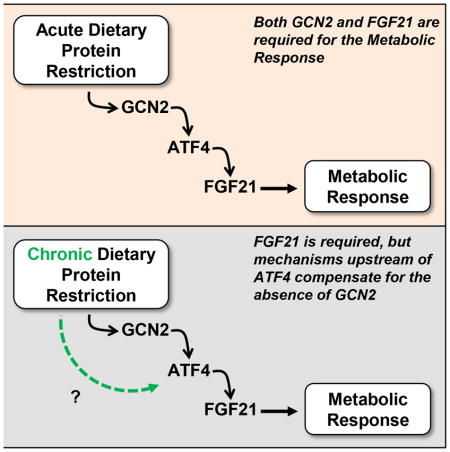
Since FGF21 is required for metabolic and behavioral responses to protein restriction, and GCN2 contributes to the increase of FGF21 in this setting, we hypothesized that GCN2-deficient mice would fail to respond to reduced dietary protein intake and thereby recapitulate the phenotype of FGF21-KO mice. The results indicate that GCN2-KO mice do recapitulate the phenotype of FGF21-KO, but only transiently, such that GCN2-KO mice begin to respond to LP diet in a fashion similar to wildtype after approximately 14 days. In contrast, FGF21-KO mice never exhibit a metabolic response to protein restriction, even for 6 months, demonstrating that FGF21 is indeed an essential signal of protein restriction.
Results
FGF21 is required for changes in food intake, energy expenditure and body weight gain
WT and FGF21-KO were placed on isocaloric control and LP diets for 27 weeks (6 months). In wildtype mice, LP diet significantly impaired body weight gain, with WT-LP mice losing ~1 gram of BW in the first week, and then largely failing to gain weight for the entire 6 month period (Figure 1A). Over the 6 months of dietary exposure the WT-CON mice gained 7.5 ± 1.3 g while WT-LP mice lost only 0.13 ± 0.8 g, a number not statistically different from zero (Figure 1A). The LP-induced reduction in BW gain was associated with a significant reduction in fat gain (Figure 1C) and a loss of lean mass (Figure 1D). However, because the absolute reduction in fat mass gain was much larger than the loss of lean mass, percent body fat was actually reduced whereas percent body lean was increased in WT-LP mice compared to WT-CON (Figure 1E–F). Despite the reduction in total protein intake, we observe no negative effect of the LP diet on motor coordination and athleticism in the rotarod task (Figure S1). Strikingly, these effects of LP diet on weight gain and adiposity were completely abolished in FGF21-KO mice, who in fact exhibited increased weight gain when placed on a LP diet (Figure 1B). FGF21-LP mice also gained significantly more total body fat (Figure 1C) and did not lose lean mass (Figure 1D), and therefore exhibited increased body adiposity (Figure 1E) compared to FGF21-CON. Finally, the effect of LP to reduce body length in WT mice was also completely abolished in FGF21-KO mice (Figure 1I), suggesting that the LP-induced reduction in weight was associated with a reduction in linear growth. In summary, the consumption of a LP diet for 27 weeks reduced growth, body weight, and total body adiposity in wildtype but not FGF21-KO mice.
Figure 1. FGF21 is persistently required for low protein-induced changes in body weight, body composition, food intake, and energy expenditure.

Body weight change in WT (A) and FGF21-KO mice (B), body fat change (C) and body lean change (D) in WT and FGF21-KO mice on isocaloric control or LP diet for 27 weeks. Percent body fat (E) and percent body lean (F) at sacrifice on week 27. Average EE (G) over the 3 day period of EE measurement after 11 weeks on diet. Average daily food intake (H) over the entire 27 week period. Final body length (I) at sacrifice on week 27 n = 8–10/group, #0.1 > P > 0.05; *P < 0.05; **P < 0.01. Data are represented as mean ± SEM
To understand the mechanism driving these changes in BW and adiposity with LP diet, we compared food intake and energy expenditure (EE) in WT and FGF21-KO mice. Our previous work indicated that WT mice and rats exhibit significant increases in food intake when exposed to LP diet for 2 weeks (Laeger et al., 2014a; Laeger et al., 2014b; Morrison et al., 2007), and here this hyperphagic response extended to the entire 27 week experiment (Figure 1H). The increase in food intake was accompanied by a persistent increase in EE, as demonstrated by the increase in EE in WT-LP mice after 11 weeks on diet (Figure 1G and S2), and by the fact that the LP-induced hyperphagia did not produce weight gain. Importantly, these long term effects of LP diet on food intake and EE were completely absent in FGF21-KO mice (Figure 1 and S2).
FGF21 is required for changes in liver metabolic gene expression
Previous work focusing on models of single amino acid restriction or deprivation suggest that amino acid restriction alters the expression of hepatic lipogenic gene expression (Anthony et al., 2013; Guo and Cavener, 2007), and there is evidence that FGF21 contributes to these effects (De Sousa-Coelho et al., 2013). Within the liver, 2 weeks of LP diet consistently reduced the expression of genes associated with lipogenesis (Scd1, Fas, and Srebp1), and this effect of LP-diet was lost in FGF21-KO mice (Figure 2A). A tendency for a reduction in Pepck expression was observed in all groups, although in no group was the effect statistically significant. Finally, consistent with our previous work, LP had no significant effect on Pparα mRNA expression in any group. The effect of LP diet to reduce Srepb1 was lost at 27 weeks, while Scd1 and Pparα mRNA expression was reduced in WT but not FGF21-KO mice (Figure 2B). Acc1 mRNA expression was induced by LP-diet in the FGF21-KO group at both 2 and 27 weeks. Taken together, these data suggest that the FGF21 is required for LP-induced changes in metabolic gene expression within the liver, particularly for genes related to lipid metabolism.
Figure 2. FGF21 is required for effects of low protein diet on hepatic metabolism.

WT and FGF21-KO were placed on isocaloric control or LP diet for 2 weeks (A) or 27 weeks (B) and mRNA expression was measured in liver. n = 10/group, #0.1 > P > 0.05; *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
FGF21 is required for changes in BAT and iWAT thermoregulatory gene expression
We next tested whether the robust and persistent increase in energy expenditure due to LP diet was associated with changes in BAT or iWAT. Consistent with the acute increase in energy expenditure induced by LP diet, Ucp1 gene expression (P < 0.01; Figure 3A) and UCP1 protein levels (P < 0.01; Figure 3B) were significantly increased after 14 days of protein restriction, although Cidea mRNA expression was not. There was no effect of LP diet on BAT UCP1 mRNA and protein at 27 weeks (Figure 3A and 3B), despite evidence for persistent increases in EE. Importantly, these effects of LP on UCP1 levels at 2 weeks were lost in FGF21-KO mice.
Figure 3. FGF21 is required for effects of protein restriction on metabolic and thermogenic gene expression in BAT and iWAT.

WT and FGF21-KO mice were placed on isocaloric control or LP diet for 2 (A, B) or 27 (A, B, C, D, E) weeks to assess thermogenic gene expression (A, C), UCP1 protein levels (B) and metabolic gene expression (E). Haematoxylin and eosin staining (D) of inguinal WAT revealed morphological changes consistent with browning in WT but not FGF21-KO mice on LP. The scale bar indicates 100 μm. n = 8–10/group, #0.1 > P > 0.05; *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
Considering that FGF21 has been linked to the browning of iWAT, we next tested whether LP promoted thermogenic gene expression and browning in iWAT. Unfortunately, iWAT was not collected at 2 weeks in WT and FGF21-KO mice. However, at 27 weeks we observe strong evidence for browning in iWAT. Levels of Ucp1 and Cidea are robustly increased by LP diet (Figure 3C), although Pgc1α was not affected. Histological assessment revealed discrete patches of morphological change that are consistent with browning, including the appearance of multilocular adipocytes (Figure 3D). Finally, these effects of LP diet on iWAT gene expression and morphology were absent in FGF21-KO mice (Figure 3C, 3D, 3E).
GCN2 is required for the acute behavioral and metabolic response to protein restriction
Our previous work suggested that GCN2 contributes to LP-induced increases in FGF21 (Laeger et al., 2014a). If FGF21 is required for the metabolic response to protein restriction, and GCN2 is essential for the induction of FGF21, then GCN2-KO mice should recapitulate the phenotype of FGF21-KO mice by not responding to protein restriction. To assess the acute response to LP diet, WT and GCN2-KO mice were placed on LP diet for 14 days. As expected, WT mice on LP diet displayed a progressive increase in energy expenditure (Figure 4A and 4C) during both the light and dark periods. This effect was evident irrespective of whether EE was expressed on a per-animal basis (Figure 4C), normalized to body weight or lean mass (Figure S3), or analyzed via ANCOVA with body weight as the covariant (Figure S3). LP diet also induced a trend toward increased food intake (P = 0.075; Figure 4D). Just as in FGF21-KO mice, there was no detectable change in either EE or food intake in GCN2-KO mice.
Figure 4. GCN2 is required for acute LP-induced changes in energy expenditure, body weight, and body composition.

Energy expenditure (EE) in WT (A) and GCN2-KO (B) mice consuming isocaloric control or LP diet. Average EE (C) over days 5 to 7 of diet consumption. Average daily food intake (D) in WT and GCN2-KO mice over the 14 days of diet exposure. Change in body weight (E), fat mass (F), and lean mass (G) in WT and GCN2-KO mice on control or LP diet for 14 days. n = 8/group, #0.1 > P > 0.05; *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
Also as expected, LP diet induced a significant loss of body weight in WT mice (P < 0.01; Figure 4E), with this weight loss being largely explained by loss of lean mass (P < 0.01; Figure 4G). GCN2-KO mice again failed to respond to LP, as they maintained their weight during LP diet (Figure 4E) and exhibited no significant change in either fat or lean mass (Figure 4F and 4G). Finally, changes in hepatic lipogenic gene expression were assessed in wildtype and GCN2-KO mice after 14 days (Figure S4), and here again the GCN2-KO mice replicate the phenotype of FGF21-KO mice by failing to exhibit a decrease Scd, Fas or Srebp1 mRNA expression. Collectively, these demonstrate that GCN2-KO mice do not exhibit acute metabolic responses to protein restriction, and as such replicate the phenotype of FGF21-KO mice.
GCN2 is not required for chronic responses to dietary protein restriction
To test whether GCN2 was also required for the chronic response to dietary protein restriction, a separate group of WT and GCN2 mice were placed on control or LP diets for 27 weeks (6 months) to replicate the prior study of FGF21-KO. As before, dietary protein restriction induced significant and persistent changes in body weight gain, food intake, energy expenditure and body composition in wildtype mice (Figure 5). Consistent with our prior data, this weight reducing effect of LP diet was not apparent in GCN2-KO mice at 1 or 2 weeks (Figure 5B). However, body weight gain began to diverge between GCN2-Control and GCN2-LP mice at 3 weeks on diet, and energy expenditure at 11 weeks on diet was significantly increased in GCN2-KO mice (Figure 5G and S2). Daily food intake averaged over the 27 week experiment was increased in GCN2-KO mice (Figure 5H), and at the end of the study LP diet exerted significant effects on body fat and lean gain and percent body fat in both wildtype and GCN2-KO mice (Figure 5C–F). Because the response to LP diet was largely intact in GCN2 mice at the end of the 27 week experiment, additional tissue endpoints in these mice were not analyzed. Taken together, these data suggest that GCN2 is required for LP-induced metabolic responses through approximately 14 days, but that in contrast to FGF21, GCN2 becomes unnecessary after more chronic dietary exposure.
Figure 5. GCN2 is not required for long-term low protein-induced changes in body weight, body composition, food intake, and energy expenditure.

Body weight change in WT (A) and GCN2-KO mice (B), body fat change (C) and body lean change (D) in WT and GCN2-KO mice on isocaloric control or LP diet for 27 weeks. Percent body fat (E) and percent body lean (F) at sacrifice on week 27. Average EE (G) over the 3 day period of EE measurement after 11 weeks on diet. Average daily food intake (H) over the entire 27 week period. n = 8/group, #0.1 > P > 0.05; *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
FGF21 levels progressively increase over time in association with a compensatory increase in ATF4 binding to the FGF21 gene promoter in GCN2-KO mice on LP diet
To explain this transient contribution of GCN2, we measured hepatic FGF21 mRNA expression and circulating FGF21 protein levels in WT and GCN2-KO mice over time. As expected, no Fgf21 mRNA could be detected in the FGF21-KO mice (Figure 6A), while hepatic Fgf21 mRNA expression was markedly increased by LP diet in wildtype mice at both 2 and 27 weeks. In GCN2-KO mice, this LP-induced increase was greatly blunted at 2 weeks (19-fold increase in WT vs 2.7-fold increase in GCN2-KO). However, at 27 weeks the effect of LP diet to increase hepatic Fgf21 mRNA expression in GCN2-KO mice was fully intact (Figure 6A), such that Fgf21 mRNA expression in GCN2-LP mice was increased relative to both GCN2-CON at 27 weeks (P < 0.01) and GCN2-LP at 2 weeks (P = 0.029). Consistent with Fgf21 mRNA expression, plasma levels of FGF21 were robustly increased in WT mice at 2 weeks (P < 0.01, Figure 6B), and FGF21 levels remained elevated in WT-LP mice for the entire 27 week experiment. This LP-induced increase in FGF21 was significantly blunted in GCN2-KO mice at 2 weeks, but blood samples collected in mice following more chronic dietary exposure (12 and 27 weeks) revealed a progressive increase in circulating FGF21 in LP-fed GCN2-KO mice, such that at 27 weeks serum FGF21 levels in GCN2-LP mice was increased relative to both GCN2-CON at 27 weeks (P < 0.01) and GCN2-LP at 2 weeks (P < 0.01). Just as the effects of LP-diet on EE and BW are transiently blunted in GCN2-KO mice, LP-induced increases in FGF21 were also transiently blunted in GCN2-KO mice.
Figure 6. The delayed response to protein restriction in GCN2-KO mice is explained by a delayed, ATF4-dependent increase in FGF21.

Liver FGF21 mRNA expression (A) in WT, FGF21-KO, and GCN2-KO after 2 and 27 weeks on isocaloric control or LP diets. Circulating FGF21 protein levels assessed by ELISA (B) in WT and GCN2-KO mice consuming control or LP diet for 2, 12, and 27 weeks. Schematic representation (C) of the mouse FGF21 promoter containing the distal (ATF4RE1) and proximal (ATF4RE2) genomic response elements recognized by ATF4. A negative control region without ATF4 binding sites is also shown. Arrows indicate the respective regions within the gene promoter targeted for amplification by qPCR. ChIP assays were performed using liver tissue from mice after 2 weeks (D) or 27 weeks (E) on LP diet. n = 8–10/group, except n=3/group for D and E. *P < 0.05; **P < 0.01. Data are represented as mean ± SEM.
Finally, we used a chromatin immunoprecipitation assay (ChIP) in liver tissue to test whether this progressive increase in FGF21 was mirrored by changes in ATF4 binding to the FGF21 promoter (Figure 6C), as ATF4 is considered to be the transcription factor that mediates GCN2-dependent regulation of FGF21 (De Sousa-Coelho et al., 2012; Schaap et al., 2013). ChIP analysis demonstrates that LP markedly increases ATF4 binding to the FGF21 promoter in WT mice at both 2 (Figure 6D) and 27 (Figure 6E) weeks. In GCN2-KO mice, this ATF4 binding was greatly blunted at 2 weeks; however, at 27 weeks ATF4 binding to the FGF21 promoter was normalized. Thus binding of ATF4 to the FGF21 promoter is highly correlated with the expression of FGF21 in response to LP diet. Taken together, these data indicate that protein restriction induces a robust and persistent increase in circulating FGF21 in wildtype mice, and that the delayed metabolic response to LP diet in GCN2-KO mice is explained by a delayed, GCN2-independent activation of ATF4 binding activity and a resultant increase in circulating FGF21.
Discussion
We previously demonstrated that dietary protein restriction induces a marked increase in circulating FGF21, and that this increase in FGF21 is required for behavioral and metabolic responses induced by two weeks of protein restriction (Laeger et al., 2014a). We also demonstrated that the acute, LP-induced increase in FGF21 (4 days) was greatly blunted in GCN2-KO mice. The present study extends these data in four important ways: First, we demonstrate that the robust changes in body weight, adiposity, energy expenditure, and food intake induced by protein restriction are accompanied by changes in metabolic gene expression in both liver, BAT and iWAT. Second, we demonstrate that these changes persist for at least 6 months of diet exposure. Third, we show that FGF21 is persistently elevated by dietary protein restriction and fully required for the metabolic effects induced by protein restriction at both acute and chronic time points. These data compellingly demonstrate that FGF21 is essential for adaptive responses to protein restriction and may be an endocrine signal that links dietary protein availability to metabolic function. Finally, our data suggest that GCN2-dependent activation of ATF4 is required for acute effects of LP diet on both FGF21 induction and metabolic responses, but that over time additional mechanisms upstream of ATF4 compensate for the absence of GCN2.
When wild-type C57BL6 mice are placed on a diet containing 5% protein, we observe a series of metabolic changes that are reminiscent of those induced by general dietary (calorie) restriction. Perhaps the most dramatic effect is the cessation of body weight gain, as over the 6 months of dietary exposure the wild-type mice on LP diet lost a miniscule 0.1 grams, whereas control mice gained 7 grams. This cessation of body weight gain was also associated with reductions in both fat and lean mass gain, and a reduction in body length. While LP diet produced a significant loss of lean mass, this loss was counterbalanced by a much larger reduction in fat gain. As a result, WT-LP mice were leaner (reduced percent fat) than their controls. It is possible that consuming a diet containing only 5% protein might exert negative effects on muscle growth and performance, and we therefore assessed athletic performance and coordination using the rotarod. Interestingly, WT-LP exhibited either no change or an improvement in rotarod performance in the two studies we conducted. Thus, despite the rather marked reduction in protein intake, our data provide no evidence that this level of protein restriction exerts marked negative effects on health or muscular function, at least through 6 months of diet exposure.
Dietary protein restriction is known to increase both food intake and energy expenditure (Gosby et al., 2014; Hasek et al., 2010; Laeger et al., 2014a; Laeger et al., 2014b; Morrison et al., 2007; Rothwell and Stock, 1987; Rothwell et al., 1983; Simpson and Raubenheimer, 1997, 2005; White et al., 2000). We observe similar effects in the current data, and demonstrate that these changes are persistent over the entire 6 month experiment. Although EE was only measured at week 1 and week 11, it was increased at both time points and food intake was increased throughout the experiment, including the last few weeks of dietary treatment. The maintenance of hyperphagia without weight gain strongly implies that EE was significantly increased for the entire 6 month period. These data suggest that the reduction in long-term body weight gain is driven largely by this robust and persistent increase in whole body energy expenditure, with the concomitant hyperphagia being an adaptive response to avoid weight loss and increase protein intake.
Remarkably, the robust and persistent effects of dietary protein restriction on feeding behavior, energy expenditure, body weight gain, and metabolic gene expression were completely absent in FGF21-KO mice. These data clearly demonstrate that FGF21 is robustly induced by the specific reduction of protein intake, that FGF21 levels remained elevated for the duration of protein restriction (through 6 months), and that FGF21 is required for adaptive responses to protein restriction. The fact that FGF21-KO mice grew normally, despite consuming the same LP diet as controls, also leads to the interesting conclusion that the LP-induced reduction in growth and BW gain were not directly due an insufficient supply of amino acids to support growth, but instead due to the FGF21-dependent metabolic response induced by protein restriction.
Previous work from our lab and others suggests that the amino acid sensor GCN2 may be a key molecular link driving the increases in FGF21 observed during amino acid restriction (De Sousa-Coelho et al., 2012; Laeger et al., 2014a; Wilson et al., 2015). If GCN2 is indeed required for LP-induced increases in FGF21, then GCN2-KO mice, like FGF21-KO mice, should be largely unresponsive to dietary protein restriction. The current manuscript supports this conclusion since neither food intake, EE or body weight gain was altered by LP diet in GCN2-KO mice through 14 days. However, subsequent long-term studies yielded a much different result. Body weight began to diverge between GCN2-CON and GCN2-LP mice at 3 weeks (in WT mice the divergence appears immediately at 1 week). Similarly, although there was no effect of LP diet to increase EE or food intake in GCN2-KO mice over the first 7 days of diet exposure, a clear increase EE and food intake was observed when GCN2-KO mice were tested at 11 weeks. Therefore, these data suggest that the contribution of GCN2 to the metabolic response to protein restriction is only transient.
Our data indicate that this transitory contribution of GCN2 is largely explained by changes in circulating FGF21. As previously shown and discussed above, dietary protein restriction induces an acute, robust and persistent increase in both hepatic Fgf21 mRNA and circulating FGF21 protein. This acute induction of FGF21 by LP diet is markedly blunted in GCN2-KO mice, and it seems likely that the lack of response to LP diet in GCN2-KO mice is explained by this blunted increase in FGF21. However, LP diet still produces a small increase in FGF21 in GCN2-KO mice at 4 (Laeger et al., 2014a) and 14 days, suggesting that GCN2 is not the sole driver of FGF21 during protein restriction. Most importantly, this GCN2-independent effect is mobilized over time, such that FGF21 levels progressively rise in GCN2-LP mice. Currently the identity of this GCN2-independent pathway(s) is unclear, but our data suggest that the compensation occurs upstream of the transcription factor ATF4. ATF4 has been previously linked to amino acid-dependent FGF21 regulation, as it was shown that ATF4 binds to the FGF21 promoter and is required for induction of FGF21 in response to amino acid restriction (De Sousa-Coelho et al., 2012; Schaap et al., 2013). Our data demonstrate that the binding of ATF4 to the FGF21 promoter in the liver is robustly and persistently increased by dietary protein restriction. In addition, a GCN2-dependent activation of ATF4 signaling appears to be a key mechanism underlying the acute induction of FGF21 following protein restriction, as ATF4 binding is blunted in the GCN2-KO mice after 2 weeks on LP diet. However, just as GCN2 is not required for the LP-induced increases in FGF21 over longer periods, GCN2 is also not required for the LP-induced binding of ATF4 to the FGF21 promoter. Thus some additional pathway, upstream of ATF4, compensates for the absence of GCN2 following long-term protein restriction. Whether this pathway contributes to FGF21 expression in wildtype mice, or is instead only recruited in the absence of GCN2, remains unclear.
Finally, these data also conceptually suggest that FGF21 levels must increase above a threshold before they exert marked effects on body weight or EE. In other words, our data in GCN2-KO mice suggest that FGF21 concentrations below approximately 2 ng/ml are insufficient to drive marked metabolic effects in mice, but that changes in EE and BW are readily observed when concentrations approach 4ng/ml. Additional work is required to precisely define this threshold concentration and to extrapolate it to other contexts, but these data suggest that fold increases in FGF21 that appear large (from 0.3 to 1.48 ng/ml or 5-fold) may still be insufficient to drive metabolic responses.
In addition to inducing changes in whole body energy expenditure, FGF21 also acts on multiple tissues to coordinate substrate metabolism and cellular function (Kharitonenkov and Adams, 2014; Owen et al., 2015). We therefore analyzed a variety of genes associated with carbohydrate and lipid metabolism in liver, iWAT, and BAT. Consistent with the reduction in fat gain and adiposity in WT-LP mice, genes associated with hepatic lipogenesis were significantly reduced in WT-LP mice after 14 days on diet, and these effects were absent in both GCN2-KO and FGF21-KO mice. These data are consistent with prior work in mice using diets devoid of a single amino acid (De Sousa-Coelho et al., 2013; Guo and Cavener, 2007), and previous work in rats demonstrating that single amino acid restriction potently alters lipid metabolism in liver and adipose tissue (Hasek et al., 2013). FGF21 also promotes thermogenic activity in BAT and WAT (Adams et al., 2012; Douris et al., 2015; Fisher et al., 2012; Lee et al., 2014), and it might therefore be expected that the chronic induction of FGF21 induces thermogenic activation of these tissues (De Sousa-Coelho et al., 2013). Long-term protein restriction significantly increased both UCP1 and Cidea mRNA within iWAT in association with morphological changes consistent with browning, and these effects were absent in FGF21-KO mice. Whether these effects on WAT and BAT are required for the persistent increase in EE and reduction in body weight gain observed with long-term dietary protein restriction remains unclear, as recent reports suggest that the effects of FGF21 on EE may not require UCP1 or WAT browning (Bernardo et al., 2015; Samms et al., 2015; Veniant et al., 2015), although UCP1 appears contribute to the metabolic effects induced by dietary restriction of just the amino acid methionine (Wanders et al., 2015).
There is ample evidence supporting the existence of regulatory mechanisms which sense nutrient depletion and coordinate adaptive metabolic and behavioral responses (Morrison and Berthoud, 2007), and it seems likely that protein intake is also regulated via a mechanism that is independent of energy intake (Morrison and Laeger, 2015; Morrison et al., 2012). Long-term dietary protein restriction induces a series of behavioral and metabolic effects that, perhaps contrary to expectations, result in ‘beneficial’ changes in energy expenditure, body weight and adiposity that are consistent with the well-described effect of dietary restriction on health and longevity (Solon-Biet et al., 2015). Our data strongly suggest that FGF21 is an endocrine hormone that plays a key biological role in the mediating these adaptive responses to protein restriction.
Experimental Procedures
Animals and Diets
All procedures involving animals were approved by the PBRC Institutional Animal Care and Use Committee. Male C57BL6 mice (Jackson Lab) were used in all studies. GCN2-deficient mice on the B6 background were purchased from Jackson Labs (Stock #:008240). FGF21-deficient mice on the B6 background were kindly provided by Dr. Steven Kliewer (UT Southwestern) as described previously (Potthoff et al., 2009). Control and LP diets were formulated and produced by Research Diets, and were designed to be isocaloric by equally varying protein and carbohydrate while keeping fat constant. Control diet contained 20% casein (by weight) as the protein source, while the LP diet contained 5% casein. On an energy basis the diets contained protein at 18% (Control) and 4% (LP). A detailed dietary composition is provided in Table S1. Animals were single housed in 12:12hr light:dark cycle with ad libitum access to food or water unless otherwise noted.
For general effects of dietary protein restriction, single housed mice were transferred from chow to the control diet for approximately 5 days, at which point a random subgroup of animals were transferred to the respective LP diet. Animals consumed control or LP diet ad libitum for the duration of the study until they were sacrificed and liver, inguinal white adipose tissue (iWAT), and brown adipose tissue (BAT) collected and snap frozen for further analysis. Mice were sacrificed during the mid-light cycle in the fed state (unless otherwise noted) using acute exposure to CO2 followed by rapid decapitation. Trunk blood was also collected at sacrifice, allowed to clot overnight at 4°C, centrifuged at 3000xg and serum collected.
Acute response to LP diet in GCN2 and FGF21-deficient mice (14 days)
GCN2-KO and WT mice (8 mice/diet/genotype) were initially adapted to the control diet while in metabolic chambers (OxyMax/CLAMS, Columbus Instruments), were transitioned to the LP diet while in the OxyMax, and remained in the OxyMax for the first 7 days of dietary exposure. Afterward mice were returned to standard caging for the final 7 days, and after 14 days on diet were sacrificed and tissue collected as described above. Body weight and food intake were collected 4-times through the 14 day experiment. Body composition was measured via TD-NMR (Bruker Minispec) on the start of experimental diets (Day 0), the final day of OxyMax metabolic analysis (Day 7) and on the day of sacrifice (Day 14). FGF21-deficient (FGF21-KO) mice were treated as above, with LP-dependent effects on metabolic gene expression within liver and BAT described in this manuscript while data related to changes in body weight, energy expenditure and food intake have been previously published for these mice (Laeger et al., 2014a).
Chronic response to LP diet in GCN2 and FGF21-deficient mice (6 months)
15 week old GCN2-KO, FGF21-KO, and WT mice were adapted to control diet and then randomly placed on either control or LP diet (8 mice/diet/genotype) for 27 weeks. Body weight and food intake were collected every 7 days during the experiment. Body composition was measured via TD-NMR (Bruker Minispec) every 14 days until the end of the experiment. After 11 weeks on diet mice were placed into metabolic chambers (OxyMax) for 3 days to assess energy expenditure, respiratory exchange ratio, and activity (beam breaks), after which mice were returned to standard caging until the end of the experiment. After 12 weeks on the diet blood was collected via the saphenous vein from GCN2-KO and corresponding WT mice to analyze FGF21. After 20 weeks on diet motor function and coordination was tested via Rotarod (Med-Associates, St Albans, YT). In this test mice are placed on a slowly accelerating rod, progressing from 4 to 40 rpm during a period of 300 sec, and the latency to fall was recorded for each animal. Mice were given 3 trials, with an inter-trial interval of 30 min. Each mouse was acclimated to the movement of the rod and then to its acceleration for 1 minute during an initial training session the day before the first test session. Finally, GCN2-KO and FGF21-KO with corresponding WT mice were sacrificed after 27 weeks and tissue collected as described above.
Immunoassay Determination of FGF21
Concentrations of FGF21 in serum were determined in mice with an ELISA according to the procedure recommended by the manufacturer (no. RD291108200R, Mouse and Rat FGF-21 ELISA, BioVendor). The minimal detectable concentration of FGF21 with this assay was 18.4 pg/ml. The intra- and inter-assay coefficients of variation for the mouse and rat FGF21 assay were both < 9.0%, respectively. For determination of serum FGF21, 50 μl of serum were diluted in 200 μl of dilution buffer before analysis.
Real-time PCR
RNA extraction and real-time PCR was conducted as described previously (Morrison et al., 2007). Total RNA was extracted from liver, iWAT, and BAT using TRIzol reagent following the manufacturer’s protocol (15596018, Invitrogen). RNA quality and quantity was determined by spectrophotometry using a NanoDrop (Thermo Scientific). cDNA synthesis was performed with M-MLV reverse transcriptase (M1701, Promega) and mRNA was quantified on the ABI 7900 platform using the SYBR green methodology in optical 384-well plates (Applied Biosystems). Primer pairs were designed using NCBI Primer-BLAST with at least one primer spanning an exon-exon boundary. Target gene expression was normalized with cylcophilin B as the endogenous control.
Haematoxylin and eosin staining (H&E)
Paraffin sections of inguinal WAT were mounted on slides and dewaxed through immersion in xylene substitute, 100% ethanol, and 95% ethanol, prior to a water immersion step. The slides were then stained 3 min in Hematoxylin, washed, immersed in Define solution 1 min, washed, immersed in bluing solution 1 min, washed, dehydrated with 95% ethanol 1 min, stained with eosin, then dehydrated through 100% ethanol and xylene substitute prior to coverslipping. All staining was done on a Leica ST5020 autostainer.
Western Blot
BAT was added frozen and immediately minced in cold whole cell lysate buffer with protease & phosphatase inhibitors, followed by sonication and centrifugation. 5–10 ug of supernatant from centrifugation was used for subsequent Western blotting on a standard 10% SDS-PAGE gel in a Bio-Rad electrophoresis system in Laemmli loading sample buffer. Blots were blocked with Odyssey blocking buffer (Licor) in nitrocellulose membrane, then incubated overnight at 4C with primary antibodies. Ucp-1 (ab10983) antibody was also obtained from AbCam, at a dilution of 1:4000, and alpha-tubulin (1:4000, Abcam ab7291; Cambridge, UK) was used as an internal control. The next morning they were washed then probed with Licor IR-secondary antibodies at 1:5000 for 1 hour at room temperature (Odyssey Secondary antibody: Donkey IRDye 800CW anti-rabbit IgG (H+L) (926–32213) and Odyssey Secondary antibody: Donkey IRDye 680LT anti-mouse IgG (H+L) (926–68022)), washed again, and then imaged on Licor Odyssey.
Chromatin Immunoprecipitation
Mouse liver tissue (30–50 mg) was thawed on ice and chopped into 1–3 mm3 pieces using a clean razor blade. Tissue was placed in a 1.5 mL tube, and 1 mL of crosslinking solution (1% formaldehyde-HEPES-NaOH, pH 7.8) was added. Tubes were rotated for 10 min at room temp. Crosslinking was stopped by adding glycine to a final concentration of 125 mM and tubes were rotated for an additional 5 min at room temp. Tissue pellets were collected by centrifugation at 500 × g for 5 min at 4°C and washed with 1 mL ice-cold PBS, followed by centrifugation at 500 × g for an additional 5 min at 4°C. Supernatants were discarded, and tissue was lysed in 0.9 mL SDS Lysis Buffer containing 1× protease inhibitors. Lysates were incubated at 4°C, with end-ove r-end mixing, for 30 min. Lysates were separated into 3× 300 μL aliquots in polystyrene sonication tubes and sonicated at 4°C using a Bioruptor Pico (Diagenode). The following sonication conditions generated fragments of DNA between 100 and 500 bp in length: 30 sec on, 30 sec off, 10 cycles. Tubes were incubated on ice for 15 min and sonication was repeated for 5 more cycles. Aliquots were recombined into one tube and centrifuged at 12,000 × g for 10 min at 4°C. Supern atants were transferred to a clean microcentrifuge tube. Protein concentration in lysates was determined by BCA assay (Thermo Scientific). 750 μg sheared, cross-linked DNA was diluted 10-fold in dilution buffer containing 1× protease inhibitors. 11 μL of ATF4 antibody, or control rabbit IgG, was added and incubated overnight at 4°C with end-over-end mixing. Immunoprecipita tion of DNA–protein–antibody complexes was performed by incubation with 40 μL Protein G Dynabeads (Invitrogen) for 1 h at 4°C with end-over-end mixin g. The magnetic beads binding to the antibody–antigen–chromatin complexes were washed sequentially for 5 mins each at 4°C with 0.5 mL buffers 1–4. 100 μL chelating resin was added, vortexed for 10 s, and heated to 100°C for 10 min. Following centrifugation at 17,000 × g for 1 min at 4°C, the supernatants were transferred to new tubes. A total of 120 μL nuclease-free water was added, vortexed, and recentrifuged. Supernatants were pooled. Inputs were processed as followed: 90 μL (10%) sonicated DNA was incubated overnight at 65°C with NaCl to a final concentration of 200 mM. Input DNA was then treated with RNase A for 30 min at 37°C and proteinase K for 1 h at 45°C, followed by purification using a QIAGEN Cleanup kit (QIAGEN). Input DNA was run on a 2% agarose gel to test for sonication efficiency. 5 μL DNA was used as a template for downstream SYBR Green–based PCR reactions. All data showing specific binding events are represented as percentage of input. Buffers and chelating resin were from the ExactaChIP Chromatin Immunoprecipitation Kit (R&D Systems). ATF4 antibody was from Cell Signaling (Cat # 11815). Primer sequences used for amplifying the distal (ATF4RE1) and proximal (ATF4RE2) ATF4 binding sites, as well as a negative control region, within the mouse FGF21 promoter were from Shimizu N et al. (Shimizu et al., 2015).
Statistical Analysis
Data were analyzed using the SAS software package (SAS V9, SAS Institute) using one-way or two-way ANOVA, or repeated measures ANOVA, using the general linear model procedure. When experiment-wide tests were significant, post-hoc comparisons were made using the LSMEANS statement with the PDIFF option, and represent least significant differences tests for pre-planned comparisons. Analysis of energy expenditure with body weight as the covariate was assessed via analysis of covariance (ANCOVA) using the general linear model procedure of SAS, with results validated against the MMPC.org ANCOVA data analysis tool. All data are expressed as mean ± SEM, with a probability value of 0.05 considered statistically significant.
Supplementary Material
Highlights.
Protein restriction increases FGF21 acutely and persistently
FGF21 is required for the metabolic and behavioral response to protein restriction
GCN2 is only transiently required for the response to protein restriction
Pathways upstream of ATF4 compensate for the absence of GCN2 and induce FGF21
Acknowledgments
The authors would like to thank the staff of the PBRC Comparative Biology Core for their skillful assistance and excellent technical support. This work was supported by NIH R01DK081563 and R01DK105032 to CDM. TL was supported by a research fellowship from the Deutsche Forschungsgemeinschaft (DFG) LA 3042/2-1. HRB was supported by R01DK047348, TWG was supported by ADA 1-12-BS-58 and NIH DK-096311, and HM was supported by R01DK092587. This project/work used facilities within the Animal Metabolism & Behavior Core, Genomics Core, and Cell Biology and Bioimaging Core at PBRC that are supported in part by COBRE (P20GM103528) and NORC (P30DK072476) center grants from the National Institutes of Health.
Footnotes
The authors have declared that no conflict of interest exists.
Author Contributions
Conceptualization, TL, CDM; Methodology, TL, SJB, JJC, DCA; Investigation, TL, DCA, SJB, LT, JH, JJC, CDM; Analysis and Interpretation, TL, HRB, TG, HM, CDM; Writing – Original Draft, TL, CDM; Writing – Review and Editing, TL, HRB, TG, JJC, HM, CDM; Funding Acquisition, CDM.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, Kharitonenkov A. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Molecular metabolism. 2012;2:31–37. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony TG, McDaniel BJ, Byerley RL, McGrath BC, Cavener DR, McNurlan MA, Wek RC. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem. 2004;279:36553–36561. doi: 10.1074/jbc.M404559200. [DOI] [PubMed] [Google Scholar]
- Anthony TG, Morrison CD, Gettys TW. Remodeling of lipid metabolism by dietary restriction of essential amino acids. Diabetes. 2013;62:2635–2644. doi: 10.2337/db12-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Bernardo B, Lu M, Bandyopadhyay G, Li P, Zhou Y, Huang J, Levin N, Tomas EM, Calle RA, Erion DM, et al. FGF21 does not require interscapular brown adipose tissue and improves liver metabolic profile in animal models of obesity and insulin-resistance. Scientific reports. 2015;5:11382. doi: 10.1038/srep11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J. 2012;443:165–171. doi: 10.1042/BJ20111748. [DOI] [PubMed] [Google Scholar]
- De Sousa-Coelho AL, Relat J, Hondares E, Perez-Marti A, Ribas F, Villarroya F, Marrero PF, Haro D. FGF21 mediates the lipid metabolism response to amino acid starvation. Journal of lipid research. 2013;54:1786–1797. doi: 10.1194/jlr.M033415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, Zarebidaki E, Adams AC, Kharitonenkov A, Flier JS, et al. Central Fibroblast Growth Factor 21 Browns White Fat via Sympathetic Action in Male Mice. Endocrinology. 2015;156:2470–2481. doi: 10.1210/en.2014-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Semenkovich CF. Essential amino acids regulate fatty acid synthase expression through an uncharged transfer RNA-dependent mechanism. J Biol Chem. 1995;270:29323–29329. doi: 10.1074/jbc.270.49.29323. [DOI] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosby AK, Conigrave AD, Raubenheimer D, Simpson SJ. Protein leverage and energy intake. Obes Rev. 2014;15:183–191. doi: 10.1111/obr.12131. [DOI] [PubMed] [Google Scholar]
- Guo F, Cavener DR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab. 2007;5:103–114. doi: 10.1016/j.cmet.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16:5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasek BE, Boudreau A, Shin J, Feng D, Hulver M, Van NT, Laque A, Stewart LK, Stone KP, Wanders D, et al. Remodeling the integration of lipid metabolism between liver and adipose tissue by dietary methionine restriction in rats. Diabetes. 2013;62:3362–3372. doi: 10.2337/db13-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasek BE, Stewart LK, Henagan TM, Boudreau A, Lenard NR, Black C, Shin J, Huypens P, Malloy VL, Plaisance EP, et al. Dietary methionine restriction enhances metabolic flexibility and increases uncoupled respiration in both fed and fasted states. Am J Physiol Regul Integr Comp Physiol. 2010;299:R728–739. doi: 10.1152/ajpregu.00837.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Adams AC. Inventing new medicines: The FGF21 story. Molecular metabolism. 2014;3:221–229. doi: 10.1016/j.molmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Kim SH, Jung HS, Park KS, Lee HY, Lee MS. Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun. 2013;440:76–81. doi: 10.1016/j.bbrc.2013.09.026. [DOI] [PubMed] [Google Scholar]
- Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Munzberg H, Hutson SM, Gettys TW, Schwartz MW, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014a;124:3913–3922. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laeger T, Reed SD, Henagan TM, Fernandez DH, Taghavi M, Addington A, Munzberg H, Martin RJ, Hutson SM, Morrison CD. Leucine acts in the brain to suppress food intake but does not function as a physiological signal of low dietary protein. Am J Physiol Regul Integr Comp Physiol. 2014b;307:R310–320. doi: 10.1152/ajpregu.00116.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014;19:302–309. doi: 10.1016/j.cmet.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD, Berthoud HR. Neurobiology of nutrition and obesity. Nutrition reviews. 2007;65:517–534. doi: 10.1301/nr.2007.dec.517-534. [DOI] [PubMed] [Google Scholar]
- Morrison CD, Laeger T. Protein-dependent regulation of feeding and metabolism. Trends Endocrinol Metab. 2015;26:256–262. doi: 10.1016/j.tem.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD, Reed SD, Henagan TM. Homeostatic regulation of protein intake: in search of a mechanism. Am J Physiol Regul Integr Comp Physiol. 2012;302:R917–928. doi: 10.1152/ajpregu.00609.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD, Xi X, White CL, Ye J, Martin RJ. Amino acids inhibit Agrp gene expression via an mTOR-dependent mechanism. Am J Physiol Endocrinol Metab. 2007;293:E165–171. doi: 10.1152/ajpendo.00675.2006.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab. 2015;26:22–29. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. Influence of carbohydrate and fat intake on diet-induced thermogenesis and brown fat activity in rats fed low protein diets. J Nutr. 1987;117:1721–1726. doi: 10.1093/jn/117.10.1721. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ, Tyzbir RS. Mechanisms of thermogenesis induced by low protein diets. Metabolism. 1983;32:257–261. doi: 10.1016/0026-0495(83)90190-7. [DOI] [PubMed] [Google Scholar]
- Samms RJ, Smith DP, Cheng CC, Antonellis PP, Perfield JW, 2nd, Kharitonenkov A, Gimeno RE, Adams AC. Discrete Aspects of FGF21 In Vivo Pharmacology Do Not Require UCP1. Cell reports. 2015;11:991–999. doi: 10.1016/j.celrep.2015.04.046. [DOI] [PubMed] [Google Scholar]
- Schaap FG, Kremer AE, Lamers WH, Jansen PL, Gaemers IC. Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie. 2013;95:692–699. doi: 10.1016/j.biochi.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, Tasaka Y, Kuribara-Souta A, Miyata K, Oike Y, et al. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nature communications. 2015;6:6693. doi: 10.1038/ncomms7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson SJ, Raubenheimer D. Geometric analysis of macronutrient selection in the rat. Appetite. 1997;28:201–213. doi: 10.1006/appe.1996.0077. [DOI] [PubMed] [Google Scholar]
- Simpson SJ, Raubenheimer D. Obesity: the protein leverage hypothesis. Obes Rev. 2005;6:133–142. doi: 10.1111/j.1467-789X.2005.00178.x. [DOI] [PubMed] [Google Scholar]
- Solon-Biet SM, Walters KA, Simanainen UK, McMahon AC, Ruohonen K, Ballard JW, Raubenheimer D, Handelsman DJ, Le Couteur DG, Simpson SJ. Macronutrient balance, reproductive function, and lifespan in aging mice. Proc Natl Acad Sci U S A. 2015;112:3481–3486. doi: 10.1073/pnas.1422041112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, Moyer C, Lloyd DJ. Pharmacologic Effects of FGF21 Are Independent of the “Browning” of White Adipose Tissue. Cell Metab. 2015;21:731–738. doi: 10.1016/j.cmet.2015.04.019. [DOI] [PubMed] [Google Scholar]
- Wanders D, Burk DH, Cortez CC, Van NT, Stone KP, Baker M, Mendoza T, Mynatt RL, Gettys TW. UCP1 is an essential mediator of the effects of methionine restriction on energy balance but not insulin sensitivity. FASEB J. 2015;29:2603–2615. doi: 10.1096/fj.14-270348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497–4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BD, Porter MH, Martin RJ. Protein selection, food intake, and body composition in response to the amount of dietary protein. Physiol Behav. 2000;69:383–389. doi: 10.1016/s0031-9384(99)00232-2. [DOI] [PubMed] [Google Scholar]
- Wilson GJ, Lennox BA, She P, Mirek ET, Al Baghdadi RJ, Fusakio ME, Dixon JL, Henderson GC, Wek RC, Anthony TG. GCN2 is required to increase fibroblast growth factor 21 and maintain hepatic triglyceride homeostasis during asparaginase treatment. Am J Physiol Endocrinol Metab. 2015;308:E283–293. doi: 10.1152/ajpendo.00361.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, Meng Q, Cheng Y, Gao X, Li J, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes. 2011;60:746–756. doi: 10.2337/db10-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22:6681–6688. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.