

Abstract
Recent studies utilizing transcriptomics, metabolomics, and bottom up proteomics have identified molecular signatures of kidney allograft pathology. Although these results make significant progress toward non-invasive differential diagnostics of dysfunction of a transplanted kidney, they provide little information on the intact, often modified, protein molecules present during progression of this pathology. Because intact proteins underpin diverse biological processes, measuring the relative abundance of their modified forms promises to advance mechanistic understanding, and might provide a new class of biomarker candidates. Here, we used top down proteomics to inventory the modified forms of whole proteins in peripheral blood mononuclear cells (PBMCs) taken at the time of kidney biopsy for 40 kidney allograft recipients either with healthy transplants or those suffering acute rejection. Supported by gas-phase fragmentation of whole protein ions during tandem mass spectrometry, we identified 344 proteins mapping to 2,905 distinct molecular forms (proteoforms). Using an initial implementation of a label-free approach to quantitative top down proteomics, we obtained evidence suggesting relative abundance changes in 111 proteoforms between the two patient groups. Collectively, our work is the first to catalog intact protein molecules in PBMCs and suggests differentially abundant proteoforms for further analysis.
Keywords: Biomarker, Mass spectrometry, Proteoform, Proteomics, Transplant
1. INTRODUCTION
Recent data from the US Department of Health and Human Services estimates over 100,000 people per year are diagnosed with end-stage kidney disease in the United States (http://www.niddk.nih.gov/health-information/health-statistics/Pages/kidney-disease-statistics-united-states.aspx#5). The current treatment of choice for this is transplantation, where the work of the non-functional kidneys is replaced with a functional one. Successful kidney transplantation has its challenges; chief among these is preventing immune-mediated rejection of the allograft. Granted immunosuppressive therapy is now routine procedure in post-transplant care, occasional incidents of acute rejection still occur and clinical complications include loss of the allograft [1, 2]. Because the mechanisms effecting the rejection process are relatively unclear, a better understanding of the molecular and cellular events associated with acute rejection is of high interest for the transplant research community and could provide novel drug targets and diagnostics for improving patient health.
Multiple studies over the past decade have sought to identify molecular signatures associated with kidney transplant dysfunction, including RNA and bottom up proteomics profiling of kidney biopsies and peripheral blood lymphocytes over a range of clinical outcomes [3–8]. On one level these studies have succeeded: mRNA profiling of kidney biopsies has enabled the classification of three different subtypes of acute rejection [7], added molecular signatures to complement current gold standard histology diagnostics [8], revealed differences in gene expression patterns between kidney biopsy tissue and circulating lymphocytes of acute rejection patients [3], and provided numerous gene signatures for future biomarker validation studies [5]. On another level, these prior studies have limitations as RNA and peptide-based studies are disconnected from the intact, mature protein molecules present within immune cells.
Functional protein molecules within a cell are the result of a multi-stage process beginning with mRNA transcription and ending in a properly processed polypeptide that can be modified co- and/or post-translationally. As variation can be introduced at each stage, the variety in protein molecular forms from a given gene are vast. Examples of where variation could be introduced include alternative splicing, differential translational start sites, proteolytic processing, and post-translational modification such as phosphorylation, acetylation, and methylation in various combinations. These combinatorial events create various “proteoforms”, a gene-centric term used to denote intact protein molecules with distinct chemical composition and site-specific post-translational modifications (see Figure 1) [9]. Appreciating this molecular complexity, a comprehensive understanding of molecular processes operative at the level of protein primary structure requires resolution and quantitation of proteoforms.
Figure 1. Proteins, isoforms, proteoforms, and the naming convention of each.
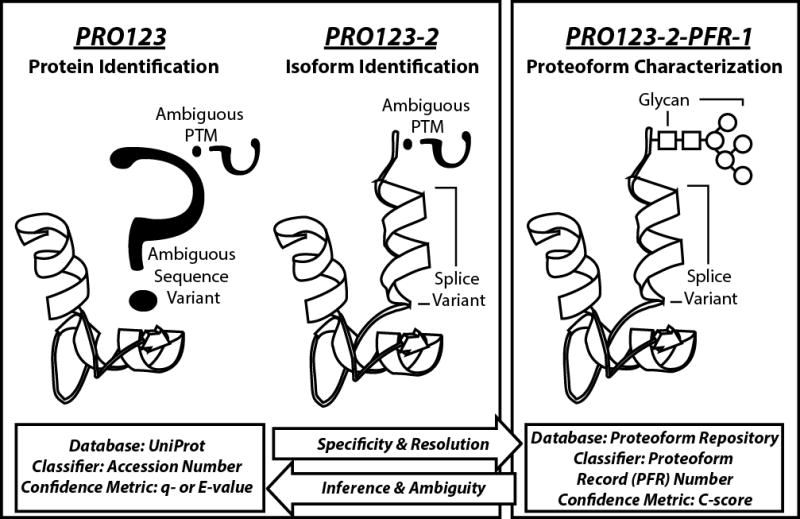
Protein molecules can be discussed using three levels of increasing specificity: protein identification (far left), isoform identification (middle), and proteoform characterization (far right). UniProtKB entries are based on accession numbers, which is gene-specific but does not distinguish between alternative transcripts or molecular variants resulting from post-translational modification (PTM). UniProt handles the alternative transcripts by introducing isoform information into the classification, but this is still unable to distinguish different forms of proteins carrying specific PTMs. The proteoform record (PFR) number carries maximal molecular specificity by referring to an intact protein molecule’s chemical formula, including type and location of PTMs. In top down proteomics, confidence metrics for protein identification by database retrieval are q- or E-values (bottom left); for proteoform characterization the metric is the C-score (bottom right). “PRO123” is a hypothetical protein; “PRO123-2” specifies a non-canonical isoform of a UniProt entry; “PFR-1” is a specific proteoform, which contains a glycan modification, and maps to the hypothetical PRO123 accession in UniProtKB.
Top down proteomics measures proteins in their intact state affording information on exact molecular weight, protein identity, and the type, location, and relative abundance of post-translational modifications [10]. Accordingly, top down proteomics has been used to explore the proteoform landscape within a wealth of biological studies, with recent iterations yielding information on over 1,000 proteins distributed into an average of ~4 proteoforms characterized in automated fashion [11, 12]. In addition, the ability to quantify proteoforms <30 kDa between two states in discovery mode using a novel label-free quantitative top down approach has been demonstrated [13], along with successful implementation of top down proteomics by a variety of laboratories [14–18]. Collectively, quantitative top down proteomics—with the ability to identify and quantify proteoforms between biological states—is the direct approach for proteoform-resolved molecular profiling.
Here, we applied top down proteomics in both qualitative and quantitative modes to begin studying peripheral blood mononuclear cells (PBMCs) from kidney transplant recipients diagnosed on a biopsy as either having clinical acute rejection (cAR) or a normal biopsy—transplant excellent (TX). Our goal was to supplement prior exploratory studies reporting on mRNA and in vitro processed proteomes with a precise map of proteoforms from similar tissue and clinical status. Our data provide the first foray of proteoform-resolved measurement of PBMCs (complete with regularized proteoform reporting) and suggest differentially abundant targets between those with clinical acute rejection and transplant excellent kidneys for follow-up in clinical research.
2. MATERIALS AND METHODS
2.1 Experimental design
The study design used here is described in the results section and is depicted in Figure 2. Specifically, patient samples were processed in two sample sets: Sample Set 1 and Sample Set 2. Each sample set consisted of 20 total kidney allograft patient samples; 10 of these were diagnosed as transplant excellent (TX) by the lack of clinical data indicative of rejection (i.e. elevated creatinine) and a protocol biopsy confirming no rejection; 10 were diagnosed as clinical acute rejection (cAR) by clinical data and confirmatory for-cause biopsy. TX patient samples were collected at times of either 3 months or 12 months post-transplant; cAR samples were collected at similar times post-transplant with the exception of a few outliers (Figure S1). For both sample sets, individual patient samples of each category (TX or cAR) were pooled into three pools to achieve the minimal starting amount of total protein, which was 300 μg at the time these data were collected in 2012. Sample Set 1 was run in qualitative mode without randomization whereas Sample Set 2 was properly randomized to allow for label-free quantitation using a linear hierarchical model previously described [13, 19]. All patient samples were collected according to institutional review board (IRB) approved protocols.
Figure 2. Study design for comparative top down proteomics of peripheral blood mononuclear cells from kidney transplant recipients.
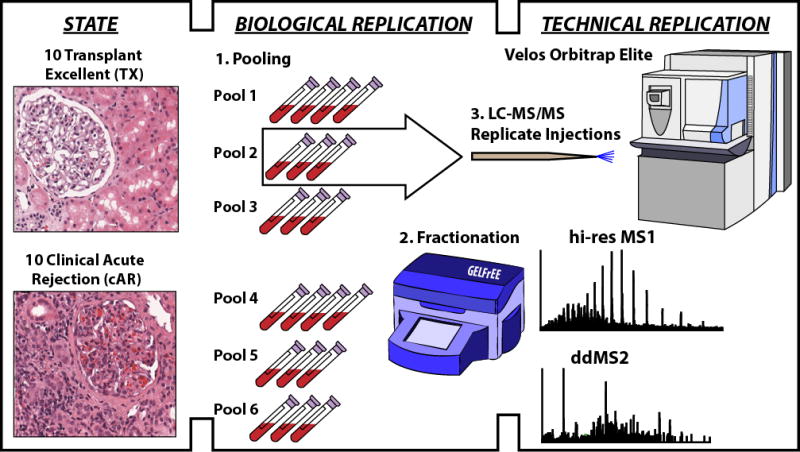
Patients were stratified into two groups by experienced pathologists independently reviewing pathology to confirm phenotypic assignments. Those with a normal functioning allograft and protocol biopsy showing no signs of cell-mediated rejection were called transplant excellent (TX); those with clinical signs of allograft dysfunction (i.e. elevated creatinine in the circulation) and a for-cause biopsy confirming cell-mediated rejection were called clinical acute rejection (cAR). In each of two phenotypic states, peripheral blood mononuclear cells (PBMCs) were collected from ten patients per state. Multiple samples from within each group were pooled to yield approximately 300 μg of total protein extracted from PBMCs. Although represented above as three and four samples per pool for illustrative purposes, actual pool size ranged from two to five samples per pool across the entire study (described in Materials and Methods). Proteins were resolved by size using GELFrEE, and the resulting fractions were analyzed by liquid chromatography-mass spectrometry in technical duplicate.
2.2 Sample processing
Upon procurement of patient peripheral blood, PBMCs were isolated from Cell Preparation Tubes (Becton Dickinson, Franklin Lakes, NJ). After collection of blood (8 mL; ~ 8×106 PBMC) the tubes were centrifuged for 20 minutes at 1500 to 1800 RCF, the pellets were washed twice with 15 mL Phosphate Buffered Saline (PBS), and the pellets were frozen. Frozen pellets were then thawed and TRIzol® Reagent (Thermo Fisher, Carlsbad, CA), which is a monophasic solution of phenol, guanidine isothiocyanate, and other proprietary components, was added. Proteins were extracted from TRIzol® Reagent by adding 1.5 mL of isopropanol to the organic phenol-chloroform phase, centrifugation at 12,000 RCF and washing the pellet three times with a wash solution consisting of 0.3 M guanidine hydrochloride in 95% ethanol. The pellets were stored in 0.3 M guanidine HCL/95% ethanol. Protein samples were washed in 100% ethanol and resuspended in 200 μL of 1% SDS. From this resuspension, 35 μL of each sample was taken for BCA protein concentration assay. After determining protein concentration per sample, which averaged around 100 μg each, samples (165 μL of protein/1% SDS) were pooled to achieve approximately 300 μg per pool; the number of samples per pool ranged from two to five. Protein in pooled samples was then precipitated by addition of acetone at a ratio of at least 3:1 acetone:sample and incubation at −80°C for at least 30 min. Precipitated protein was fractionated using gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) following the manufacturer’s protocol (GELFREE 8100 Fractionation System, Expedeon, San Diego, CA). Sample Set 1 was fractionated using a 12% GELFrEE cartridge; Sample Set 2 used a 10% GELFrEE cartridge. Following fractionation, SDS was removed by precipitating with methanol/chloroform/water as previously described [20] and protein was resuspended in buffer A (95% H2O, 5% acetonitrile, 0.2% formic acid) for LC-MS/MS analysis.
2.3 Liquid chromatography-mass spectrometry
Protein fractions were analyzed in technical duplicate using nano-flow reverse phase chromatography with PLRP-S as stationary phase and electrospray ionization as described [11]. Data were collected with an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) operated in data-dependent MSn mode with higher energy collisional dissociation (HCD) of the top two most abundant masses from the precursor scan using 15 m/z isolation windows. MS1 and MS2 data were collected in high resolution (60,000 – 120,000 resolving power at m/z 400) in the Orbitrap analyzer.
2.4 Data processing
Creation of targets
For top down data, .raw files were first converted to the mzML format using in-house conversion software. Next, each file was “crawled” to generate the linked sets of MS1 and MS2 data. Crawling was performed using in-house version of the ProSightPC crawler logic. This software created targets by grouping MS1 precursors across scans with a 0.1 Thomson tolerance within a 2.0 min retention time window. Next, summed MS1 and MS2 scans were deconvoluted using Thermo Scientific Xtract software with a signal to noise threshold of 3 or greater, a remainder threshold of 20, and a minimum fit threshold of 15. .raw files converted to mzML format supporting this manuscript will be accessible at http://www.topdownproteomics.org/data.
Protein Identification and Proteoform Characterization
The linked output from deconvoluted MS1 and MS2 scans were then searched using a three-tiered search tree in ProSightPC 3.0. The first search was an Absolute Mass search with MS1 tolerance = 2.2 Da, MS2 tolerance of 10 ppm, against a highly annotated version of Human UniProt (July 2014). The second search was a ProSight Biomarker search with MS1 tolerance of 10 ppm, MS2 tolerance of 10ppm, against a highly annotated version of Human UniProt (July 2014). Lastly there was a second Absolute Mass search with MS1 tolerance = 200 Da, MS2 tolerance of 10ppm, using Delta M mode, against a highly annotated version of Human UniProt (July 2014). This last search allows the detection of proteoforms with unknown modifications [21].
The p-scores used to measure the confidence in the identification of a given proteoform returned by each of the three searches were then subjected to a local FDR estimation to control for multiple testing. An estimate of the FDR was inferred based on modeling the individual proteoform’s null distribution of the negative LOG of the p-score. The first step in estimating the null distribution was to create a decoy database containing one scrambled proteoform for each proteoform in the forward database. Next all observed spectra were interrogated against the scrambled database and the best non-zero score for each observed target was collected and the resulting set of scores were fit to a gamma distribution. This distribution was used to determine the negative LOG p-score cutoff for accepting a forward hit. A global FDR was then determined using the methods developed by Higdon et al. for bottom up proteomics [22]. All proteoforms with a 10% global FDR were then passed to the quantitative analysis.
2.5 Statistical analysis of Quantitative Data
The list of differentially abundant proteoforms was generated using a custom SAS script (SAS Institute, Cary NC) that employed PROC MIXED to perform an ANOVA analysis of the normalized intensities between each of the treatment levels. For normalization, intensities were standardized across each proteoform over all the MS runs belonging to Sample Set 2. A hierarchical linear model was used that allowed variation in the technical replicates to be nested within their biological replicates, before testing for an overall fixed effect difference within the two “treatments” (i.e., cAR vs. TX in this study).
3. RESULTS AND DISCUSSION
Our study design was crafted to compare the proteomes of circulating peripheral blood mononuclear cells (PBMCs) from kidney transplant recipients that were diagnosed as either clinical acute rejection (cAR) or transplant excellent (TX). cAR is characterized by elevated creatinine levels, indicative of kidney dysfunction, and a tissue biopsy confirming acute rejection (Figure 2); TX means that there was no elevation in creatinine levels and protocol tissue biopsy showed no signs of acute rejection (Figure 2). Our study consisted of 40 total patient samples, of which 20 were TX and 20 were cAR. The samples were analyzed in two sets: Sample Set 1 and Sample Set 2. Each sample set contained 10 TX and 10 cAR. Sample Set 1 was analyzed in qualitative mode without randomization; Sample Set 2 was analyzed quantitatively using randomization and linear hierarchical statistical modeling [13]. At the time these data were collected the minimum amount of protein starting material was 300 μg. Because each patient PBMC sample yielded on average around 100 μg total protein, samples were pooled to obtain the required protein starting amount. Protein lysates were fractionated by protein size using GELFrEE and the proteoforms below 30 kDa were analyzed intact by liquid chromatography tandem mass spectrometry using an Orbitrap Elite (Figure 2).
Using a 1% FDR cutoff at the protein level we identified 283 proteins in Sample Set 1 (Table S1) and 264 proteins in Sample Set 2 (Table S2); combining the two protein datasets yielded 344 unique proteins identified. Using a 1% FDR cutoff at the proteoform level we identified 1,940 proteoforms in Sample Set 1 (Table S3) and 2,108 proteoforms in Sample Set 2 (Table S4); combining the two proteoform datasets yielded 2,905 unique proteoforms. As a measure of statistical confidence in proteoform assignment, we included a recently implemented C-score metric [23] with each entry (Tables S3 and S4). Proteoforms with a C-score of 40 and above are characterized with high confidence based on an intact mass matching within 10 ppm and fragment ion spectra distinguishing one proteoform from others of the same intact mass (e.g. two proteoforms resulting from phosphorylation at different sites). The number of proteoforms characterized with high confidence (i.e., C-score >40) was 552 and 639 in Sample Sets 1 and 2, respectively, and all these were assigned permanent PFR numbers for accessing in the publically-available repository (http://repository.topdownproteomics.org/). Those interested may download from the top down repository the list of high confidence proteoforms associated with this study. Collectively, these results provide new, regularized reporting metrics for proteoforms and the first inventory of them in human PBMCs.
Because collection of data for Sample Set 2 (10 cAR, 10 TX) was superior in key measures of technical variation and randomized sample run order (critical in label-free workflows [13]), we utilized hierarchical linear modeling to look for evidence of differential proteoform abundance between cAR and TX states. The data are presented as a volcano plot in Figure 3A where each proteoform is a data point, the x-axis is the relative abundance difference between states, and the y-axis is a measure of confidence that the difference is statistically significant. There were 30 proteoforms with statistically significant abundance differences below a 5% FDR for quantification, which corresponds to a -Log instantaneous q-value of 1.3 and above on the volcano plot (Figure 3A). Of these 30 differentially abundant proteoforms, 27 were identified below a 1% FDR for identification and are thus highly confident identifications (Table 1); the remaining three were identified below a 10% FDR for identification. Of the 27 confidently identified proteoforms with statistically significant differential abundance, 26 were proteoforms of histone H2A elevated in TX and one was a proteoform of 60S ribosomal protein L36 elevated in cAR (Table 1).
Figure 3. Relative proteoform abundances between clinical acute rejection (cAR) and transplant excellent (TX) patients.
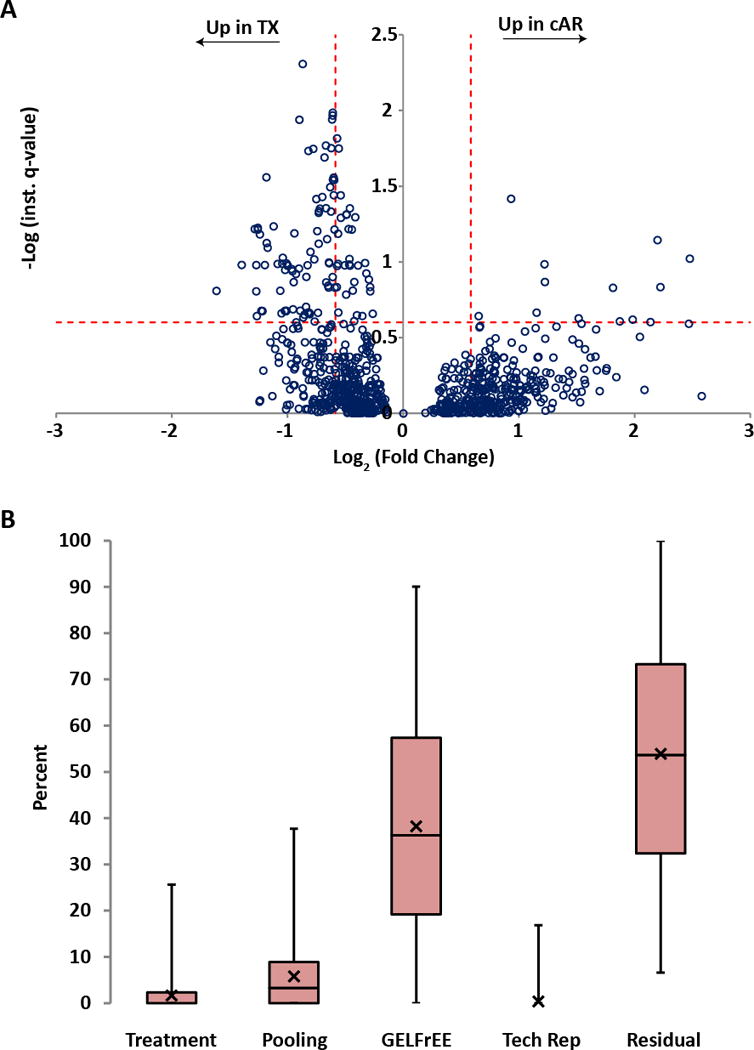
(A) To each proteoform identified in the dataset (open circle), a linear hierarchical model was applied to assign sources of variation in signal intensity from across the experiment. The y-axis (instantaneous q-value) is a measure of statistical confidence that the variation in signal intensity of a given proteoform is due to the treatment effect (TX vs. cAR) rather than due to patient variability, GELFrEE fractionation, technical variability, or noise. The x-axis is the fold change in proteoform abundance between patient groups. The horizontal dotted line represents a quantitative false discovery rate of 25%, that is, at this instantaneous q-value, signal intensity differences are 75% likely to be due to the treatment effect. The vertical dotted lines represent effect sizes of 1.5-fold above and below a no-change value. (B) For each proteoform quantified, variation in signal intensity was assigned, as a percent of the total for that proteoform, to either Treatment (TX vs. cAR), Pooling (biological replicates, which were pooled patient samples), GELFrEE (fractionation), Tech Rep (replicate LC-MS injections), or Residual (variation not explained by any of the aforementioned sources). The data are displayed as box-and-whisker plots, where the first quartile extends from the bottom line to the bottom of the box; the second quartile extends from the bottom of the box to the median, which is represented as a horizontal line within the box; the third quartile extends from the median to the top of the box; the fourth quartile extends from the top of the box to the top of the line. The average for each category is represented as an X. To illustrate, if there were only one proteoform quantified, the chart would have one data point in each category with the percentages across all categories summing to one hundred.
Table 1.
Proteoforms* with evidence for differential abundance between TX and cAR
| PFR | Accession | Description | Monoisotopic Mass (Da) | Up in: | Log2 (fold change) | −Log (inst. q-value) | Quant. FDR# (%) |
|---|---|---|---|---|---|---|---|
| 18216 | Q99878 | Histone H2A type 1-J | 14,049.82 | TX | −0.87 | 2.31 | 0.5 |
| 7325 | Q93077 | Histone H2A type 1-C | 14,073.90 | TX | −0.61 | 1.99 | 1.0 |
| 17853 | Q93077 | Histone H2A type 1-C | 14,073.90 | TX | −0.61 | 1.97 | 1.1 |
| 17676 | P0C0S8 | Histone H2A type 1 | 14,073.88 | TX | −0.61 | 1.94 | 1.1 |
| 17694 | P20671 | Histone H2A type 1-D | 14,048.85 | TX | −0.90 | 1.94 | 1.2 |
| 12488 | Q96KK5 | Histone H2A type 1-H | 13,808.80 | TX | −0.57 | 1.82 | 1.5 |
| 16201 | P20671 | Histone H2A type 1-D | 14,090.86 | TX | −0.67 | 1.77 | 1.7 |
| 13576 | P0C0S8 | Histone H2A type 1 | 14,073.88 | TX | −0.56 | 1.75 | 1.8 |
| 16234 | Q99878 | Histone H2A type 1-J | 14,091.87 | TX | −0.78 | 1.75 | 1.8 |
| 16095 | Q16777 | Histone H2A type 2-C | 14,091.89 | TX | −0.82 | 1.73 | 1.8 |
| 16952 | P0C0S8 | Histone H2A type 1 | 14,087.90 | TX | −0.68 | 1.69 | 2.0 |
| 2459 | Q7L7L0 | Histone H2A type 3 | 14,089.89 | TX | −0.60 | 1.56 | 2.8 |
| 17779 | P04908 | Histone H2A type 1-B/E | 14,089.89 | TX | −0.61 | 1.54 | 2.9 |
| 16220 | Q93077 | Histone H2A type 1-C | 14,087.88 | TX | −0.60 | 1.54 | 2.9 |
| 16418 | P20671 | Histone H2A type 1-D | 14,089.92 | TX | −0.63 | 1.49 | 3.2 |
| 16221 | P20671 | Histone H2A type 1-D | 14,089.88 | TX | −0.60 | 1.44 | 3.6 |
| 16686 | Q7L7L0 | Histone H2A type 3 | 14,075.88 | TX | −0.54 | 1.44 | 3.6 |
| 17992 | Q8IUE6 | Histone H2A type 2-B | 13,939.82 | TX | −0.70 | 1.43 | 3.7 |
| 5935 | Q9Y3U8 | 60S ribosomal protein L36 | 12,114.87 | cAR | 0.93 | 1.42 | 3.8 |
| 17882 | Q93077 | Histone H2A type 1-C | 14,045.86 | TX | −0.75 | 1.42 | 3.8 |
| 17361 | Q8IUE6 | Histone H2A type 2-B | 13,939.86 | TX | −0.67 | 1.35 | 4.4 |
| 16714 | P20671 | Histone H2A type 1-D | 2,323.35 | TX | −0.46 | 1.35 | 4.4 |
| 18092 | Q8IUE6 | Histone H2A type 2-B | 14,066.81 | TX | −0.72 | 1.35 | 4.4 |
| 16232 | Q99878 | Histone H2A type 1-J | 13,852.83 | TX | −0.73 | 1.34 | 4.6 |
| 17873 | Q99878 | Histone H2A type 1-J | 13,904.80 | TX | −0.62 | 1.33 | 4.7 |
| 16677 | Q99878 | Histone H2A type 1-J | 13,810.82 | TX | −0.49 | 1.31 | 4.9 |
| 18021 | Q8IUE6 | Histone H2A type 2-B | 13,936.75 | TX | −0.49 | 1.31 | 4.9 |
| 16704 | Q8IUE6 | Histone H2A type 2-B | 13,897.81 | TX | −0.41 | 1.30 | 5.1 |
| 18274 | P58876 | Histone H2B type 1-D | 13,880.55 | TX | −1.26 | 1.23 | 5.9 |
| 16167 | P62807 | Histone H2B type 1-C/E/F/G/I | 13,880.55 | TX | −1.28 | 1.22 | 6.1 |
| 16209 | Q99878 | Histone H2A type 1-J | 13,894.84 | TX | −0.44 | 1.21 | 6.1 |
| 17828 | P58876 | Histone H2B type 1-D | 13,880.55 | TX | −1.26 | 1.21 | 6.1 |
| 17144 | Q16777 | Histone H2A type 2-C | 13,904.83 | TX | −0.59 | 1.21 | 6.1 |
| 18059 | Q96KK5 | Histone H2A type 1-H | 13,939.84 | TX | −0.74 | 1.20 | 6.3 |
| 18993 | Q5VSD8 | Putative uncharacterized protein LOC401522 | 8,238.26 | TX | −0.94 | 1.19 | 6.5 |
| 16378 | Q99878 | Histone H2A type 1-J | 13,932.79 | TX | −0.56 | 1.19 | 6.5 |
| 16155 | Q7L7L0 | Histone H2A type 3 | 14,051.94 | TX | −0.66 | 1.15 | 7.1 |
| 17998 | P51858-1 | Hepatoma-derived growth factor | 5,177.25 | cAR | 2.20 | 1.14 | 7.2 |
| 16649 | P62805 | Histone H4 | 11,496.37 | TX | −1.18 | 1.13 | 7.5 |
| 17775 | Q93077 | Histone H2A type 1-C | 14,077.95 | TX | −0.73 | 1.12 | 7.6 |
| 17838 | P62805 | Histone H4 | 11,496.37 | TX | −1.17 | 1.09 | 8.1 |
| 17657 | P62491-1 | Ras-related protein Rab-11A | 24,833.88 | TX | −0.80 | 1.07 | 8.6 |
| 16639 | Q16777 | Histone H2A type 2-C | 14,105.87 | TX | −1.04 | 1.03 | 9.4 |
| 18964 | P33241-1 | Lymphocyte-specific protein 1 | 6,464.89 | cAR | 2.48 | 1.02 | 9.5 |
| 18006 | Q8IUE6 | Histone H2A type 2-B | 13,883.83 | TX | −0.77 | 1.02 | 9.6 |
| 16329 | P04908 | Histone H2A type 1-B/E | 14,051.94 | TX | −0.64 | 1.00 | 10.1 |
| 13216 | P62805 | Histone H4 | 11,341.39 | TX | −0.33 | 0.99 | 10.2 |
| 17437 | Q99878 | Histone H2A type 1-J | 14,050.80 | TX | −0.64 | 0.99 | 10.2 |
| 17758 | Q6FI13 | Histone H2A type 2-A | 14,105.86 | TX | −1.01 | 0.99 | 10.2 |
| 1905 | Q93079 | Histone H2B type 1-H | 13,883.54 | TX | −1.04 | 0.99 | 10.3 |
| 1664 | Q99877 | Histone H2B type 1-N | 13,862.48 | TX | −1.09 | 0.99 | 10.3 |
| 11839 | P25815 | Protein S100-P | 10,393.22 | cAR | 1.22 | 0.98 | 10.4 |
| 16770 | Q8IUE6 | Histone H2A type 2-B | 13,977.78 | TX | −0.47 | 0.98 | 10.4 |
| 18044 | P62807 | Histone H2B type 1-C/E/F/G/I | 13,880.55 | TX | −1.40 | 0.98 | 10.5 |
| 16146 | P62805 | Histone H4 | 11,397.38 | TX | −1.20 | 0.98 | 10.5 |
| 17968 | Q99878 | Histone H2A type 1-J | 13,881.81 | TX | −1.27 | 0.98 | 10.5 |
| 17813 | Q93077 | Histone H2A type 1-C | 14,133.98 | TX | −0.83 | 0.98 | 10.5 |
| 16027 | Q93077 | Histone H2A type 1-C | 14,091.97 | TX | −1.00 | 0.98 | 10.5 |
| 11821 | Q96KK5 | Histone H2A type 1-H | 13,897.83 | TX | −0.39 | 0.98 | 10.6 |
| 18210 | Q8IUE6 | Histone H2A type 2-B | 13,977.82 | TX | −0.46 | 0.98 | 10.6 |
| 18089 | Q99878 | Histone H2A type 1-J | 14,105.85 | TX | −1.00 | 0.97 | 10.6 |
| 17262 | Q96KK5 | Histone H2A type 1-H | 13,902.78 | TX | −0.57 | 0.97 | 10.6 |
| 18232 | Q6FI13 | Histone H2A type 2-A | 14,129.88 | TX | −0.90 | 0.96 | 11.1 |
| 17204 | P04406-1 | Glyceraldehyde-3-phosphate dehydrogenase | 3,526.98 | TX | −0.96 | 0.95 | 11.1 |
| 18235 | Q9BTM1-1 | Histone H2A.J | 14,133.80 | TX | −1.02 | 0.95 | 11.3 |
| 2674 | Q93077 | Histone H2A type 1-C | 14,096.94 | TX | −0.96 | 0.94 | 11.5 |
| 16544 | Q16777 | Histone H2A type 2-C | 13,994.84 | TX | −0.32 | 0.92 | 11.9 |
| 17707 | Q7L7L0 | Histone H2A type 3 | 14,131.90 | TX | −0.93 | 0.92 | 12.0 |
| 17713 | Q7L7L0 | Histone H2A type 3 | 14,093.95 | TX | −0.84 | 0.90 | 12.5 |
| 17128 | P09382 | Galectin-1 | 14,617.17 | TX | −0.61 | 0.90 | 12.6 |
| 16065 | P62805 | Histone H4 | 11,341.39 | TX | −0.30 | 0.88 | 13.0 |
| 17509 | Q8IUE6 | Histone H2A type 2-B | 13,897.85 | TX | −0.42 | 0.87 | 13.6 |
| 17705 | Q93077 | Histone H2A type 1-C | 14,105.95 | TX | −0.95 | 0.87 | 13.6 |
| 17695 | P20671 | Histone H2A type 1-D | 14,117.91 | TX | −0.67 | 0.87 | 13.6 |
| 17215 | Q99878 | Histone H2A type 1-J | 13,908.85 | TX | −0.65 | 0.84 | 14.3 |
| 18258 | P62805 | Histone H4 | 11,341.39 | TX | −0.29 | 0.83 | 14.6 |
| 18998 | Q9UHH9-2 | Inositol hexakisphosphate kinase 2 | 11,041.62 | cAR | 2.22 | 0.83 | 14.7 |
| 18577 | P20671 | Histone H2A type 1-D | 14,047.87 | TX | −0.57 | 0.83 | 14.7 |
| 17195 | P0C0S8 | Histone H2A type 1 | 14,021.95 | TX | −0.59 | 0.83 | 14.8 |
| 18202 | Q9Y2Q5-1 | Ragulator complex protein LAMTOR2 | 13,540.86 | TX | −0.64 | 0.83 | 14.8 |
| 18667 | P02671-1 | Fibrinogen alpha chain | 9,329.29 | cAR | 1.81 | 0.83 | 14.8 |
| 16560 | Q99877 | Histone H2B type 1-N | 13,866.53 | TX | −1.06 | 0.81 | 15.5 |
| 6983 | Q9Y6X1 | Stress-associated endoplasmic reticulum protein 1 | 7,237.89 | TX | −1.27 | 0.81 | 15.6 |
| 12325 | P62805 | Histone H4 | 11,299.38 | TX | −0.28 | 0.81 | 15.6 |
| 18038 | Q8IUE6 | Histone H2A type 2-B | 13,977.78 | TX | −0.49 | 0.78 | 16.4 |
| 15948 | P58876 | Histone H2B type 1-D | 13,838.54 | TX | −0.84 | 0.70 | 19.8 |
| 17892 | P62807 | Histone H2B type 1-C/E/F/G/I | 13,876.49 | TX | −0.95 | 0.69 | 20.6 |
| 17732 | Q6FI13 | Histone H2A type 2-A | 14,105.86 | TX | −0.89 | 0.68 | 21.0 |
| 18103 | Q7L7L0 | Histone H2A type 3 | 14,107.96 | TX | −1.01 | 0.68 | 21.0 |
| 15867 | P58876 | Histone H2B type 1-D | 13,880.55 | TX | −1.23 | 0.68 | 21.0 |
| 16673 | Q99878 | Histone H2A type 1-J | 13,997.88 | TX | −1.02 | 0.68 | 21.1 |
| 16907 | P04908 | Histone H2A type 1-B/E | 14,107.96 | TX | −1.01 | 0.68 | 21.1 |
| 17868 | P58876 | Histone H2B type 1-D | 13,880.55 | TX | −1.22 | 0.67 | 21.1 |
| 14140 | P62807 | Histone H2B type 1-C/E/F/G/I | 13,838.54 | TX | −0.82 | 0.67 | 21.5 |
| 17831 | Q6FI13 | Histone H2A type 2-A | 13,970.85 | TX | −0.56 | 0.67 | 21.6 |
| 18231 | Q71DI3 | Histone H3.2 | 15,424.47 | cAR | 1.15 | 0.67 | 21.6 |
| 1101 | P56381 | ATP synthase subunit epsilon; mitochondrial | 5,645.07 | TX | −0.74 | 0.66 | 21.7 |
| 16756 | O75964 | ATP synthase subunit g; mitochondrial | 11,332.21 | TX | −0.81 | 0.66 | 21.7 |
| 16324 | Q93079 | Histone H2B type 1-H | 13,878.53 | TX | −1.05 | 0.66 | 21.8 |
| 12647 | P62805 | Histone H4 | 11,341.39 | TX | −0.26 | 0.66 | 22.0 |
| 18174 | P20671 | Histone H2A type 1-D | 14,131.93 | TX | −0.81 | 0.66 | 22.1 |
| 18322 | Q8IUE6 | Histone H2A type 2-B | 14,150.83 | TX | −0.85 | 0.65 | 22.3 |
| 16529 | Q96KK5 | Histone H2A type 1-H | 13,888.80 | TX | −0.35 | 0.64 | 22.7 |
| 17798 | Q93079 | Histone H2B type 1-H | 13,878.53 | TX | −1.27 | 0.64 | 22.8 |
| 17280 | Q93077 | Histone H2A type 1-C | 14,110.95 | TX | −0.86 | 0.63 | 23.3 |
| 18750 | P20930 | Filaggrin | 2,603.44 | cAR | 1.52 | 0.63 | 23.6 |
| 17901 | P05386-1 | 60S acidic ribosomal protein P1 | 3,672.78 | cAR | 1.98 | 0.62 | 24.0 |
| 7670 | P04908 | Histone H2A type 1-B/E | 14,023.94 | TX | −0.42 | 0.61 | 24.6 |
| 18960 | Q00839-1 | Heterogeneous nuclear ribonucleoprotein U | 5,857.62 | cAR | 1.87 | 0.61 | 24.7 |
| 16947 | P06703 | Protein S100-A6 | 10,187.38 | TX | −0.76 | 0.61 | 24.7 |
| 18705 | P02671-1 | Fibrinogen alpha chain | 6,079.76 | cAR | 2.14 | 0.60 | 24.9 |
Proteoforms passed a 1% FDR cutoff for identification.
See Table S4 for proteoform characterization information (e.g. polypeptide sequence, modifications, C-scores).
Quantitative FDR refers to the false positive rate for calling a proteoform differentially abundant between TX and cAR
An advantage of hierarchical modeling is the ability to assign variation in proteoform signal intensity to its proper source[13]. Figure 3B shows the relative contribution of the five sources of variation in this study. Next to the “Residual” category, which is variation unassignable to any of the other sources, the largest source of variation was from proteome fractionation by GELFrEE prior to LC-MS/MS (Figure 3B). Because since the time of collecting these data we have moved to analyzing the 0 – 30 kDa proteome in a single fraction, future studies could be void of this source of variation. To increase the number of proteoforms making the list for future validation studies, and in light of the likelihood that proteome fractionation is weakening the statistical power of the analysis by introducing technical variation (Figure 3B), we lowered the stringency of the FDR cutoff for quantification from 5% to 25% (Figure 3A, horizontal line). Using this more relaxed cutoff, there were 111 proteoforms with evidence for differential abundance identified below a 1% FDR for identification: 11 were elevated in cAR, 100 were elevated in TX, 91 of which were histone proteoforms (Table 1). The complete list of proteoforms statistically analyzed for differential abundance can be found in supplemental material, which includes those additional proteoforms with FDRs for identification between 1% and 10% (Table S5).
Five proteoforms with evidence for differential abundance from hierarchical modeling were manually examined: unmodified protein S100-P (PFR: 11839), unmodified isoform 2 of inositol hexakisphosphate kinase 2 (PFR: 18998), a 6.5 kDa proteolytic fragment of lymphocyte-specific protein 1 (PFR: 18964), unmodified stress-associated ER protein 1 (PFR: 6983), and N-terminally acetylated isoform 1 of Ragulator complex protein LAMTOR2 (PFR: 18202) (Table 1). Total MS1 intensity values for each were summed across the entire Sample Set 2 experiment, including across sample pools within patient groups (cAR or TX), across GELFrEE fractions, and across technical replicates; the data are presented as box-and-whisker plots in Figure 4. Although the evidence for differential abundance for these proteoforms did not pass the 5% FDR cutoff for quantification, but rather passed the more relaxed 25% cutoff, the summed intensities across the experiment are consistent with what modeling suggests (Figure 4). MS1 intensities for the proteoforms of protein S100-P, inositol hexakisphosphate kinase 2, and lymphocyte-specific protein 1 were elevated in cAR patients compared to TX while stress-associated ER protein 1 and Ragulator complex protein LAMTOR2 proteoforms were elevated in TX patients compared to cAR (Figure 4). Manual examination of MS1 and MS2 spectra verified accurate proteoform assignment by confirming deconvolutable MS1 spectra that align within tolerance with theoretical isotopic distributions and confirm MS2 fragment ions for proteoform identification (Figure 5).
Figure 4. Relative abundance of five proteoforms of interest.
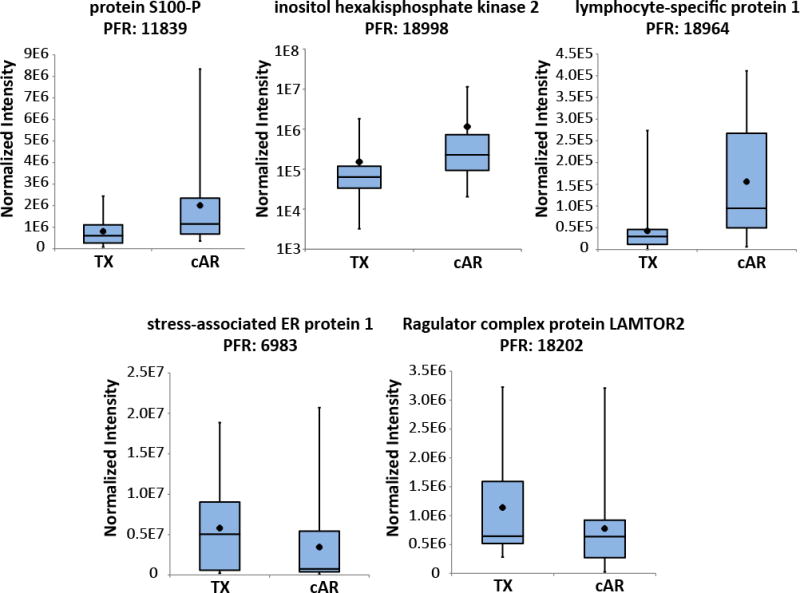
For each proteoform, intensity values normalized to the total ion current of each LC-MS injection were combined across GELFrEE fractions and biological and technical replicates within a patient group (TX or cAR). The data are represented as box-and-whisker plots, which are described in the legend to Figure 3B. The range of each dataset and apparent variability is due to the fact that intensity values from every GELFrEE fraction collected for each patient pool were considered for each proteoform. See Table 1 for quantitative quality metrics associated with these proteoforms.
Figure 5. Tandem MS data for the five proteoforms of interest.
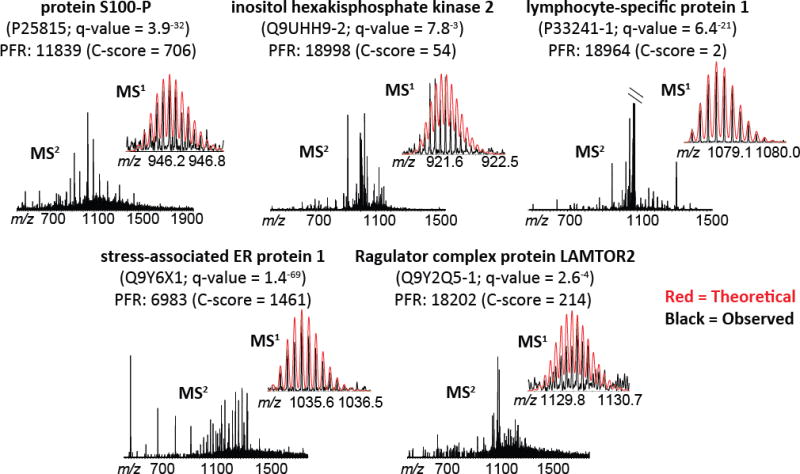
Raw spectra are shown, with theoretical MS1 overlaid for comparison (theoretical = red; observed = black). Protein descriptions and the proteoform record (PFR) number are included for each set of spectra, along with the UniProt Accession number, q-value for identification, and C-score for characterization. In the case of lymphocyte-specific protein 1 (PFRI: 18964; upper right), the MS2 spectrum was cropped to 6% of the base peak to show fragment ions; the base peak is unfragmented precursor. Refer to Table S4 for the quality metrics for identification and characterization associated with these spectra. mMass open source software [26–28] was used for modeling theoretical isotopic distributions and comparing to observed data.
Because PBMCs are the effector cells of the rejection process, a snapshot of their proteoforms in a clinical context is a significant step toward a clearer picture of allograft pathophysiology. Our data capture the information contained within the intact protein molecule—including splice variants and post-translational modifications—and, therefore, provide high molecular specificity and resolution for protein studies. Any subsequent studies targeting proteins for analysis to better understand PBMC-mediated transplant outcomes will benefit from the proteoform profiles of PBMCs generated in this work.
Our results are also semi-quantitative; although we used a limited set of patient samples and pooled them, hierarchical linear modeling coupled to quantitative top down methodology produced a list of 111 proteoforms with evidence for differential abundance below a 25% quantitative FDR (Table 1). Of these 111, the five we examined manually showed abundance changes consistent with the output of statistical modeling (Figure 4). A previous study using similar samples found upregulation at the RNA level of the Stress-associated ER protein 1 gene in TX and upregulation of a ribosomal protein gene, 60S ribosomal protein L35a, in cAR [5]. Our data at the proteoform level suggest elevation of a proteoform of Stress-associated ER protein 1 in TX and two proteoforms of ribosomal genes in cAR (Table 1). Although a linear relationship between RNA and proteoform cannot be assumed—due to the many levels of regulation operative on proteins following translation, all of which will influence proteoform level—the fact that the direction of change in these three proteoforms is consistent with the direction of change at the RNA level may suggest an overall pathway effect. Though a pathway effect is possible, top down proteomics studies such as these are advantageous as they enable the possibility of proteoform-specific effects be explored[24, 25].
4. CONCLUDING REMARKS
Collectively, our data provide the first proteoform-resolved look at the proteome of PBMCs of kidney transplant recipients. In addition, employing our label-free quantitative top down proteomics workflow we forward a set of proteoforms with evidence for differential abundance between cAR and TX states that can serve as targets for follow-up analysis. Given that PBMCs are a diverse population of cell types, and assuming validation studies confirm the trends found here, a valuable next step will be to map which cell types are responsible for elevated proteoform levels. Such results could advance mechanistic understanding and may provide new options for non-invasive biomarkers of the rejection process.
Supplementary Material
Figure S1. Timing of sample collection post-transplant. Patients are represented as single data-points and the timing of sample collection (in months post-transplant) is displayed by patient group and sample set.
{kind=link}
Table S1 = Sample Set 1 proteins using 1% FDR cutoff.
Table S2 = Sample Set 2 proteins using 1% FDR cutoff.
Table S3 = Sample Set 1 proteoforms using 1% FDR cutoff.
Table S4 = Sample Set 2 proteoforms using 1% FDR cutoff.
Table S5 = Sample Set 2 total proteoforms statistically analyzed for differential abundance.
SIGNIFICANCE OF THE STUDY.
Transplantation is currently the treatment of choice for end-stage kidney disease. While transplant offers the potential for significant improvement in quality of life, acute rejection and ultimate failure of the transplanted kidney still occurs in spite of routine immunosuppressive therapy. It is hoped that better understanding of the molecular events underpinning acute rejection will enable new and improved therapeutic strategies to mitigate the acute rejection burden. Here, we employed top down proteomics to begin exploration of the intact proteins within circulating peripheral blood mononuclear cells of kidney transplant recipients with acute rejection and functionally healthy grafts. Our study provides the first proteoform-resolved dataset of circulating immune cells in the context of transplant, adding valuable molecular specificity for the field. Our data also suggest differentially abundant protein molecules between clinical states providing targets for follow-up analysis.
Acknowledgments
This work was supported by the National Institutes of Health P41 GM108569, R01 GM067193 to NLK, P01 AI112522 to NLK and MMA, and U19 A1063603 to DRS. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Institutes of Health, the National Resource for Translational and Developmental Proteomics, or Northwestern University. Computational resources at Indiana University were used for statistical analysis.
ABBREVIATIONS
- MS1
precursor scan
- MS2
fragmentation scan
- PBMC
peripheral blood mononuclear cell
- cAR
clinical acute rejection
- TX
transplant excellent
- PFR
proteoform record
- ppm
parts per million
- GELFrEE
gel-eluted liquid fraction entrapment electrophoresis
Footnotes
The authors declare the following competing financial interest(s): Qualitative protein analyses were performed in ProSightPC, a product commercialized by Thermo Fisher Scientific with assistance from some members of the research group.
References
- 1.Matas AJ, Gillingham KJ, Payne WD, Najarian JS. The impact of an acute rejection episode on long-term renal allograft survival (t1/2) Transplantation. 1994;57:857–859. doi: 10.1097/00007890-199403270-00015. [DOI] [PubMed] [Google Scholar]
- 2.Pallardo Mateu LM, S CA, Capdevila Plaza L, Franco Esteve A. Acute rejection and late renal transplant failure: Risk factors and prognosis. Nephrol Dial Transplant. 2004;19:iii38–42. doi: 10.1093/ndt/gfh1013. [DOI] [PubMed] [Google Scholar]
- 3.Flechner SM, Kurian SM, Head SR, Sharp SM, et al. Kidney transplant rejection and tissue injury by gene profiling of biopsies and peripheral blood lymphocytes. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2004;4:1475–1489. doi: 10.1111/j.1600-6143.2004.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurian SM, Heilman R, Mondala TS, Nakorchevsky A, et al. Biomarkers for early and late stage chronic allograft nephropathy by proteogenomic profiling of peripheral blood. PloS one. 2009;4:e6212. doi: 10.1371/journal.pone.0006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurian SM, Williams AN, Gelbart T, Campbell D, et al. Molecular classifiers for acute kidney transplant rejection in peripheral blood by whole genome gene expression profiling. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2014;14:1164–1172. doi: 10.1111/ajt.12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakorchevsky A, Hewel JA, Kurian SM, Mondala TS, et al. Molecular mechanisms of chronic kidney transplant rejection via large-scale proteogenomic analysis of tissue biopsies. Journal of the American Society of Nephrology : JASN. 2010;21:362–373. doi: 10.1681/ASN.2009060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarwal M, Chua MS, Kambham N, Hsieh SC, et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. The New England journal of medicine. 2003;349:125–138. doi: 10.1056/NEJMoa035588. [DOI] [PubMed] [Google Scholar]
- 8.Reeve J, Einecke G, Mengel M, Sis B, et al. Diagnosing rejection in renal transplants: a comparison of molecular- and histopathology-based approaches. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2009;9:1802–1810. doi: 10.1111/j.1600-6143.2009.02694.x. [DOI] [PubMed] [Google Scholar]
- 9.Smith LM, Kelleher NL. Consortium for Top Down, P., Proteoform: a single term describing protein complexity. Nature methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catherman AD, Skinner OS, Kelleher NL. Top Down proteomics: facts and perspectives. Biochemical and biophysical research communications. 2014;445:683–693. doi: 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catherman AD, Durbin KR, Ahlf DR, Early BP, et al. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Molecular & cellular proteomics : MCP. 2013;12:3465–3473. doi: 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran JC, Zamdborg L, Ahlf DR, Lee JE, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ntai I, Kim K, Fellers RT, Skinner OS, et al. Applying label-free quantitation to top down proteomics. Analytical chemistry. 2014;86:4961–4968. doi: 10.1021/ac500395k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu S, Brown JN, Tolic N, Meng D, et al. Quantitative analysis of human salivary gland-derived intact proteome using top-down mass spectrometry. Proteomics. 2014;14:1211–1222. doi: 10.1002/pmic.201300378. [DOI] [PubMed] [Google Scholar]
- 15.Kellie JF, Higgs RE, Ryder JW, Major A, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Scientific reports. 2014;4:5797. doi: 10.1038/srep05797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iavarone F, Melis M, Platania G, Cabras T, et al. Characterization of salivary proteins of schizophrenic and bipolar disorder patients by top-down proteomics. Journal of proteomics. 2014;103:15–22. doi: 10.1016/j.jprot.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Edwards RL, Griffiths P, Bunch J, Cooper HJ. Compound heterozygotes and beta-thalassemia: top-down mass spectrometry for detection of hemoglobinopathies. Proteomics. 2014;14:1232–1238. doi: 10.1002/pmic.201300316. [DOI] [PubMed] [Google Scholar]
- 18.Barnidge DR, Dasari S, Botz CM, Murray DH, et al. Using mass spectrometry to monitor monoclonal immunoglobulins in patients with a monoclonal gammopathy. Journal of proteome research. 2014;13:1419–1427. doi: 10.1021/pr400985k. [DOI] [PubMed] [Google Scholar]
- 19.Ntai I, LeDuc RD, Fellers RT, Erdmann-Gilmore P, et al. Integrated Bottom-up and Top-down Proteomics of Patient-derived Breast Tumor Xenografts. Molecular & cellular proteomics : MCP. 2015 doi: 10.1074/mcp.M114.047480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical biochemistry. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 21.Leduc RD, Kelleher NL. Using ProSight PTM and related tools for targeted protein identification and characterization with high mass accuracy tandem MS data. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis … [et al.] 2007;Chapter 13(Unit 13 16) doi: 10.1002/0471250953.bi1306s19. [DOI] [PubMed] [Google Scholar]
- 22.Higdon R, Haynes W, Kolker E. Meta-analysis for protein identification: a case study on yeast data. Omics : a journal of integrative biology. 2010;14:309–314. doi: 10.1089/omi.2010.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeDuc RD, Fellers RT, Early BP, Greer JB, et al. The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. Journal of proteome research. 2014;13:3231–3240. doi: 10.1021/pr401277r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holland C, Schmid M, Zimny-Arndt U, Rohloff J, et al. Quantitative phosphoproteomics reveals link between Helicobacter pylori infection and RNA splicing modulation in host cells. Proteomics. 2011;11:2798–2811. doi: 10.1002/pmic.201000793. [DOI] [PubMed] [Google Scholar]
- 25.Thiede B, Koehler CJ, Strozynski M, Treumann A, et al. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture(SILAC), two-dimensional gel electrophoresis(2DE) and nano-liquid chromatograpohy coupled to an LTQ-OrbitrapMass spectrometer. Molecular & cellular proteomics : MCP. 2013;12:529–538. doi: 10.1074/mcp.M112.019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niedermeyer TH, Strohalm M. mMass as a software tool for the annotation of cyclic peptide tandem mass spectra. PloS one. 2012;7:e44913. doi: 10.1371/journal.pone.0044913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strohalm M, Hassman M, Kosata B, Kodicek M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid communications in mass spectrometry : RCM. 2008;22:905–908. doi: 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- 28.Strohalm M, Kavan D, Novak P, Volny M, Havlicek V. mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Analytical chemistry. 2010;82:4648–4651. doi: 10.1021/ac100818g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Timing of sample collection post-transplant. Patients are represented as single data-points and the timing of sample collection (in months post-transplant) is displayed by patient group and sample set.
Table S1 = Sample Set 1 proteins using 1% FDR cutoff.
Table S2 = Sample Set 2 proteins using 1% FDR cutoff.
Table S3 = Sample Set 1 proteoforms using 1% FDR cutoff.
Table S4 = Sample Set 2 proteoforms using 1% FDR cutoff.
Table S5 = Sample Set 2 total proteoforms statistically analyzed for differential abundance.