

Summary
Anti-Müllerian hormone (AMH) and its type II receptor AMHR2, both previously thought to primarily function in gonadal tissue, were unexpectedly identified as potent regulators of TGF-β/BMP signaling and epithelial-mesenchymal transition (EMT) in lung cancer. AMH is a TGF-β/BMP superfamily member, and AMHR2 heterodimerizes with type I receptors (ALK2, ALK3) also used by the type II receptor for BMP (BMPR2). AMH signaling regulates expression of BMPR2, ALK2 and ALK3, supports AKT-NFκB and SMAD survival signaling, and influences BMP-dependent signaling in NSCLC. AMH and AMHR2 are selectively expressed in epithelial versus mesenchymal cells, and loss of AMH/AMHR2 induces EMT. Independent induction of EMT reduces expression of AMH and AMHR2. Importantly, EMT associated with depletion of AMH or AMHR2 results in chemoresistance, but sensitizes cells to the HSP90 inhibitor ganetespib. Recognition of this AMH/AMHR2 axis helps to further elucidate TGF-β/BMP resistance-associated signaling and suggests new strategies for therapeutic targeting of EMT.
eTOC blurb
Beck et al. identify active signaling by the TGF-β/BMP superfamily member anti-Müllerian hormone (AMH) and its receptor AMHR2 in non-small cell lung cancer (NSCLC), demonstrating a role for AMH/AMHR2 in influencing the basal and BMP-dependent SMAD signaling that constrains epithelial-mesenchymal transition (EMT), and regulating drug resistance.
Introduction
Lung cancer is the leading cause of cancer related mortality (Stewart et al., 2014). In about 70% of lung cancer patients, the malignancy presents with locally advanced or metastatic elements, requiring systemic therapies (Molina et al., 2008). Treatment of lung and other cancers is increasingly based on consideration of underlying molecular mechanisms identified through genomic and transcriptomic profiling. Although this approach has dramatically improved outcomes for some patients, intrinsic and acquired drug resistance remain major challenges, associated with intratumoral clonal heterogeneity, elevated expression and activity of proteins that contribute to survival, and drug-resistant populations of cancer stem cells (Pattabiraman and Weinberg, 2014). Further, some drug resistance is conferred by proteins that are either expressed at very low levels, or which are upregulated post-transcriptionally, making it difficult to discern relation to resistance except through functional testing. In part because of this difficulty in identifying responsive patient populations, drugs broadly targeting the processes driving therapeutic resistance have attracted considerable interest for clinical evaluation (Proia and Bates, 2014).
In non-small cell lung cancer (NSCLC), the molecular chaperone heat shock protein 90 (HSP90) helps counteract the high rates of protein misfolding and aggregation that characterize rapidly and abnormally proliferating cells (Kamal et al., 2003). HSP90 binding supports the activity of numerous client proteins (including EGFR, ERBB2/HER2, c-MET, RAF, EML4-ALK, and SRC family kinases) that are critical constituents of oncogenic and drug resistance pathways (Echeverria et al., 2011; Taipale et al., 2012). Elevated expression of HSP90 in NSCLC is linked to poor prognosis and drug resistance (Biaoxue et al., 2012; Nagaraju et al., 2014). Several studies suggested that inhibition of HSP90 might have therapeutic efficacy in some subtypes of lung and other cancers (Proia and Bates, 2014; Socinski et al., 2013). For example, the HSP90 inhibitor ganetespib had potent activity in NSCLC characterized by the driver oncogene EML4-ALK (Sang et al., 2013). In contrast, tumors with KRAS mutations, detected in 20–30% of NSCLC (Cancer Genome Atlas Research, 2014; Imielinski et al., 2012) and associated with poor prognosis in NSCLC and other tumor types, are currently not clinically actionable using ganetespib or other targeted approaches.
We were interested in exploring the biological machinery involved in tumor resistance to HSP90 inhibition versus standard of care agents. In this study, we used an RNA interference (RNAi)-based approach to compare the functional requirements for the resistance of RAS-mutated (RASMUT) and EML4-ALK expressing NSCLC cell lines to ganetespib. Based on this work, we report here the identification and characterization of a previously undefined autocrine signaling axis in a subset of NSCLC tumors, involving anti-Müllerian hormone (AMH; also known as Müllerian inhibiting substance, MIS), and its type II receptor, AMHR2, as important for response both to ganetespib and to the approved chemotherapeutic cisplatin. AMH is a little-studied member of the transforming growth factor (TGF-β)/bone morphogenetic protein (BMP) family of secreted extracellular growth regulators (Massague, 2012). TGF-β and BMP are master regulators of epithelial-mesenchymal transition (EMT), a process occurring during tumor progression, in which tumor cells undergo transformative changes to acquire mesenchymal features (Thiery et al., 2009; Ye and Weinberg, 2015). EMT has been directly linked to chemoresistance and stem cell identity for many solid tumors (Fischer et al., 2015; Zheng et al., 2015). TGF-β has a well-documented activity in promoting EMT during cancer progression, while BMP typically opposes these activities: the balance between TGF-β and BMP activity plays a critical role in regulation of tumor cell plasticity and treatment resistance (Massague, 2008; Ye and Weinberg, 2015). Nevertheless, in spite of extensive study, the full range of functional crosstalk and feedback loops connecting members of this family of ligands and their receptors is not completely understood. Our findings provide new insight into NSCLC biology and TGF-β/BMP signaling, and suggest potential approaches to therapeutically target EMT.
Results
RNAi screening identifies AMH and AMHR2 in NSCLC
We used RNAi to screen for genes conferring resistance to ganetespib. Based on the observation that synthetic lethality is most often identified among genes with linked functions that provide protective redundancy (Hartwell et al., 1999), we developed a targeted siRNA library for depletion of 655 proteins defined as relevant to NSCLC survival and EMT signaling and enriched for druggable kinases and HSP90 clients (Table S1). Transfection conditions were optimized (Figure S1A–E) and ganetespib response curves were established (Figure S1F) in NSCLC cell lines. RNAi screening was performed two times independently in A549 (KRAS mutated, c.34G>A (G12S), TP53 wild type), and sensitizing hits were identified (Figure 1A). All hits were validated by confirmation that at least two of four independent siRNAs reproduced the sensitization phenotype. As a separate confirmation, for a number of hits, we used quantitative RT-PCR to show that individual sensitizing siRNAs reduced the abundance of the target mRNA (Figure S1G, H; Table S2).
Figure 1. RNAi screening of NSCLC cell lines identifies AMH and AMHR2.

(A) Representation of top hits from RNAi screen of NSCLC cell lines for sensitization to ganetespib. Left columns (value), sensitization index (SI) values ((test siRNA)/(negative control siRNA)) for ganetespib treated cells, divided by the (test siRNA)/(negative control siRNA) for vehicle treated cells; sensitizing (S) SI (≤0.85), yellow, indicates sensitizing siRNAs; SI = 1, black, indicates no activity; antagonizing (A) SI (≥1.15), blue, indicates siRNAs conferring resistance. Right columns (cutoff), all hits (SI ≤0.85 and FDR ≤25%) are indicated by red squares. (*), green boxes indicate hits for A549 cells treated with cisplatin (SI ≤0.85 and FDR ≤25%). See also Figure S1 for complete listing of screening results. (B) Enriched pathways for genes depicted in (A) as determined using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) classification system. Genes in pathways demarcated with a black diamond are highlighted with colored squares in (A). See also Figure S2 for non-overlapping hits in each of the highlighted categories. (C) AMH and AMHR2 mRNA expression in lung adenocarcinoma (ADCA) and squamous cell carcinoma (SCC) specimens reported in TCGA, indicating % of cases with z-scores of 1–2 (gray) or >2 (black). See also Figure S2 for AMH and AMHR2 expression in all cancer types reported by the TCGA. (D) AMH and AMHR2 expression in patient NSCLC samples and adjacent normal tissue with a fold difference of >1.0. Data are presented as mean ± standard error of the mean (SEM); one-way analysis of variance (ANOVA) was used to assess significance; *, p<0.05; **, p<0.01. See also Tables S1, S3 and S5.
The final group of 152 validated siRNA pools (Table S3) was screened using two additional RASMUT cell lines, H1299 and H460, and two cell lines bearing the EML4-ALK translocation (H3122 and H2228; Figure 1A and Figure S1I). In addition, validated siRNAs were re-screened in the A549 cell line to determine which siRNAs sensitized cells to an unrelated cytotoxic agent, cisplatin, used at an IC20 dose (Figure S1F), and to establish if sensitization of screened genes was specific to ganetespib (Figure 1A and Figure S1I). This analysis identified 14 genes associated with ganetespib resistance in 4/5 cell lines, of which 11 were specific to ganetespib, and an additional 60 genes that were active in 3/5 of the cell lines, or at least 2/3 RASMUT cell lines, of which 41 were specific for ganetespib. This high value gene set included a number of genes clearly related to RAS signaling in NSCLC, including the receptor tyrosine kinases (RTKs) ERBB3, KDR, and FGFR1, as well as IRS1, NOTCH, BCAR1 and other proteins that influence survival signaling. We also identified a number of genes not previously linked to NSCLC or therapy resistance.
To gain further insight into relevant resistance mechanisms, PANTHER gene function pathway analysis (Mi et al., 2013) was performed for a top group of 73 validated sensitizing hits, defined as those active in at least 3 out of 5 NSCLC cell lines, or at least 2 out of 3 RAS-mutated NSCLC cell lines. This analysis identified hits concentrated in pathways known to be relevant to NSCLC and survival signaling, including the angiogenesis pathway (P = 9.54E-10) and the VEGF and EGF receptor signaling pathways (P = 5.83E-09 and 1.17E-07, respectively), as well as cell adhesion-related pathways involving integrins and cadherins (Figure 1A and 1B). Unexpectedly, the gonadotropin releasing hormone (GRH) receptor pathway was the single most enriched pathway (P = 2.56E-13; Figure 1B, Figure S2A, Table S4). This focused attention on two genes identified as among the top sensitizing hits in all three RASMUT cell lines and previously uncharacterized for a role in NSCLC: AMH, a member of the TGF-β/BMP superfamily, and its receptor AMHR2.
AMH has long been thought to be expressed and functional primarily in embryonic gonad development, to regulate regression of the Müllerian ducts, and in the female ovary, to control oocyte maturation. However, interrogation of The Cancer Genome Atlas (TCGA) datasets (Table S5) indicated that AMH and AMHR2 expression is elevated in 5–8% of lung cancers (Figure 1C), and in several other cancer types (Figure S2B), and that overexpression did not correlate to a specific genotype for RAS or EML4-ALK mutation (Table S5). Analysis of TCGA datasets (Table S5) indicated that the mRNAs for AMH and AMHR2 were elevated relative to matched normal lung tissue in a subset of lung tumors (Figure 1D). Furthermore, TCGA data also indicated that genomic changes for AMH and AMHR2 are limited in lung cancer (Figure S3A, S3B), suggesting an epigenomic basis for elevated gene expression.
Autocrine AMH survival signaling in NSCLC
Signaling by TGF-β and BMP regulates EMT, invasion, and survival in NSCLC (Hsu et al., 2011; Nolan et al., 2015). TGF-β and BMP binding to a heterodimeric receptor complex with type I and type II components activates downstream signaling, with direct effectors that include SMAD1, 5, and 8 as well as multiple additional proteins that support survival signaling, such as NFκB, β-catenin and others (Figure 2A). In contrast to TGF-β and BMP, few studies have addressed AMH signaling, with none addressing potential action in lung cancer. However, based on work in studies of gonadal development (di Clemente et al., 2003; Rey and Picard, 1998), the type II receptor for AMH, AMHR2, is known to heterodimerize with three type I receptors (variously ALK2, ALK3 or ALK6; (di Clemente et al., 2010; di Clemente et al., 2003)) also utilized by BMP2 and the BMP type II receptor (BMPR2) in NSCLC (Langenfeld et al., 2013). Similar close pathway interactions between TGF-β and BMP often result in context specific and at times unpredictable signaling outputs (Oshimori and Fuchs, 2012; Scheel et al., 2011). Thus, our screening results suggested the intriguing idea that in lung cancer, the much-studied TGF-β/BMP signaling system might have a hitherto unappreciated input in the form of AMH/AMHR2 signaling.
Figure 2. AMH and AMHR2 regulate survival signaling in NSCLC.

(A) Schematic of TGF-β, BMP and AMH signaling. (B) qRT-PCR for AMH, normalized to total levels of AMH in testis (positive control), for two NSCLC cell lines (A549, H1299), two ovarian cancer cell lines (O5 = OVCAR5, O8 = OVCAR8) and normal lung tissue. (C, D) Western blots indicating depletion of AMH (C) or AMHR2 (D) for two independent siRNAs in A549 cells, with quantification of results normalized to GAPDH loading controls. (E) FACS using AMHR2-specific antibodies and non-permeabilized (blue; membranous (M)) or permeabilized (green; total (T)) cells after depletion of AMHR2. Averaged median fluorescence intensity (MFI) values from multiple independent experiments for each condition normalized to membraneous AMHR2 (control (C) siRNA) are show. The percentage of membranous AMHR2 (gray) is the MFI for M divided by the MFI for T. (F) Representative Western blots and quantifications for total and phosphorylated SMAD1/5/8 following siRNA depletion of AMH and AMHR2 in A549 cells. (G) Representative Western blots for total and phosphorylated SMAD1/5/8 following doxycycline (Dox) induced overexpression of AMH in A549 cells. (H) Representative Western blots for total and phosphorylated SMAD1/5/8 following 1 hour stimulation of A549 and SMAT1 cells with recombinant AMH or BMP2 (positive signaling control). (I) Representative Western blots for indicated total and phosphorylated survival-signaling proteins, following siRNA depletion of AMH or AMHR2; quantification is shown in Figure S4. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM. See also Figure S4.
As AMH expression in NSCLC was unexpected, we first characterized its expression. The AMH transcript was of low abundance in NSCLC cell lines compared to testicular tissue (a positive control), albeit comparable in expression to that seen in epithelial ovarian carcinoma cell lines and higher than that in normal lung tissues (Figure 2B). We confirmed low abundance but detectable proteins corresponding to full-length AMH and AMHR2 were expressed in RASMUT A549 and H1299 cells, and were depleted by multiple independent siRNAs (Figure 2C, 2D, S4A, and S4B). Interestingly, a recent study of AMHR2 in COS7 cells indicated a substantial pool of the protein resided in intracellular compartments, rather than at the cell surface (Hirschhorn et al., 2015). FACS analysis of permeabilized versus non-permeabilized A549 and H1299 cells demonstrated a similar distribution, with ~3–4% of AMHR2 on the cell surface, and a substantially larger intracellular pool (Figure 2E).
We then evaluated the role of AMH and AMHR2 in influencing the basal activity of canonical TGF-β/BMP effectors. SiRNA depletion of either AMH or AMHR2 reduced basal phosphorylation of the proximal BMP effector SMAD1/5/8 (Figures 2F, S4C, and S4D). Reciprocally, exogenous tet-inducible overexpression of AMH increased the pool of active SMAD1/5/8 (Figure 2G and S4E). In addition, confirming activity of membrane-associated AMHR2, treatment with exogenous AMH induced SMAD1/5/8 phosphorylation, albeit at a low level than with overexpression (Figure 2H and S4F).
Depletion of AMH and AMHR2 also reduced the basal activity of multiple core survival signaling effectors of BMP and TGF-β (Feng and Derynck, 2005; Massague, 2012; Zhang, 2009), including AKT and its activator PDK1 (pThr308AKT and pSer241PDK1), as well as pSer176/180IKKαβ and pSer235/236S6 (typically activated by AKT), and the IKK downstream signaling proteins pSer32/36IκB and pSer536NFκB (Figures 2I and S4G). In contrast, activity of the proliferation-associated protein ERK1/2, and expression of the death effector proteins PUMA and BIM were not affected by depletion of AMH or AMHR2 (Figure S4H).
AMH and AMHR2 directly influence TGF-β/BMP-dependent signaling through SMAD3 and BMPR2
We next asked if modulation of AMH signaling influenced expression or activity of receptors specifically related to BMP or TGF-β. For this, we transiently depleted AMH or AMHR2 and stimulated cells with BMP2 or TGF-β for 0, 15 or 45 minutes. While depletion of these proteins did not affect BMP2 induction of phospho-SMAD1/5/8, BMP2 stimulation in the context of depleted AMH or AMHR2 caused a significant increase in phosphorylation of SMAD3 (Figure 3A, 3B, S5A, and S5B). In contrast, depletion of AMH or AMHR2 reduced TGFβ1-dependent phosphorylation of SMAD3 (Figure S5C and S5D). For neither BMP2 nor TGFβ1 did AMH/AMHR2 status impact transient induced phosphorylation of SMAD1/5/8 or SMAD2. One possible mechanism for these altered response patterns would be if depression of AMH signaling caused reactive changes in expression of the type I or type II receptors for BMP or TGF-β ligands. Indeed, we found that depletion of AMH specifically increased expression of the type II receptor BMPR2, and the type I receptors ALK2 and ALK3, while expression of TGFBR2 or ALK6 was not impacted (Figure 3C, S5E and S5F). In further support of this observation, tet-inducible overexpression of AMH significantly decreased detectable levels of BMPR2, but not TGFBR2 (Figure 3D).
Figure 3. AMH and AMHR2 impact BMP signaling.

(A) Representative Western blots for siRNA depleted and BMP2 stimulated A549 cells and (B) quantification of signals normalized to GAPDH loading controls. (C) Western blots for the indicated proteins following stable depletion of AMH using two independent shRNAs (1 and 2) and quantification thereof. (D) Western blots for BMPR2 and TGFBR2 following Dox induced overexpression of AMH in A549 and H1299 cells. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM. See also Figure S5.
Depletion of AMH or AMHR2 induces EMT and increasing sensitivity to HSP90 inhibition and resistance to cisplatin
As TGF-β and BMP dynamically interact to regulate epithelial versus mesenchymal cell identity (Oshimori and Fuchs, 2012; Scheel et al., 2011), the fact that AMH influenced signaling in a manner that enhanced BMP responsiveness suggested AMH/AMHR2 might also influence this balance. SiRNA depletion of AMH or AMHR2 reduced the epithelial phenotype in A549 and H1299 cells, as reflected by downregulation of cadherins (CDH1 and CDH3) and the tight junction protein ZO1/TJP1 (Figure 4A; (Beck et al., 2014; Thiery et al., 2009)). AMH or AMHR2 knockdown also elevated expression of the mesenchymal markers ACTA2/αSMA, ZEB1, vimentin (VIM) and β-catenin (CTNNB1) (Figure 4A and 4B; (Thiery et al., 2009; Ye and Weinberg, 2015)), and caused cells to assume a more mesenchymal phenotype, characterized by larger cells, spindle-shaped that formed fewer cell-cell contacts and lacked cadherin staining at cell junctions (Figure 4C and S5G). Stable AMH depletion (Figure 5A and S5H) also significantly increased invasion into Matrigel (Figure 5B, and S5I), compatible with a mesenchymal transition, and increased resistance to anoikis (Figure 5C; (Frisch et al., 2013)). Importantly, AMH depletion increased the proportion of cells expressing high levels of CD133 and CD44, associated with mesenchymal and stem cell-like characteristics (Figure 5D and E; (Leung et al., 2010; Pattabiraman and Weinberg, 2014)). Conversely, cells overexpressing AMH had decreased expression of VIM and increased expression of CDH1/3 (Figure 5F and S5J), were less invasive (Figure 5G and S5K), and were sensitized to anoikis (Figure 5H). However, treatment of cells for 24–36 hours with exogenous AMH or BMP used at concentrations that induced SMAD1/5/8 phosphorylation did not significantly reduce expression of VIM, although TGFβ1 treatment induced VIM, suggesting higher doses or a longer time of exposure were required to reduce EMT (Figure S5L).
Figure 4. AMH and AMHR2 promote epithelial identity in NSCLC cells.

(A, B) Representative Western blots (A) and quantification (B) normalized to GAPDH loading controls indicating expression of proteins associated with an epithelial (ZO1 and CDH1) or a mesenchymal (ZEB1, CTNNB1, VIM and ACTA2) state, 48 hours after siRNA depletion of AMH and AMHR2. (C) Immunofluorescence visualization of expression of E-cadherin (CDH1), vimentin (VIM), or DNA (DAPI) 48 hours after siRNA depletion of AMH or AMHR2, or GL2 control (siCtrl). Scale bar, 30 μm. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM. See also Figure S5.
Figure 5. EMT reduces AMH/AMHR2 expression, sensitizes cells to HSP90 inhibition and contributes to chemoresistance.

(A) Western analysis of A549-derivative cell lines constructed with two independent AMH-depleting shRNAs or vector control (Ctrl). (B) Representative images for invasion assays using A549 cells following shRNA depletion of AMH, with quantification of relative invasion through Matrigel following depletion of AMH or negative control. Scale bar, 100 μm. (C) Relative caspase 3/7 activity in cells plated on ultra-low attachment plates to induce anoikis; values normalized to control shRNA (C). (D, E) Representative FACS spectra for CD44-specific (D) and CD133-specific (E) antibodies using shCtrl and shAMH1 A549-derivatives; quantification of multiple independent experiments for A549 and H1299 cells is also shown. (F) Western blots for CDH1 and VIM for two clones with or without Dox induced AMH overexpression. (G) Representative images for invasion assays using A549 cells with or without Dox induced overexpression of AMH, with quantification of relative invasion through Matrigel for A549 and H1299 cells. Scale bar, 100 μm. (H) Relative caspase 3/7 activity for cells with or without Dox induced overexpression of AMH; cells were plated on ultra-low attachment plates. (I) Representative Western blots and quantifications normalized to GAPDH loading controls for A549 cells 24–48 hours after siRNA depletion of E-cadherin (CDH1) with two independent siRNAs (1 and 2). *, p<0.05; **, p<0.01; ***, p<0.001. (J) A549 and H1299 cells transfected with siCDH1 or siCDH3 and treated with vehicle (V) or two different concentrations of ganetespib for 72 hours. # indicates synergy (SI <0.85; p<0.001). (K) Same as (J) except cells were treated with cisplatin instead of ganetespib. * indicates antagonism (SI >1.15; p<0.001). (L) Same as (K) except cells were transfected with siAMH or siAMHR2. * Indicates antagonism (SI >1.15; p<0.001). (M) Normalized viability of cells following 72 hours of treated with increasing concentrations of cisplatin, with (red; +Dox) or without (black; -Dox) Dox induced AMH overexpression. Data are presented as mean ± SEM; Student’s t-test was used to assess significance. See also Figure S6.
We further tested the hypothesis that AMH signaling is linked to the epithelial state by inducing EMT through direct depletion of cadherin (CDH1 or CHD3) and assessing AMH and AMHR2 expression. Expression of AMH and AMHR2 decreased with transition to a more mesenchymal phenotype (Figure 5I and S6A–E), supporting this idea. The PANTHER analysis of ganetespib hits from the initial siRNA screen had indicated enrichment for cell adhesion-related pathways (Figure 1A, B, and S2A), with siCDH3 sensitizing multiple NSCLC cell lines to ganetespib. This result raised the possibility that the mesenchymal state per se might contribute to sensitization to ganetespib, in spite of the normal association of EMT with drug resistance (Shintani et al., 2011; Thiery et al., 2009). Indeed, depletion of CDH1 or CDH3 significantly sensitized A549 and H1299 cells to ganetespib (Figure 5J). Conversely, AMH-, AMHR2-, CDH1- or CDH3-depleted mesenchymal cells were resistant to cisplatin (Figures 5K and 5L), a drug commonly used in the neoadjuvant and adjuvant setting for NSCLC and previously reported to be less effective against mesenchymal NSCLC cells (Shintani et al., 2011). Interestingly, Tet-induced overexpression of AMH strongly sensitized cells to cisplatin (Figure 5M), but did not impact response to ganetespib (Figure S6F).
Inhibition of HSP90 opposes EMT and induces AMH and AMHR2 expression
Previous studies suggested HSP90 supports EMT in some cell types (Nagaraju et al., 2014; Nolan et al., 2015). We found that depletion of cadherins, AMH, or AMHR2 induced expression of HSP90 in parallel with inducing EMT (Figures S5C and S6G), compatible with this idea. To explore this relationship in NSCLC, we treated cells with ganetespib for up to 72 hours. Inhibition of HSP90 triggered mesenchymal-epithelial transition (MET) as shown by reduced expression of ZEB1, CNNB1, VIM and ACTA2 and by induction of ZO1 and CDH1 or CDH3 expression (Figure 6A). Furthermore, ganetespib significantly reduced invasion into Matrigel (Figure 6B and S6H), especially for cells with depleted AMH (Figure 6C and S6H). Compatible with this MET, inhibition of HSP90 induced AMH mRNA (Figure 6D) and protein within 24 hours of treatment (Figure 6E). HSP90 inhibition also significantly induced AMHR2 expression after 24–48 hours (Figure 6E). In contrast to changes in expression, HSP90 inhibition had a limited effect on AMHR2 localization, with the slight decrease in percent of the cellular pool at the surface due to the greater magnitude of the ganetespib-induced increase in cytoplasmic AMHR2 (Figures 6F and 6G). Among the type I receptors, ALK2 and ALK3 were abundant both intracellularly and at the plasma membrane, while ALK6, was strictly associated with the plasma membrane (Figure 6H, 6I, S6I and S6J); findings similar to previous reports in non-lung cancer cell lines (Lee et al., 2009; Song et al., 2010). Interestingly, expression of ALK3 significantly increased after treatment with ganetespib for 24–48 hours, potentially reflecting co-regulation with AMHR2 (Figure 6H, 6I, S6I and S6J). Together, these results indicated that inhibition of HSP90 induces a MET process characterized in part by elevated AMH, AMHR2 and ALK3 expression in NSCLC models.
Figure 6. HSP90 inhibition induces MET and increases AMH/AMHR2 expression.

(A) Quantification (normalized to β-actin loading controls) of Western analysis of indicated proteins for A549 and H1299 cells 0–72 hours after treatment with 100 nM of ganetespib. (B, C) Quantification of relative cell invasion through Matrigel for A549 cells and H1299 treated with vehicle or 100 nM of ganetespib, +/− depletion of AMH; see also Figure S6. (D) Relative mRNA expression (normalized to the POLR2F housekeeping gene) for AMH, HSP90, HSP70 and a TRAP1 unaffected control after 24 hours of treatment with vehicle (V) or 100 nM ganetespib (G). (E) Representative Western blots and quantification for AMH and AMHR2 expression in A549 and H1299 cells treated with 100 nM ganetespib for 0–72 hours. Increased HSP70 expression indicates response to HSP90 inhibition. (F, G) FACS of non-permeabilized (blue; membranous (M)) or permeabilized (green; total (T)) cells using AMHR2-specific antibodies after treatment with 150 nM ganetespib. Averaged median fluorescence intensity (MFI) values from multiple independent experiments for each condition normalized to membranous AMHR2 for control (Gan. 0) are shown. (H) Representative images of Western blots following treatment with 150 nM ganetespib; different cellular components are shown: total (T), cytosolic (C), and plasma membrane (M) fractions. (I) Quantifications for (H): protein expression for (M) was normalized to a CAV1 loading control, and for (C) to GAPDH and total (T) protein expression for vehicle treated cells; normalized results for M and C were used to calculate % plasma membrane localization (M/C × 100; gray). *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM; Student’s t-test was used to assess significance.
AMH provides a proliferative advantage in vivo
To assess the in vivo relevance of AMH signaling in NSCLC tumorigenesis and ganetespib response, A549 and H1299 cells with shRNA-depleted AMH or vector controls (Figure 5A and S5H) were used for xenograft analysis. Although in vitro siRNA depletion of AMH or AMHR2 had limited impact on cell proliferation (Figure S7A), AMH-depleted cells grew significantly slower than vector controls in vivo (Figure 7A), resulting in smaller tumors after 5 weeks of treatment (Figure 7B and S7B), with reduced Ki-67 staining (Figure 7C and S7C). Ganetespib treatment significantly reduced tumor volume, particularly in AMH-depleted tumors (Figure 7B, and S7B). Ganetespib treatment did not further reduce Ki-67 staining, likely reflecting the fact that specimens were analyzed one week after the last dose (Figure 7C and S7C). Tumors with depleted AMH expressed significantly less CDH1 than parental tumors, suggesting a more mesenchymal identity (Figure 7D and S7D), as predicted by in vitro data. Also paralleling in vitro results, ganetespib treatment partially reverted AMH-depleted tumors to a more epithelial-like state (Figure 7D and S7D). In contrast, AMH overexpression accelerated the growth of tumors in vivo, and caused ganetespib resistance (Figure 7E); moreover, AMH-overexpressing tumors had elevated levels of Ki-67 and CDH1, indicating a more proliferative and epithelial phenotype (Figure 7F, 7G, S7E and S7F).
Figure 7. AMH depletion reduces tumor growth and sensitizes tumors to HSP90 inhibition.

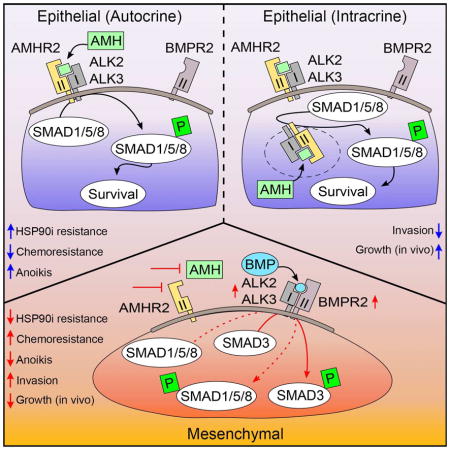
(A) Days required for tumors to reach 70–100 mm3 after inoculation with 1 × 107 shAMH- or control-depleted (shCtrl) A549 or H1299 cells. (B) Relative tumor volume for A549 or H1299 xenografts with or without stable AMH depletion, receiving weekly dosing with ganetespib (100 mg/kg) or vehicle. (C, D) Quantified Ki-67 and CDH1 staining for all tumors. See Figure S7 for representative images. (E) Relative tumor volume for A549 or H1299 xenografts with or without Dox activated overexpression of AMH, receiving weekly dosing with ganetespib (100 mg/kg) or vehicle. (F, G) Quantified Ki-67 and CDH1 staining for all tumors. See Figure S7 for representative images. (H) Disease free survival of patients with lung adenocarcinoma expressing (TCGA RNA-seq. data) either high or low levels of AMH and/or AMHR2 (high AMH/AMHR2 (red) = z-score of >0.9). Left pie chart: samples with high (z-score of >0.9; red) or low (blue) expression of AMH, AMHR2 or both. Right pie charts: data for metastatic status (top chart) and post-surgery residual tumor (bottom chart); the percentages of cases with high (red) or low (blue) AMH/AMHR2 expression for each of the two clinical determinants are shown. M0 = no distant metastasis detected; M1 = metastasis to distant organs detected; R0 = negative margins (microscopically); R1/2 = positive margins (microscopically/gross examination). (I) Proposed model. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM; Student’s t-test was used to assess significance.
Intriguingly, in further analysis of TCGA data, high expression of AMH and/or AMHR2 in 517 NSCLC samples predicted significantly increased disease free survival (P = 1.2E–02; Figure 7H). Because of the many links between AMH and epithelial identity, we asked if AMH and/or AMHR2 expression correlated with postoperative residual tumor or distal metastasis in the 517 NSCLC cases. TCGA included data on metastatic status and residual tumor detection for 344 and 340 of the 517 cases, respectively (Figure 7H and Table S6). 23 of 344 patients (6.7%) had a validated distal metastasis (M1) and 16 out of 340 (4.7%) had positive surgical margins (R1/2). We detected a trend compatible with the interpretation that high AMH/AMHR2 levels are associated with reduced metastatic disease and positive tumor margins, as only 9% of metastatic cases and 6% of cases with positive tumor margins had high levels of AMH/AMHR2 (Figure 7I and Table S6), in contrast to expected values of 14.5 %, based on the representation of high AMH/AMHR2 specimens in the analyzed set of tumors.
Discussion
In this study, we identify AMH signaling as an important contributor to epithelial plasticity, survival signaling and selective drug resistance in NSCLC. Signaling by TGF-β/BMP family proteins is strongly associated with, and is a determinant of prognosis for, lung malignancies (Borczuk et al., 2011; Kretzschmar et al., 1999). Importantly, this signaling is significantly influenced by context, with TGF-β signaling altering from tumor-suppressive to metastasis-promoting in response to the contributions of diverse cell intrinsic and cell extrinsic factors (Massague, 2012). By identifying a previously undetected input that is intertwined with central TGF-β/BMP effector cascades to support the epithelial state, this work offers new insights into mechanisms of NSCLC pathogenesis, which have direct possible implications for the therapeutic management of lung cancers.
Studies of AMH during gonadal development have documented production by Sertoli cells in males and granulosa cells in females (Josso and di Clemente, 2003; Takahashi et al., 1986; Tran et al., 1977). Catlin and colleagues first determined that fetal lung tissue was also responsive to recombinant AMH, suggesting an intact receptor system: fetal signaling proteins relevant to control of tissue differentiation are frequently re-expressed and utilized in cancers (Catlin et al., 1990; Catlin et al., 1997; Naxerova et al., 2008). AMH and AMHR2 have been investigated as potential therapeutic targets in epithelial ovarian cancer (EOC) and granulosa cell tumors (GCT; (Kim et al., 2014)), since reflecting their lineage of origin, some of these tumors continue to express AMHR2 (Anttonen et al., 2011; La Marca and Volpe, 2007). In EOC, exogenously administered AMH significantly inhibits proliferation and induces cell cycle arrest, mimicking the anti-proliferative activity of AMH in the reproductive cycle (Kim et al., 2014; Molina et al., 2008), and inhibits cell invasion (Chang et al., 2011). AMH-dependent inhibitory signaling proceeds through activation of SMAD1/5/8 (Alarcon et al., 2009; Meirelles et al., 2012), and depletion of SMAD1 or SMAD5 in the ovary or testes caused metastatic gonadal tumor development in mouse models (Pangas et al., 2008). Beyond this proximal effector, there has been relatively little study of AMH-dependent cancer signaling, and no report of a role of AMH in influencing TGF-β, BMP, or NF-κB in NSCLC.
The previously unsuspected signaling role we describe here for AMH in NSCLC differs significantly from that reported in EOC, with AMH sustaining epithelial identity and preventing invasion in part by regulating activity of ALK2, ALK3, BMPR2, and SMAD3. For canonical TGF-β signaling, SMAD2 and 3 are typically thought to be the dominant signal propagators (Massague, 2012). For BMP, SMAD 1, 5 and 8 usually take on this role (Figure 2A; (Shi and Massague, 2003)). We, in this study, and many others previously (Feng and Derynck, 2005; Massague, 2012; Oshimori and Fuchs, 2012; Pickup et al., 2013; Scheel et al., 2011), have shown that context strongly influences crosstalk between TGF-β and BMP signaling, and that although generally associated with TGF-β signaling (Massague, 2012), BMP induces SMAD3 phosphorylation in some settings (Holtzhausen et al., 2014). Mechanistically, our data indicate that depletion of AMH and AMHR2 contributes to the selective activation of SMAD3 by BMP ligands, likely at least in part due to selective upregulation of BMPR2, ALK2, and ALK3 (Figure 3). Interestingly, overexpression of ALK2, most promiscuously used by TGF-β, BMP, and AMH, has also been shown to induce non-canonical crosstalk between pathways (Renlund et al., 2007), suggesting AMH/AMHR2 expression may, through competition, similarly affect available pools of subunits for multiple competing heterodimeric signaling systems. The predominantly intracellular localization of AMHR2, paralleling a recent report in COS7 cells (Hirschhorn et al., 2015), suggests that some activity of the AMH/AMHR2 complex may be exerted at the level of expression and trafficking control for the ALK2,3 and BMPR2 receptors, and suggests a potential intracrine role (Re, 2014) for AMH/AMHR2 that was previously unknown. Future work is needed to fully elucidate the functional role of intracellular AMHR2. Nevertheless, these findings strongly suggest that evaluation of AMH and AMHR2 expression may be necessary in some cases to fully appreciate the signaling regulation, crosstalk and impact on EMT of TGF-β and BMP.
Intriguingly, the pro-survival and proliferative activity of AMH alone is not adequate to cause sensitization to non-specific cytotoxic agents; rather, the sensitizing effect unmasked for ganetespib is linked to the balance between epithelial and mesenchymal status. This is compatible with recent studies that have shown a specific role of EMT transitions in protecting cells growing under abnormally detached conditions from anoikis (Cieply et al., 2015). Particularly in light of recent findings suggesting EMT is more important for chemoresistance than metastasis in NSCLC and other cancer (Fischer et al., 2015; Zheng et al., 2015), it is conceivable that patient tumors with low AMH/AMHR2 define a class of particularly chemoresistant cases that may significantly benefit from combination therapy that includes an HSP90 inhibitor.
Overall, this study emphasizes the importance of context and tissue type for accurate determination of protein activity (Bissell and LaBarge, 2005; Massague, 2012). This may be particularly relevant considering the continued clinical development of therapeutics to target TGF-β superfamily members and the association of EMT with chemoresistance (Akhurst and Hata, 2012; Fischer et al., 2015; Thiery et al., 2009; Zheng et al., 2015). Targeting AMH signaling has been proposed as a potential treatment for EOC, either through administration of exogenous AMH (Kim et al., 2014), adeno-associated virus (AAV)-delivery of AMH (Pepin et al., 2015), or use of AMHR2-targeting antibodies (Kersual et al., 2014). The present study suggests that modulating AMH homeostasis may also have effect in non-gynecological tumors. The findings in this study are broadly informative for ongoing efforts to target the TGF-β/BMP/AMH superfamily, for HSP90 inhibitors, and in targeting tumors chemoresistant due to EMT.
Experimental Procedures
Please also refer to the Supplemental Experimental Procedures section.
High-throughput RNAi screening and validation
Methods were modified from previously published work (Astsaturov et al., 2010). PLK1 (positive control) and GL2 (negative control) siRNAs (Thermo Fisher Scientific; Waltham, MA, USA) were used to optimize transfection conditions and achieve Z′ values of 0.5 or greater (Zhang et al., 1999)), using a standard reverse transfection procedure for Dharmafect 1 (Thermo Fisher Scientific; Waltham, MA, USA); in the siRNA library used for screening, two independent siRNA duplexes (Qiagen, Valencia, CA) arrayed in 96-well plates targeted each gene of interest, with systematically placed PLK1 and GL2 controls.
Pathway enrichment, TCGA data and survival data
The PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System was used to identify enriched pathways, using the statistical overrepresentation test and PANTHER pathway categories (Table S4; http://www.pantherdb.org/, (Mi et al., 2013)). The TCGA results shown in this study are based on data generated by the TCGA Research Network (http://cancergenome.nih.gov/). TCGA data sets were downloaded from cBioPortal and are listed in Table S5 or published (Cancer Genome Atlas Research, 2014).
Statistical analysis
In analysis of RNAi screening data, viability for each target gene was normalized to the average control siRNA (GL2) viability per plate. Sensitization index (SI) was calculated for each siRNA as SI = (Vdrug/GL2drug)/(Vvehicle/GL2vehicle), where V was the viability in wells transfected with targeting duplexes and GL2 was the average viability of cells transfected with negative control siRNA on the same plate. Hits were identified based on statistically significant P values and SI values below 0.85. P values controlled for the false discovery rate (FDR) with Benjamini-Hochberg step-up method. All hits with FDR values of ≤25% were considered significant. Only hits statistically (FDR ≤25%) and biologically significant (SI ≤0.85) were further validated. P-values were calculated using one-way ANOVA (GraphPad Prism version 6.00 for Mac; GraphPad Software; La Jolla, CA, USA) or Student’s t-test as indicated for each specific experiment.
Supplementary Material
Highlights.
TGF-β superfamily member AMH regulates tumor growth and drug resistance in NSCLC
AMH and AMHR2 activity influences SMAD, AKT, and NFκB signaling in NSCLC cells
Loss of AMH/AMHR2 promotes EMT through direct modulation of TGF-β/BMP receptors
EMT promotes chemoresistance, but sensitizes NSCLC cells to HSP90 inhibition
Acknowledgments
We thank Drs. Jonathan Chernoff, Jeffrey Peterson, Mauricio Reginato and Mark Lemmon for critical reading of the manuscript. We also thank current and former members of the Golemis lab, and the facilities and our colleagues at Fox Chase Cancer Center. Jean-Marc Barret and Jean-François Prost from GamaMabs Pharma deserve special thanks for generously sharing reagents and ideas. The authors were supported by U54 CA149147, R21 CA181287, R21 CA191425 and P50 CA083638 from the NIH (to EAG), the Ruth L. Kirschstein NRSA F30 fellowship (F30 CA180607) from the NIH (to TNB), NCI Core Grant P30 CA006927 (to Fox Chase Cancer Center), the Russian Ministry of Science and Education, as task #17.1891.2014/K (to VAK), and the Russian Government to support the Program for Competitive Growth of Kazan Federal University (to IGS). DAP is employed by Synta Pharmaceuticals.
Footnotes
Author Contributions
Conceptualization, TNB, IGS, and EAG; Methodology, TNB, IGS and EAG; Investigation, TNB, VAK, AEK, RG, NRS, MAH, TMK, EN, MBE, YZ and IGS; Writing – Original Draft, TNB and EAG; Writing – Review & Editing, TNB, YB, DAP, and EAG; Funding Acquisition, TNB and EAG; Resources, DP, PKD, DAP and EAG; Supervision, IGS and EAG.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttonen M, Farkkila A, Tauriala H, Kauppinen M, Maclaughlin DT, Unkila-Kallio L, Butzow R, Heikinheimo M. Anti-Mullerian hormone inhibits growth of AMH type II receptor-positive human ovarian granulosa cell tumor cells by activating apoptosis. Laboratory investigation; a journal of technical methods and pathology. 2011;91:1605–1614. doi: 10.1038/labinvest.2011.116. [DOI] [PubMed] [Google Scholar]
- Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, Devarajan K, Silverman JS, Tikhmyanova N, Skobeleva N, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Science signaling. 2010;3:ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck TN, Chikwem AJ, Solanki NR, Golemis EA. Bioinformatic approaches to augment study of epithelial-to-mesenchymal transition in lung cancer. Physiological genomics. 2014;46:699–724. doi: 10.1152/physiolgenomics.00062.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biaoxue R, Xiling J, Shuanying Y, Wei Z, Xiguang C, Jinsui W, Min Z. Upregulation of Hsp90-beta and annexin A1 correlates with poor survival and lymphatic metastasis in lung cancer patients. Journal of experimental & clinical cancer research : CR. 2012;31:70. doi: 10.1186/1756-9966-31-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, LaBarge MA. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borczuk AC, Sole M, Lu P, Chen J, Wilgus ML, Friedman RA, Albelda SM, Powell CA. Progression of human bronchioloalveolar carcinoma to invasive adenocarcinoma is modeled in a transgenic mouse model of K-ras-induced lung cancer by loss of the TGF-beta type II receptor. Cancer research. 2011;71:6665–6675. doi: 10.1158/0008-5472.CAN-11-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlin EA, Powell SM, Manganaro TF, Hudson PL, Ragin RC, Epstein J, Donahoe PK. Sex-specific fetal lung development and mullerian inhibiting substance. The American review of respiratory disease. 1990;141:466–470. doi: 10.1164/ajrccm/141.2.466. [DOI] [PubMed] [Google Scholar]
- Catlin EA, Tonnu VC, Ebb RG, Pacheco BA, Manganaro TF, Ezzell RM, Donahoe PK, Teixeira J. Mullerian inhibiting substance inhibits branching morphogenesis and induces apoptosis in fetal rat lung. Endocrinology. 1997;138:790–796. doi: 10.1210/endo.138.2.4906. [DOI] [PubMed] [Google Scholar]
- Chang HL, Pieretti-Vanmarcke R, Nicolaou F, Li X, Wei X, MacLaughlin DT, Donahoe PK. Mullerian inhibiting substance inhibits invasion and migration of epithelial cancer cell lines. Gynecol Oncol. 2011;120:128–134. doi: 10.1016/j.ygyno.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B, Koontz C, Frisch SM. CD44S-hyaluronan interactions protect cells resulting from EMT against anoikis. Matrix Biol. 2015;48:55–65. doi: 10.1016/j.matbio.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Clemente N, Jamin SP, Lugovskoy A, Carmillo P, Ehrenfels C, Picard JY, Whitty A, Josso N, Pepinsky RB, Cate RL. Processing of anti-mullerian hormone regulates receptor activation by a mechanism distinct from TGF-beta. Molecular endocrinology. 2010;24:2193–2206. doi: 10.1210/me.2010-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Clemente N, Josso N, Gouedard L, Belville C. Components of the anti-Mullerian hormone signaling pathway in gonads. Molecular and cellular endocrinology. 2003;211:9–14. doi: 10.1016/j.mce.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An Interaction Network Predicted from Public Data as a Discovery Tool: Application to the Hsp90 Molecular Chaperone Machine. Plos One. 2011;6 doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Bi. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–29. doi: 10.1242/jcs.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Hirschhorn T, di Clemente N, Amsalem AR, Pepinsky RB, Picard JY, Smorodinsky NI, Cate RL, Ehrlich M. Constitutive negative regulation in the processing of the anti-Mullerian hormone receptor II. J Cell Sci. 2015;128:1352–1364. doi: 10.1242/jcs.160143. [DOI] [PubMed] [Google Scholar]
- Holtzhausen A, Golzio C, How T, Lee YH, Schiemann WP, Katsanis N, Blobe GC. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. Faseb J. 2014;28:1248–1267. doi: 10.1096/fj.13-239178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YL, Huang MS, Yang CJ, Hung JY, Wu LY, Kuo PL. Lung Tumor-associated Osteoblast-derived Bone Morphogenetic Protein-2 Increased Epithelial-to-Mesenchymal Transition of Cancer by Runx2/Snail Signaling Pathway. Journal of Biological Chemistry. 2011;286:37335–37346. doi: 10.1074/jbc.M111.256156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josso N, di Clemente N. Transduction pathway of anti-Mullerian hormone, a sex-specific member of the TGF-beta family. Trends Endocrin Met. 2003;14:91–97. doi: 10.1016/s1043-2760(03)00005-5. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kersual N, Garambois V, Chardes T, Pouget JP, Salhi I, Bascoul-Mollevi C, Bibeau F, Busson M, Vie H, Clemenceau B, et al. The human Mullerian inhibiting substance type II receptor as immunotherapy target for ovarian cancer. Validation using the mAb 12G4. mAbs. 2014;6:1314–1326. doi: 10.4161/mabs.29316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, MacLaughlin DT, Donahoe PK. Mullerian inhibiting substance/anti-Mullerian hormone: A novel treatment for gynecologic tumors. Obstetrics & gynecology science. 2014;57:343–357. doi: 10.5468/ogs.2014.57.5.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGF beta/Smad signaling by oncogenic Ras. Genes & development. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Marca A, Volpe A. The Anti-Mullerian hormone and ovarian cancer. Hum Reprod Update. 2007;13:265–273. doi: 10.1093/humupd/dml060. [DOI] [PubMed] [Google Scholar]
- Langenfeld E, Hong CC, Lanke G, Langenfeld J. Bone morphogenetic protein type I receptor antagonists decrease growth and induce cell death of lung cancer cell lines. Plos One. 2013;8:e61256. doi: 10.1371/journal.pone.0061256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NY, Kirkbride KC, Sheu RD, Blobe GC. The transforming growth factor-beta type III receptor mediates distinct subcellular trafficking and downstream signaling of activin-like kinase (ALK)3 and ALK6 receptors. Mol Biol Cell. 2009;20:4362–4370. doi: 10.1091/mbc.E09-07-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y, Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. Plos One. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalling in context. Nature reviews Molecular cell biology. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles K, Benedict LA, Dombkowski D, Pepin D, Preffer FI, Teixeira J, Tanwar PS, Young RH, MacLaughlin DT, Donahoe PK, et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2358–2363. doi: 10.1073/pnas.1120733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic proceedings Mayo Clinic. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju GP, Long TE, Park W, Landry JC, Taliaferro-Smith L, Farris AB, Diaz R, El-Rayes BF. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Molecular carcinogenesis. 2014;54:1147–1158. doi: 10.1002/mc.22185. [DOI] [PubMed] [Google Scholar]
- Naxerova K, Bult CJ, Peaston A, Fancher K, Knowles BB, Kasif S, Kohane IS. Analysis of gene expression in a developmental context emphasizes distinct biological leitmotifs in human cancers. Genome Biol. 2008;9:R108. doi: 10.1186/gb-2008-9-7-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. The Journal of biological chemistry. 2015;290:8271–8282. doi: 10.1074/jbc.M115.637496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimori N, Fuchs E. Paracrine TGF-beta Signaling Counterbalances BMP-Mediated Repression in Hair Follicle Stem Cell Activation. Cell Stem Cell. 2012;10:63–75. doi: 10.1016/j.stem.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangas SA, Li X, Umans L, Zwijsen A, Huylebroeck D, Gutierrez C, Wang D, Martin JF, Jamin SP, Behringer RR, et al. Conditional deletion of Smad1 and Smad5 in somatic cells of male and female gonads leads to metastatic tumor development in mice. Mol Cell Biol. 2008;28:248–257. doi: 10.1128/MCB.01404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells [mdash] what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin D, Sosulski A, Zhang LH, Wang D, Vathipadiekal V, Hendren K, Coletti CM, Yu A, Castro CM, Birrer MJ, et al. AAV9 delivering a modified human Mullerian inhibiting substance as a gene therapy in patient-derived xenografts of ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E4418–E4427. doi: 10.1073/pnas.1510604112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nature reviews Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia DA, Bates RC. Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer research. 2014;74:1294–1300. doi: 10.1158/0008-5472.CAN-13-3263. [DOI] [PubMed] [Google Scholar]
- Re RN. Thirty years of intracrinology. Ochsner J. 2014;14:673–680. [PMC free article] [PubMed] [Google Scholar]
- Renlund N, O’Neill FH, Zhang L, Sidis Y, Teixeira J. Activin receptor-like kinase-2 inhibits activin signaling by blocking the binding of activin to its type II receptor. J Endocrinol. 2007;195:95–103. doi: 10.1677/JOE-07-0281. [DOI] [PubMed] [Google Scholar]
- Rey R, Picard JY. Embryology and endocrinology of genital development. Bailliere Clin Endoc. 1998;12:17–33. doi: 10.1016/s0950-351x(98)80427-8. [DOI] [PubMed] [Google Scholar]
- Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer discovery. 2013;3:430–443. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et al. Epithelial to Mesenchymal Transition Is a Determinant of Sensitivity to Chemoradiotherapy in Non-Small Cell Lung Cancer. Ann Thorac Surg. 2011;92:1794–1804. doi: 10.1016/j.athoracsur.2011.07.032. [DOI] [PubMed] [Google Scholar]
- Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, Ryoo HM. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. The Journal of biological chemistry. 2010;285:22542–22553. doi: 10.1074/jbc.M109.094557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BW, Wild CP, editors. World Cancer Report 2014. Lyon, France: International Agency for Research on Cancer; 2014. [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Hayashi M, Manganaro TF, Donahoe PK. The Ontogeny of Mullerian Inhibiting Substance in Granulosa-Cells of the Bovine Ovarian Follicle. Biol Reprod. 1986;35:447–453. doi: 10.1095/biolreprod35.2.447. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Tran D, Meusydessolle N, Josso N. Anti-Mullerian Hormone Is a Functional Marker of Fetal Sertoli Cells. Nature. 1977;269:411–412. doi: 10.1038/269411a0. [DOI] [PubMed] [Google Scholar]
- Ye X, Weinberg RA. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.