

Abstract
Given the current environment in most developed countries, it is a challenge to maintain a good balance between calories consumed and calories burned, although maintenance of metabolic balance is key to good health. Therefore, understanding how metabolic regulation is achieved and how the dysregulation of metabolism affects health is an area of intense research. Most studies are focused on the hypothalamus, which is a brain area that acts as a key regulator of metabolism. Among the nuclei that comprise the hypothalamus, the arcuate nucleus is one of the major mediators in the regulation of food intake. The regulation of energy balance is also a key factor to ensure the maintenance of any species because of the dependence of reproduction on energy stores. Adequate levels of energy reserves are necessary for proper functioning of the hypothalamus-pituitary-gonadal axis. This review discusses valuable data presented in the 2015 edition of the International Workshop of Neuroendocrinology concerning the fundamental nature of the hormonal regulation of the hypothalamus and the impact on energy balance and reproduction.
Keywords: energy balance, hypothalamus, reproduction
Introduction
Given the current environment in most developed countries, it is a challenge to maintain a good balance between calories consumed and calories burned, although maintenance of metabolic balance is key to good health. The worldwide increase in the incidence of overweight and obesity demonstrates the difficulty that people have maintaining proper energy balance. In 2014, 39% of adults age 18 or over were overweight (BMI>25kg/m2), while 11% of men and 15% of women were obese (BMI>32kg/m2) (1). That means an estimate of 1.9 billion overweight adults worldwide, of which 600 million are obese. The risks associated with overweight include cardiovascular diseases and type 2 diabetes mellitus, among others. Therefore, understanding how metabolic regulation is achieved and how the dysregulation of metabolism affects health is of great importance.
The hypothalamus is a brain area that acts as a key regulator of metabolism and mediates numerous processes including food intake, body temperature, sexual behavior and reproduction, circadian rhythms and emotional responses. The hypothalamus, comprised of many distinct neuronal nuclei, integrates neural, endocrine and metabolic signals. Among these nuclei, the importance of the arcuate nucleus (ARC) in the regulation of energy balance is well known (2). Its position is adjacent to the median eminence allowing the ARC to sense circulating signals such as leptin, ghrelin and insulin, among others. Through changes in activity of its neuronal populations and through many diverse and widespread outputs, the ARC is a major participant in the regulation of energy metabolism.
The regulation of energy balance is not only important for the survival of one individual. It is also a key factor to ensure the maintenance of any species because of the dependence of reproduction on energy stores (3). Adequate levels of energy reserves are necessary for proper functioning of the hypothalamus-pituitary-gonadal axis. The neuroendocrine players that link energy balance with the reproductive system include hormones and neuropeptides that act on hypothalamic gonadotropin releasing hormone neurons. As an example, leptin is one of the hormones recognized as a modulator of the reproductive axis, as well as a modulator of energy balance. Thus, the importance of energy metabolism and its regulation to guarantee reproduction is well established.
This review will discuss valuable data presented in the 2015 edition of the International Workshop of Neuroendocrinology concerning the fundamental nature of the neuroendocrine regulation of hypothalamic neurons and the impact on energy balance and reproduction.
Main hypothalamic systems regulating metabolism
POMC and AgRP neurons in energy balance regulation: beyond the peptides
The central nervous system (CNS) plays a major role in maintaining a balance between the energy required for survival and the energy provided by feeding. In order to fully understand the central regulation of energy balance, it is essential to understand the complete compliment of neurotransmitters involved. Clearly, the peptide transmitters released from hypothalamic agouti-related peptide (AgRP) and proopiomelanocortin (POMC) neurons are important regulators of energy balance (4,5). However, it has become increasingly clear that these neurons also utilize amino acid transmitters that may also impact energy balance regulation. Therefore, many recent studies have focused on the production, release and consequence of the amino acid transmitters GABA and glutamate from AgRP and POMC neurons.
Indications that AgRP and POMC neurons likely use a mix of peptide and amino acid transmitters date back several decades to immuno-electron microscopy studies showing both dense-core and small-clear vesicles in the axon terminals of these neurons (4,6). A role for amino acid transmitters in food intake and metabolism was suggested through pharmacologic studies carried out from the 1970s through the 1990s. However, experiments exploring the potential significance of GABA or glutamate release from neurons in energy balance circuits were not possible until advances were made in genetic manipulation. Using genetic approaches to delete the orexigenic peptides NPY, AgRP or both, it was found that energy balance could be largely maintained in the absence of these peptides (7,8), whereas ablation of the AgRP/NPY neurons in adult mice blunted food intake and caused rapid wasting (9,10). This surprising difference between peptide deletion and neuronal ablation was eventually attributed to the loss of GABA co-release when the neurons were ablated as a whole (11), sparking renewed interest in amino acid transmitters in energy balance circuits. Optogenetic and other approaches have been used to further show the importance of GABA release from AgRP neurons in the stimulation of food intake and the downstream targets of AgRP neuron-derived GABA release are beginning to be revealed (11,12).
The release of GABA from AgRP neurons can be dynamically regulated. Fasting causes increased GABA release, whereas leptin decreases GABA release from AgRP neurons onto POMC neurons (13). While inhibition of POMC neurons may not be a necessary contributor to increased food intake upon robust AgRP neuron stimulation (14), the high degree of connectedness from AgRP to POMC neurons makes this a convenient synapse to examine changes in GABA release from AgRP neurons. It is tempting to infer that altered release at one target site likely reflects similar changes in other target regions, but this may not be the case. Neurons can release different sets of chemical transmitters from distinct fibers (15,16) and terminals may preferentially express one transmitter or another from a given cell-type (17,18). Therefore, it will be necessary to determine how changes in the activity of POMC or AgRP neurons affects GABA release in specific target sites to fully understand the dynamic regulation imposed by these neurons under select conditions.
Unlike AgRP neurons that appear to use only GABA as their amino acid transmitter, POMC neurons appear heterogeneous in their amino acid phenotype with ∼50% of POMC neurons being GABAergic and ∼10-40% glutamatergic (19,20). Interestingly, the amino acid transmitter phenotype of POMC neurons seems to be plastic throughout development with a large proportion of POMC neurons showing glutamatergic markers during the early postnatal period and tapering off into adulthood (21). The genetic deletion of the vesicular glutamate transporter vGlut2 to prevent glutamate release specifically from POMC neurons causes a modest sex-specific increase in body weight in male mice maintained on a high-fat diet (21). However, this phenotype may reflect a developmental effect since vGlut2 was constitutively deleted from POMC neurons, and potentially from a subset of other neurons that transcribe the POMC gene briefly during development (22). Therefore, further studies are needed to determine the role of glutamate release from POMC neurons in adulthood and to begin to explore the functional consequence of GABA release from POMC neurons. Additionally, determining if GABAergic and glutamatergic POMC neurons represent otherwise distinct subpopulations of POMC neurons and whether the developmental reduction in glutamatergic phenotype is important in energy balance regulation will add to a more complete understanding and inform future studies that may manipulate or examine POMC neurons in a subtype specific manner.
By identifying and understanding the actions of the array of transmitters involved in energy balance regulation, it will be possible to better identify potential points of dysfunction and perhaps therapeutic targets. The recent advances described above add essential information regarding the complex actions of POMC and AgRP neurons. Although there is much more to learn regarding the roles of amino acid transmitters in energy balance, the recognition that POMC and AgRP neurons use both peptide and amino acid transmitters to effect a variety of responses on distinct timescales in a state-dependent manner reflects a significant increase in the understanding of this system.
Hormones regulating hypothalamic systems controlling metabolism
Ghrelin and the regulation of feeding: an important player for energy homeostasis
To achieve the regulation of energy balance, CNS circuits must be able to interact with the endocrine system, which provides peripheral signals that indicate body energy status. Among them, ghrelin is the only mammalian peptide hormone known to increase food intake. Ghrelin acts primarily on CNS centers to affect not only homeostatic-driven food intake but also to regulate hedonic-driven feeding.
Ghrelin is a 28-amino acid octanoylated peptide secreted by cells located within the gastrointestinal tract. It was first discovered as the endogenous ligand of the growth hormone secretagogue receptor 1a (GHSR1a), with the ability to stimulate the secretion of growth hormone (GH) from the anterior pituitary gland (23). Further studies in the area of ghrelin led to the discovery of a role for this hormone in the regulation of several processes including food intake, glucose metabolism, gastrointestinal tract motility and stress- and anxiety-related behaviors, among others (24). Plasma ghrelin levels rise before meals and decrease after the ingestion of food (25). This pattern of variation promoted the idea of ghrelin as a “meal initiation” signal, which held up for many years, although this simplistic view is now beginning to be displaced (26).
Ghrelin acts on the CNS by binding to the GHSR1a, which is a G-protein coupled receptor highly expressed in brain centers associated with food intake (27). The main neuronal targets that mediate ghrelin's orexigenic action are the ARC of the hypothalamus and the dorsal vagal complex (DVC) of the brainstem (28). The extent to which ghrelin is able to reach these sites of action is a matter of debate (29). These two brain areas display the important feature of having a circumventricular organ associated: the median eminence, lying adjacent to the ARC, and the area postrema, which is part of the DVC. Circumventricular organs are specialized brain regions that lack the normal blood-brain barrier and present fenestrated capillaries that allow peripheral signals to reach their neuronal targets (30). In the case of the ARC, it is supposed that ghrelin is able to freely diffuse throw the median eminence and reach GHSR1a-expressing neurons (31). Regarding to the DVC, ghrelin could directly activate GHSR1a-expressing neurons of the area postrema, which in turn regulate their targets in brainstem and hypothalamus (32).
The ARC contains two major neuronal populations with opposite effects on food intake: the orexigenic AgRP/NPY-expressing neurons and the anorexigenic POMC-expressing neurons. The importance of the ARC as a mediator of ghrelin's orexigenic action emerges from the fact that the absence of AgRP/NPY neurons eliminates ghrelin triggered increase in food intake (33). Another piece of evidence that supports a key role of the ARC mediating the orexigenic effects of ghrelin is that AgRP/NPY neurons express high levels of GHSR1a mRNA (34). The AgRP/NPY neurons send projections to other hypothalamic nuclei such as the paraventricular nucleus (PVN), the dorsomedial nucleus (DMH), and the lateral hypothalamic area (LHA), all involved in the control of feeding. These hypothalamic nuclei also express the GHSR1a mRNA and show ghrelin binding when biotin- and fluorescent-labeled ghrelin binding assays are performed (31,35,36). Then, the direct and/or indirect participation of these brain areas could be important for ghrelin's regulation of food intake.
The DVC is another important brain area that mediates ghrelin's orexigenic action. This brain region is made up by three nuclei: the nucleus of the solitary tract, the area postrema and the dorsal motor nucleus of the vagus. The expression of GHSR1a mRNA has been described in all three components of the DVC (27), suggesting that ghrelin can directly act on them. Indeed, it has been shown that administration of ghrelin directly on the DVC promotes food intake (37). Additionally, intra-cerebroventricular infusion of ghrelin activates c-Fos (a marker of neuronal activation) expression in the area postrema and the nucleus of the solitary tract (38). Nevertheless, one study shows that peripheral administration of ghrelin to mice that selectively express GHSR1a in the DVC does not result in an increase in food intake (39). This evidence suggests that the DVC is a target of ghrelin to regulate food intake but it is not sufficient to mediate ghrelin's orexigenic action.
Considering that ghrelin is the only peptide hormone known to increase food intake, its relevance in the regulation of energy homeostasis is highlighted. Although the brain targets for ghrelin's orexigenic action are well established, the exact molecular mechanisms by which this hormone regulates feeding are not completely understood. Unraveling these mechanisms would be of extreme importance to consider them as potential therapeutic targets in the treatment of pathologies that affect food intake.
Oxysterols and LXRs
Overweight and obesity are primarily linked to poor eating habits. Busy lifestyle induces an increase in the availability and consumption of fast foods. This type of food contains great amounts of animal fats, which contain a mixture of triglycerides, cholesterol and phospholipids. Cholesterol is necessary to guarantee the integrity and fluidity of cell plasma membrane. It is produced by all animal cells and is also incorporated with the diet. At present, the quantity of cholesterol consumed represents an excess relative to the needs of human body. This implicates that the organism must be able to metabolize it and one important way is the oxidation of cholesterol to oxysterols.
The oxysterols are involved in different mechanisms related to the removal of cholesterol from cells (40). These compounds are capable of binding to specific proteins called liver X receptors (LXRs) acting as their endogenous ligands (41–43). The functional LXRs exist as two isoforms: LXRα and LXRβ. The first is mainly expressed in the liver and to a lesser extent in the gut, adipose tissue, kidney, spleen and macrophages, whereas LXRβ is expressed in almost all tissues (40). Upon exposure to excessive accumulation of intracellular cholesterol oxides, LXRs activate a program of gene expression for limiting the pathogenic accumulation of cholesterol (44). In the intestine, activation of LXRs decreases cholesterol absorption from the diet by promoting the expression of excretion transporters such as ABCA1, ABCG5 and ABCG8 (45). In macrophages, LXRs cause a rapid increase in the expression of genes involved in the formation of high density lipoprotein and reverse cholesterol transport (46). In the liver, LXRs activation promotes the direct conversion of excessive cholesterol to bile acids through the regulation of the limiting enzyme 7-alpha hydroxylase (CYP7a) (40).
In addition to the regulation of cholesterol homeostasis in multiple tissues, LXRs are also intimately involved in the control of hepatic lipid metabolism and in the physiological regulation of carbohydrate metabolism (47). Studies demonstrated that LXR agonists improve glucose tolerance in a mouse model of diet-induced insulin resistance. Treatment with synthetic LXR ligands alters the expression of genes in the liver and adipose tissue and causes a decrease in hepatic glucose production and increases glucose uptake by adipocytes. Furthermore, activation of LXRs indirectly suppresses the expression of hepatic gluconeogenesis enzymes (phosphoenolpyruvatecarboxykinase and glucose 6-phosphatase), whereas in adipose tissue LXRs regulate the expression of the insulin-sensitive glucose transporter GLUT4 (48). Further studies suggest that LXRβ in particular plays an important role in pancreatic insulin secretion and LXR activators promote insulin secretion (47). LXR ligands have also showed to be effective in other studies of insulin resistance and type 2 diabetes mellitus, in which spontaneously diabetic or age-developed glucose intolerant rodent strains (db/db mice, ob/ob mice, fa/fa Zucker rats) were used to highlight the potential of LXR agonists as insulin sensitizers (49,50).
While the metabolic functions of the LXRs in peripheral organs have been widely investigated, little is known about the expression and functionality of LXRs in the brain. The activation of LXRs facilitates the excretion of cholesterol in the cerebellum and hippocampus (51). Recent studies show that the expression of LXRα and LXRβ in the hypothalamus is sensitive to triglycerides and serum insulin levels. Animals with glucose intolerance show an upregulation of LXRβ and a downregulation of LXRα in the hypothalamus. In addition, a correlation between this LXR expression and triglycerides or insulin levels was described indicating the importance of both subtypes in the risk of developing metabolic diseases (52). LXRβ expression in the hypothalamus correlates negatively with the area under the curve in glucose tolerance tests in control animals, while a positive correlation is found in rats with abnormal glucose tolerance (52,53). The endogenous receptor agonists can also contribute to modulate LXR expression. The brain produces most of the 24(S)-hydroxycholesterol present in the body. This metabolite acts as an efficient LXR agonist (54) and is produced by the cholesterol-24-hydroxylase (CYP46A1). This enzyme converts cholesterol from degraded neurons into 24(S)-hydroxycholesterol to allow the removal of cholesterol from the brain and is induced by oxidative stress (55). Glucose has also been described to induce the expression of LXR target genes at physiological concentrations, although this data is controversial (56,57).
The hypothalamus coordinates several complex homeostatic mechanisms and LXRs seem to be involved in some of them. The anatomical location of both receptor subtypes in the hypothalamus has been described using confocal microscopy (Figure 1). LXRα was found in the periventricular nuclei, medial preoptic area (mPOA) and in the VMH while LXRβ was found in mPOA and the ARC (52). These nuclei contain neurons reactive to nutrient-related signals that induce neurochemical responses to regulate energy homeostasis (58). On the other hand, recent results show that in vitro treatment with glucose or insulin may alter LXR expression in hypothalamic cells. Glucose concentrations higher than 5.5 mM decrease LXRβ expression, while insulin treatment produces a similar effect only in the presence of 8.5 mM glucose. In both conditions, LXRα expression is unaffected (59). In vitro treatment with lipids also modifies the expression of this receptor. Incubation with cholic acid (4 h) and cholesterol increases the expression of LXRα, and cholic acid also promotes the expression of ABCA1. These results suggest that hypothalamic LXRβ is mainly sensitive to carbohydrate changes (47) while LXRα responds to lipid changes (60).
Figure 1.
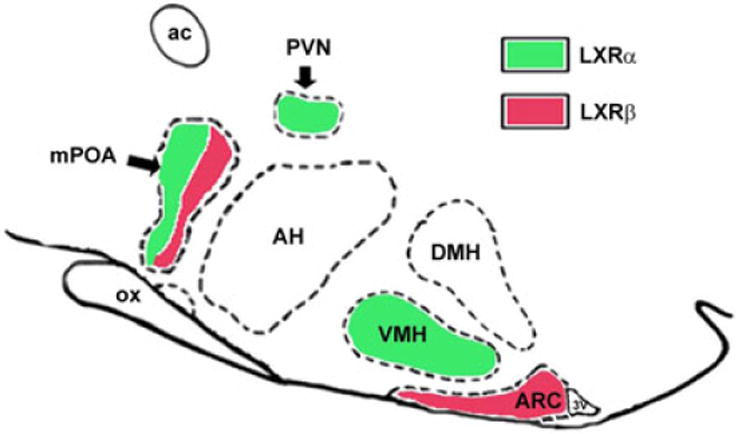
Representative diagram of LXR expression in hypothalamic nuclei of rats: LXRs localization in the hypothalamic nuclei was evaluated by immunocytochemistry using specific antibodies. LXRα signal was observed in the paraventricular (PVN) and ventromedial (VMH) nuclei while LXRβ signal was found in the arcuate (ARC) nucleus, both LXR immunosignals were detected in the median preoptic area (mPOA) expressed in different cell types. (J Endocrinol. 2012; 215:51–58) DMH: dorsomedial nucleus of hypothalamus; AH: anterior hypothalamic area; ac: anterior commissure, ox: optic chiasm 3V: third ventricle.
The data presented above indicate that oxysterols and LXRs are important players in the regulation of cholesterol metabolism in several organs. In addition, they mediate cholesterol removal from neurons in the CNS. There is also evidence that LXR expression in the hypothalamus is sensitive to nutrient levels, which suggests that they are involved in the regulation of energy balance. Thus, future studies directed to understand the role that oxysterols and LXRs play in the regulation of energy metabolism would be of great interest.
Food intake and metabolism: intertwined regulation by dopamine and prolactin
As mentioned in the introduction of this article, maintaining a proper energy balance is a key factor to guarantee reproduction. Metabolic adaptations to store energy during pregnancy in preparation for future demands is a biological allostatic hallmark in evolution. Females display a strong hyperphagia during pregnancy and lactation. The hormone prolactin may be a major factor mediating this hyperphagia (61,62), probably sustained by leptin resistant hypothalamic centers controlling food intake (63).
Prolactin acts on peripheral tissues by activating a cytokine receptor (PRLR) of which there are long and short isoforms (64). Prolactin can reach the brain through an active reuptake mechanism similar to the transport mechanisms described for leptin and insulin (65). In the brain, PRLR has been localized in the striatum as well as in a number of hypothalamic nuclei associated with food intake and metabolism, including the ARC, VMH, PVN and the DMH (61). The presence of PRLR in brain areas associated with the regulation of energy balance and food intake, as well as in white and brown adipose tissue, liver and pancreas raises the possibility that prolactin is involved in the regulation of energy balance acting at different levels (66) (Figure 2).
Figure 2.
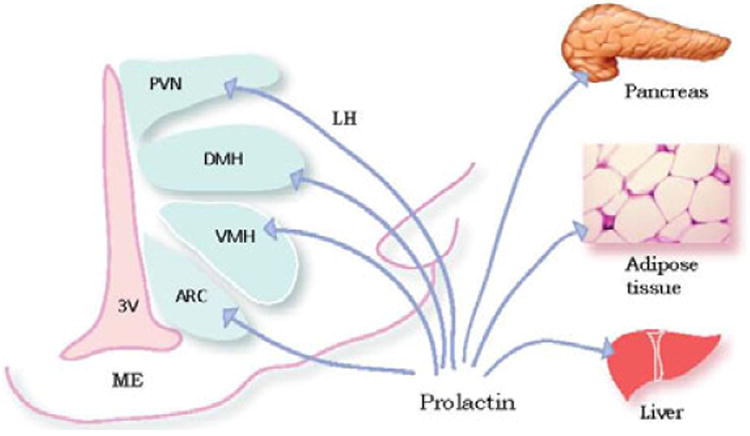
PRLR in brain and tissues. In the rat brain, PRLR have been localized in a number of hypothalamic nuclei associated with food intake and metabolism, including the arcuate nucleus (AN), ventromedial hypothalamus (VMH), paraventricular hypothalamic nucleus (PVN), and the dorsomedial nucleus of hypothalamus (DMH). PRLR have also been described in pancreas, adipose tissue, and liver. ME: median eminence, 3V: third ventricle.
Consistent with the hypothesis that prolactin has a significant role in the regulation of body weight, prolactin administration stimulates food intake (67,68) while PRLR-deficient mice exhibit lower body weight and reduced fat mass (69). Nevertheles, female mice lacking dopamine D2 receptors (D2Rs), Drd2-/- mice, exhibit chronic hyperprolactinemia and pituitary lactotrope hyperplasia (70,71) but similar body weight compared to wild-type females and only a minimal increase in food intake (72). But Drd2-/- mice may not be an optimal model to study the effects of chronic hyperprolactinemia on energy balance given the fundamental importance of central D2Rs in food intake, reward mechanisms related to feeding behavior (73,74) and growth hormone releasing hormone-growth hormone regulation (75).
To unravel the role of elevated prolactin levels, with intact central D2Rs, lactotrope specific D2R knockout (lacDrd2KO) female mice provide a unique model. In lacDrd2KO female mice serum prolactin levels are chronically elevated, mice are subfertile and have altered estrous cycles (76). Consistent with the presence of functional brain D2Rs, haloperidol- induced catatonia test is normal and the GH axis is preserved (77). In lacDrd2KO female mice, there is a marked increase in body weight, food intake and adiposity. In correlation with adiposity accretion, serum leptin is markedly elevated but hypothalamic anorexigenic peptides do not indicate leptin resistance. Hypothalamic POMC mRNA levels, as well as intermediate pituitary levels of α-melanocyte stimulating hormone (α-MSH), which are anorexigenic, are normal. Furthermore, mRNA levels of the orexigenic NPY, which are usually downregulated by leptin, are increased.
Similarly, high prolactin levels in pregnancy or lactation induce a state of leptin resistance to meet the metabolic demands of the dams (78). Both suckling and prolactin increase NPY expression in the DMH suggesting that prolactin might stimulate food intake by potentiating the effects of NPY input on the PVN (61).
On the other hand, in the global Drd2-/- knockout mouse, loss of central D2Rs mediates a decrease in prepro-orexin (Ppo) mRNA levels and an increase in α-MSH levels, and these two anorexigenic events may offset to some extent the effect of prolactin on food intake. Therefore, functional central D2R signaling in the lacDrd2KO mouse maintains POMC and Ppo mRNA levels and the central orexigenic effect of prolactin is fully evidenced (Figure 3).
Figure 3.

Effect of global (Drd2-/-) and lactotrope specific D2R knockout (LacDrd2KO) mouse models on anorexigenic and orexigenic factors in the pituitary and hypothalamus. In Drd2-/- females, two anorexigenic events (increase in αMSH, and a decrease of orexin precursors) may offset the orexigenic acton of prolactin, while in lacDrd2KO mice both Pomc and Ppo are not modified, and prolactin activates Npy expression, resulting in increased food intake.
These data indicate that central D2Rs, which are key elements in food intake homeostasis, interact with prolactin levels.
In lacDrd2KO female mice heavier gonadal and retroperitoneal fat pads, larger adipocytes, and heavier livers were found. Increased adiposity correlated with higher serum triglycerides and non-esterified fatty acids, with no changes in cholesterol or adiponectin. In adipose tissue, prolactin has been shown to upregulate the expression of its receptor, stimulate adipocyte differentiation and inhibit lipolysis (79,80). Adipose PRLR was not increased in the selective mutant, but lipolysis was decreased (76) and this may explain the increased adipocyte size found. Interestingly, the expression of a lipogenic enzyme, lipoprotein lipase, was also decreased in correlation with increased serum triglycerides. Livers were heavier in lacDrd2KO mutants. Abundant fat droplets were observed, as well as higher triglyceride content and PRLR mRNA levels. High fat content in the liver could not be attributed to changes in the expression of lipogenic or lipolytic enzymes, but to alteration of glucose homeostasis. In this respect, hyperprolactinemic lacDrd2KO mice had glucose intolerance, and a blunted insulin response to glucose (76).
In conclusion, selective ablation of D2Rs from lactotropes evokes persistent hyperprolactinemia which induces a state of leptin resistance and increases hypothalamic NPY levels, in correlation with a hyperphagic state. Increased food intake, together with prolactin acting at different organs modifies energy metabolism. There is an increase in adiposity, higher serum non-esterified fatty acids and triglycerides. At the pancreatic level, insulin response to glucose is impaired, which results in glucose intolerance, high serum glucose and hyperinsulinemia. Altered glucose metabolism may be causal of the increased lipid content in the liver. These results highlight the role of prolactin as a metabolic hormone acting on different organs to reinforce its role during pregnancy, which is to store energy for future demands.
Impact of altered metabolism on reproductive function
Metabolic control of reproduction: focus on leptin signaling
Processes involved in successful reproduction, including sexual maturation, production of gametes, pregnancy and lactation are energetically demanding (81–83). As a consequence, conditions of low energy availability or high energy utilization result in decreased activity of the reproductive axis. For example, gonadotropin secretion and ovulation are compromised in females in negative energy balance caused by inadequate feeding or excessive energy expenditure (84,85). The interaction between these complex systems (metabolism and reproduction) is orchestrated by hypothalamic neurons that sense changes in circulating levels of metabolic cues and adapt the system to the individual nutritional condition. Among these cues, leptin is essential for the regulation of the reproductive axis.
Humans and mice with loss-of-function mutations in leptin (LEP/Lep) or leptin receptor (LEPR/Lepr) genes are obese and infertile (86–88). Both have low circulating gonadotropin levels, incomplete development of the reproductive tract and no pubertal maturation. Leptin is an adipocyte hormone secreted into circulation in proportion to fat mass. It binds to cognate receptors expressed in many organs and tissues. The LepR is a class 1 cytokine receptor found in six isoforms (88–90). The LepR long form (LepRb) is the signaling isoform and contains a Box 3 motif associated with downstream phosphorylation of tyrosine residues. The best associated pathway described is the JAK/STAT signaling pathway (91–94). Deletion of leptin-induced STAT3 signaling (Tyr1138, LRbS1138s/s mice) recapitulates the hyperphagic obesity and the well-described changes in the melanocortin system of the LepR deficient (db/db) mice (95). However, disruption of STAT3 signaling in this mutant line has little effect on glycemic control and fertility (95). The s/s female mice show sexual maturation, development of the reproductive tract and ovarian signs of ovulation suggesting that leptin's effect on reproductive function is independent from STAT3 signaling pathway (95).
Disruption of LepRb tyrosine residue 1107 (Tyr1107) blocks leptin-induced phosphorylation of STAT5. These mice develop mild obesity and show very small changes in estrous cycle duration (96). However, conditional deletion of STAT5 in LepR cells causes no metabolic or reproductive deficits (97). Deletion of both STAT3 and STAT5 signaling produces mice with a phenotype similar to those with deletion of STAT3 alone i.e. increased body weight and adiposity. On the other hand, mutation in Tyr985 residue of LepRb causes a lean phenotype potentially due to increased leptin sensitivity due to blockade of SOCS3 and phosphatases associated with feedback inhibition of leptin signaling (98). No reproductive phenotype was observed in Tyr985 mutant mice.
Leptin also recruits the phosphoinositide 3-kinase (PI3K) signaling pathways (92,99–101). In hypothalamic slices, the acute effects of leptin on cell activity and feeding require intact PI3K (99,100,102–105). However, the molecular mechanisms associated with these responses are not clear, but studies have suggested that phosphorylation of insulin receptor substrate-2 (IRS-2) is upstream of leptin-induced PI3K (106,107). IRS-2 is expressed in hypothalamic neurons and IRS-2 knockout mice show metabolic dysfunction and infertility (108). Females have low sex hormones and deficient reproductive tract development. However, mice with conditional deletion of IRS-2 in LepR cells are fertile, but whether pubertal development, cyclicity and hormone levels are normal have not been reported (109).
PI3K is found as multiple classes of enzymes. Leptin recruits the class 1a PI3K comprised of heterodimers of one regulatory and one catalytic subunit. The regulatory subunits are collectively called p85s, and the catalytic subunits, p110s (110,111). The p110α and p110β catalytic subunits are widely expressed, and global deletion of either one is incompatible with life (112–115). However, 50% loss-of-function of p110α activity decreases insulin and leptin responsiveness causing hyperphagia, glucose intolerance and increased fat mass (116). Both catalytic subunits are expressed in LepR neurons of the hypothalamus (104,117). However, whether the lack of leptin-induced PI3K signaling results in metabolic or reproductive deficits has not been reported.
In summary, the role of leptin in reproductive control is well established. However, the molecular pathways associated with leptin action as permissive factor for pubertal maturation and as a signal of energy sufficiency for successful reproduction is still unsettled.
Sexual dimorphism in the control of metabolism
Sex steroids regulate metabolism
The differences in metabolic function between males and females point out the role of sex steroids in the regulation of energy balance and body composition. In this regard, androgens and estrogens are primary regulators of metabolism in both sexes. The action of sex steroids is focused on the hypothalamic nuclei that regulate food intake and energy balance, but they also regulate metabolism in peripheral tissue (muscle, liver and adipose tissue; Figure 4). Estrogens exert their effects through binding to the nuclear estrogen receptors (ER) isoforms α (ERα) and β (ERβ), or the membrane G protein-coupled estrogen receptor (GPER30), dictating the activation of genomic or non-genomic pathways, respectively. Meanwhile, androgens bind to androgen receptors (AR) located in the nucleus or the cytoplasm of cells, both exerting their actions in the nucleus.
Figure 4.
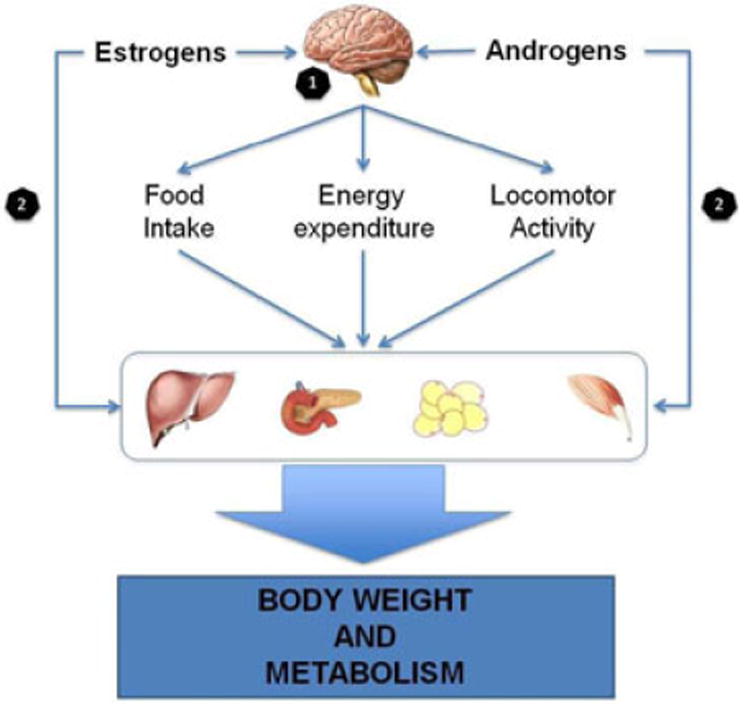
Estrogens and androgens regulate metabolic function. (1) Androgens and estrogens act on the hypothalamus, mainly in ARC and VMH, regulating food intake, energy expenditure and locomotor activity. This action impacts on the metabolic function of liver, pancreas, adipose tissue and muscle, leading to the regulation of body weight and whole-body metabolic function. Moreover, (2) androgens and estrogens can directly modulate the function of those tissues.
Androgens modulate metabolism in females and males
Multiple animal models and clinical studies have demonstrated that androgens play important roles in the control of metabolic function in both sexes. In males, androgens stimulate lean mass growth and inhibit fat accumulation (114). Therefore, it is not surprising that testosterone deficiency induces obesity, accumulation of visceral adipose tissue (VAT) and increases the risk of developing insulin resistance and diabetes mellitus (115). In females, androgen excess provokes a similar condition to androgen deficiency in males, including abdominal obesity, a pro-inflammatory profile and insulin resistance (116). The mechanisms associated with androgen deficiency-induced insulin-resistance probably include modifications in the muscle transcriptome, mainly a reduction in the expression of the transcription factor peroxisome proliferator-activated receptor-gamma coactivator alpha (PGC1α), which plays important roles in the stimulation of mitochondrial biogenesis and skeletal muscle oxidative fibers (117). Moreover, testosterone and dihydrotestosterone (DHT) can modulate adipogenesis in subcutaneous adipose tissue (SAT) and VAT in both sexes. However, androgens can limit the number of mature adipose cells in females. In contrast, testosterone in males induces the proliferation of visceral pre-adipocytes (118).
It is difficult to isolate the role of androgens from the role of estrogens on metabolic function because androstenedione and testosterone are converted to estrone and estradiol, respectively, by the P450 aromatase. DHT, which cannot be metabolized to estrogen, can be reduced by the aldo-ketoreductase family 1C to androstenediol, which has estrogen-like activity through ERβ (119). However, AR knockout (ARKO) male mice develop late-onset obesity with an increase in both SAT and VAT. ARKO female mice lack these changes but present a reduction in energy expenditure (120), which demonstrates that androgens are directly involved in the control of body weight and metabolism.
Interestingly, ARs are more abundantly expressed in the brains of males than females (90), mainly in the VMH, ARC, anteroventral periventricular nucleus, mPOA and bed nucleus of the stria terminalis. In this regard, it has been observed that hypothalamic ARs are associated with the activation of STAT3 leptin-induced signaling in ARC neurons (121). In addition, in vitro studies demonstrate that ARs are necessary to maintain hypothalamic insulin sensitivity, which is mediated by the inhibition of NF-κB (122). In female mice, androgens-induced increase of visceral fat mass seems to be mediated by a decrease in hypothalamic POMC expression and POMC neuronal innervation to the DMH, resulting in the failure of leptin to activate brown adipose tissue thermogenesis and energy expenditure (123). These antecedents indicate that the hypothalamic ARs contribute to the suppression of food intake and the control of whole-body metabolism in both males and females.
Estrogens modulate metabolism in females and males
The metabolic role of estrogens is better understood in female physiology. It is clear that the decline of ovarian function induces important changes in body composition, increasing the accumulation of total body fat, abdominal obesity and reducing energy expenditure (124). Interestingly, similar findings have been observed in female rats exposed to an inhibitor of the P450 aromatase. These animals present elevated androgens but reduced estrogen levels and an increase in adiposity, larger adipose cells and insulin resistance (125).
Animal models show that estrogens improve insulin sensitivity, body composition and lipid profile in both sexes (126). Receptor-specific KO models have demonstrated that both receptors are involved in metabolic function. However, they can have different and sometimes antagonistic actions, with ERα probably more relevant than ERβ signaling (127). The deletion of ERα induces insulin resistance, dyslipidemia, β-pancreatic cell dysfunction and impaired glucose tolerance. In the same way, the selective activation of ERα or ERβ by specific pharmacological agonists, propylpyrazoletriol and diarylpropionitrile respectively, has demonstrated similar effects (128). It seems that ERβ could have anti-obesogenic actions during high-fat diet challenge in mice, which is associated to the inhibition of PPARγ-induced adipogenesis, as it has been demonstrated in ERβ KO mice (129,130). In this regard, it has been observed that the total and plasma membrane fraction of GLUT4 is strongly reduced in skeletal muscle of ERα KO mice but not affected in ERβ KO mice (131).
In hypothalamus, ERα expression is markedly higher than ERβ in VMH, ARC, PVN, POA and LHA. Of interest, the hypothalamic expression of estrogen and androgen receptors is dependent on sex and age, indicating the importance of metabolism on reproductive function (132). Brain deletion of ERα induces hyperphagia and decreased energy expenditure and locomotor activity, leading to fat accumulation in visceral depots (133). Although these functions are determined by different hypothalamic areas, the direct injection of estradiol into the PVN, ARC and VMH are the most effective in reducing food intake, body weight and increasing locomotor activity, especially in females (134). In this regard, the specific loss of ERα in ARC POMC neurons increases food intake but does not directly affect energy expenditure (133), whereas the deletion of ERα in VMH neurons decreases energy expenditure but does not affect food intake (133,135). In addition to the metabolic effects exerted by their nuclear receptors, it has been observed that the deletion of estrogen membrane receptor GPER30 increases body weight (136). In turn, the activation of the GPER30 alone is able to trigger the STAT3 signaling pathway. Interestingly, ERα KO mice exhibit altered leptin-induced STAT3 activation (137). Overall, these antecedents suggest a crosstalk between nuclear and/or membrane estrogen receptors and leptin-induced STAT3 signaling in the control of food intake and energy expenditure.
In summary, it is clear that sex steroids are central regulators of metabolic function. Probably, androgens and estrogens act coordinately at different organs such as brain, skeletal muscle, adipose tissue and liver. The different profile in sex steroids between females and males results in sex-dependent patterns of body composition, insulin sensitivity and energy expenditure.
Concluding Remarks
The importance of the regulation of energy metabolism is highlighted by the fact that survival and reproduction strongly depend on energy levels. It is clear that the CNS regulation of energy homeostasis is principally mediated by the hypothalamus, which contains neuronal populations with the ability to sense nutrient-related signals and affect food intake. This review presented recent studies indicating that hypothalamic AgRP and POMC neurons utilize not only peptide transmitters to exert their roles but also amino acid transmitters (GABA and glutamate), and this utilization can be dynamically regulated depending on body energy status. The regulatory role of the hypothalamus is influenced by the action of peripheral hormones and metabolites produced by different organs such as adipose tissue, gonads and gastrointestinal tract. In this article, the role of ghrelin, prolactin and oxysterols as participants in the regulation of metabolism was discussed. Although these three metabolites affect energy homeostasis and have their neuronal targets mainly located in the hypothalamus, their role becomes relevant in different states. Ghrelin is important when negative energy balance is present, stimulating food intake and preparing the body for the ingestion of food. Prolactin has a prominent role when females face pregnancy and lactation, promoting food intake with the final objective of storing energy for the future demands of the offspring. Oxysterols and their receptors are involved in the excretion of cholesterol when excessive cholesterol is present in cells, but recent studies suggested that they are also implicated in the hypothalamic regulation of carbohydrates and lipid metabolism.
The hypothalamus is also important in the regulation of reproductive function, which is also influenced by body energy status. Leptin acts as a link between energy status (it is secreted in proportion to fat mass) with the reproductive axis acting on hypothalamic gonadotropin neurons. This article presented recent work that aimed to establish the molecular mechanism by which leptin modulates sexual maturation and fertility. It is also known that sex steroids are determinants of reproductive function and the present review introduced valuable data showing the intertwined regulation of energy metabolism by estrogens and androgens.
Altogether, the whole concert of hormones and metabolites that regulate energy metabolism act on interrelated pathways forming a complex network. Proper functioning of this network finally determines the capacity of an organism to survive and to breed. This review discussed some of the advances made in areas that are part of this complex network, in the framework of an environment that predisposes humans to energy balance disorders. The efforts made in these research areas contribute to the overall objective of understanding the elaborate regulation of energy metabolism.
Acknowledgments
This work was supported by the following grants: MPC was supported by CONICET; MM was supported by FONDECYT 11130250; PIP-860 by CONICET, 06CM09 by Catholic University of Cuyo and PICTO-0158 by Catholic University of Cuyo and Rene Baron Foundation supported the work of HC; NIH R01-DK078749 and an award from the Monfort Family Foundation to STH; DBV was supported by CONICET, Argentinean National Agency of Research Promotion and Rene Baron Foundation; CFE was supported by the grant NIH R01-HD69702.
MPC would like to thank Dr. Mario Perello for all the comments that were extremely helpful for the composition of this manuscript.
Footnotes
The authors of the manuscript have no conflicts of interest to declare.
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/jne.12395
References
- 1.World Health Organization. World Health Statistics. 2015 [Google Scholar]
- 2.Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012 Oct;15(10):1350–5. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol - Endocrinol Metab. 2008 May 1;294(5):E827–32. doi: 10.1152/ajpendo.00670.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercer AJ, Hentges ST, Meshul CK, Low MJ. Unraveling the central proopiomelanocortin neural circuits. Neuroendocr Sci. 2013;7:19. doi: 10.3389/fnins.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobrino Crespo C, Perianes Cachero A, Puebla Jiménez L, Barrios V, Arilla Ferreiro E. Peptides and food intake. Diabetes. 2014;5:58. doi: 10.3389/fendo.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001 May 24;411(6836):480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 7.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996 May 30;381(6581):415–8. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 8.Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, et al. Neither Agouti-Related Protein nor Neuropeptide Y Is Critically Required for the Regulation of Energy Homeostasis in Mice. Mol Cell Biol. 2002 Jul 15;22(14):5027–35. doi: 10.1128/MCB.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, et al. Postembryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J. 2005 Aug 11; doi: 10.1096/fj.04-3434fje. [DOI] [PubMed] [Google Scholar]
- 10.Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, et al. Agouti-related peptide–expressing neurons are mandatory for feeding. Nat Neurosci. 2005 Oct;8(10):1289–91. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 11.Wu Q, Palmiter RD. GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur J Pharmacol. 2011 Jun 11;660(1):21–7. doi: 10.1016/j.ejphar.2010.10.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krashes MJ, Shah BP, Koda S, Lowell BB. Rapid versus Delayed Stimulation of Feeding by the Endogenously Released AgRP Neuron Mediators GABA, NPY, and AgRP. Cell Metab. 2013 Oct 1;18(4):588–95. doi: 10.1016/j.cmet.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dicken MS, Hughes AR, Hentges ST. Gad1 mRNA as a reliable indicator of altered GABA release from orexigenic neurons in the hypothalamus. Eur J Neurosci. 2015 Nov 1;42(9):2644–53. doi: 10.1111/ejn.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012 Aug 9;488(7410):172–7. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher JM, Sossin W, Newcomb R, Scheller RH. Multiple neuropeptides derived from a common precursor are differentially packaged and transported. Cell. 1988 Sep 9;54(6):813–22. doi: 10.1016/s0092-8674(88)91131-2. [DOI] [PubMed] [Google Scholar]
- 16.Sossin WS, Sweet-Cordero A, Scheller RH. Dale's hypothesis revisited: different neuropeptides derived from a common prohormone are targeted to different processes. Proc Natl Acad Sci U S A. 1990 Jun;87(12):4845–8. doi: 10.1073/pnas.87.12.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015 Mar 5;519(7541):45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schöne C, Burdakov D. Glutamate and GABA as rapid effectors of hypothalamic “peptidergic” neurons. Front Behav Neurosci. 2012;6:81. doi: 10.3389/fnbeh.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jarvie BC, Hentges ST. Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol. 2012 Dec 1;520(17):3863–76. doi: 10.1002/cne.23127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wittmann G, Hrabovszky E, Lechan RM. Distinct glutamatergic and GABAergic subsets of hypothalamic proopiomelanocortin neurons revealed by in situ hybridization in male rats and mice. J Comp Neurol. 2013 Oct 1;521(14):3287–302. doi: 10.1002/cne.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennison CS, King CM, Dicken MS, Hentges ST. Age-dependent changes in amino acid phenotype and the role of glutamate release from hypothalamic proopiomelanocortin neurons. J Comp Neurol. 2015 Sep 1;:n/a–n/a. doi: 10.1002/cne.23900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Padilla SL, Carmody JS, Zeltser LM. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nat Med. 2010 Apr;16(4):403–5. doi: 10.1038/nm.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999 Dec 9;402(6762):656–60. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 24.Kojima M, Kangawa K. Ghrelin: Structure and Function. Physiol Rev. 2005 Jan 4;85(2):495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- 25.Cummings DE. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol Behav. 2006 Aug 30;89(1):71–84. doi: 10.1016/j.physbeh.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Müller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin Mol Metab. 2015 Jun;4(6):437–60. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006 Jan 20;494(3):528–48. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mason BL, Wang Q, Zigman JM. The Central Nervous System Sites Mediating the Orexigenic Actions of Ghrelin. Annu Rev Physiol. 2014;76(1):519–33. doi: 10.1146/annurev-physiol-021113-170310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabral A, De Francesco PN, Perello M. Brain circuits mediating the orexigenic action of peripheral ghrelin: narrow gates for a vast kingdom. Neuroendocr Sci. 2015;6:44. doi: 10.3389/fendo.2015.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyda TD, Smith PM, Ferguson AV. Gastrointestinal hormone actions in the central regulation of energy metabolism: potential sensory roles for the circumventricular organs. Int J Obes. 2009;33(S1):S16–21. doi: 10.1038/ijo.2009.11. [DOI] [PubMed] [Google Scholar]
- 31.Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci. 2013 Jan 22;110(4):1512–7. doi: 10.1073/pnas.1212137110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fry M, Ferguson AV. The sensory circumventricular organs: brain targets for circulating signals controlling ingestive behavior. Physiol Behav. 2007 Jul 24;91(4):413–23. doi: 10.1016/j.physbeh.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology. 2004 Jun;145(6):2607–12. doi: 10.1210/en.2003-1596. [DOI] [PubMed] [Google Scholar]
- 34.Willesen MG, Kristensen P, Rømer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology. 1999 Nov;70(5):306–16. doi: 10.1159/000054491. [DOI] [PubMed] [Google Scholar]
- 35.Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003 Feb 20;37(4):649–61. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 36.Cabral A, Fernandez G, Perello M. Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience. 2013 Dec 3;253:406–15. doi: 10.1016/j.neuroscience.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faulconbridge LF, Cummings DE, Kaplan JM, Grill HJ. Hyperphagic effects of brainstem ghrelin administration. Diabetes. 2003 Sep;52(9):2260–5. doi: 10.2337/diabetes.52.9.2260. [DOI] [PubMed] [Google Scholar]
- 38.Lawrence CB, Snape AC, Baudoin FMH, Luckman SM. Acute Central Ghrelin and GH Secretagogues Induce Feeding and Activate Brain Appetite Centers. Endocrinology. 2002 Jan 1;143(1):155–62. doi: 10.1210/endo.143.1.8561. [DOI] [PubMed] [Google Scholar]
- 39.Scott MM, Perello M, Chuang JC, Sakata I, Gautron L, Lee CE, et al. Hindbrain Ghrelin Receptor Signaling Is Sufficient to Maintain Fasting Glucose. PLoS ONE. 2012 Aug 31;7(8):e44089. doi: 10.1371/journal.pone.0044089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Björkhem I. Five decades with oxysterols. Biochimie. 2013 Mar;95(3):448–54. doi: 10.1016/j.biochi.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, et al. Activation of the Nuclear Receptor LXR by Oxysterols Defines a New Hormone Response Pathway. J Biol Chem. 1997 Jul 2;272(6):3137–40. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- 42.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, et al. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc Natl Acad Sci. 1999 May 1;96(1):266–71. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jakobsson T, Treuter E, Gustafsson JÅ, Steffensen KR. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012 Jul 1;33(7):394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Tontonoz P, Mangelsdorf DJ. Liver X Receptor Signaling Pathways in Cardiovascular Disease. Mol Endocrinol. 2003 Jun 1;17(6):985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 45.Bonamassa B, Moschetta A. Atherosclerosis: lessons from LXR and the intestine. Trends Endocrinol Metab. 2013 Mar 1;24(3):120–8. doi: 10.1016/j.tem.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci. 2002 Mar 9;99(18):11896–901. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang SH, Burant CF, et al. LXRβ Is Required for Adipocyte Growth, Glucose Homeostasis, and β Cell Function. J Biol Chem. 2005 Jun 17;280(24):23024–31. doi: 10.1074/jbc.M412564200. [DOI] [PubMed] [Google Scholar]
- 48.Cao G, Liang Y, Broderick CL, Oldham BA, Beyer TP, Schmidt RJ, et al. Antidiabetic Action of a Liver X Receptor Agonist Mediated By Inhibition of Hepatic Gluconeogenesis. J Biol Chem. 2003 Oct 1;278(2):1131–6. doi: 10.1074/jbc.M210208200. [DOI] [PubMed] [Google Scholar]
- 49.Chisholm JW, Hong J, Mills SA, Lawn RM. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res. 2003 Jan 11;44(11):2039–48. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Grefhorst A, van Dijk TH, Hammer A, van der Sluijs FH, Havinga R, Havekes LM, et al. Differential effects of pharmacological liver X receptor activation on hepatic and peripheral insulin sensitivity in lean and ob/ob mice. Am J Physiol - Endocrinol Metab. 2005 Nov 1;289(5):E829–38. doi: 10.1152/ajpendo.00165.2005. [DOI] [PubMed] [Google Scholar]
- 51.Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, et al. Regulation of Cholesterol Homeostasis by the Liver X Receptors in the Central Nervous System. Mol Endocrinol. 2002 Jun 1;16(6):1378–85. doi: 10.1210/mend.16.6.0835. [DOI] [PubMed] [Google Scholar]
- 52.Kruse MS, Rey M, Vega MC, Coirini H. Alterations of LXRα and LXRβ expression in the hypothalamus of glucose-intolerant rats. J Endocrinol. 2012 Jan 10;215(1):51–8. doi: 10.1530/JOE-12-0088. [DOI] [PubMed] [Google Scholar]
- 53.Kruse MS, Vega MC, Rey M, Coirini H. Sex differences in LXR expression in normal offspring and in rats born to diabetic dams. J Endocrinol. 2014 Jan 7;222(1):53–60. doi: 10.1530/JOE-14-0054. [DOI] [PubMed] [Google Scholar]
- 54.Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998 Jan 8;39(8):1594–600. [PubMed] [Google Scholar]
- 55.Ohyama Y, Meaney S, Heverin M, Ekström L, Brafman A, Shafir M, et al. Studies on the Transcriptional Regulation of Cholesterol 24-Hydroxylase (CYP46A1) Marked Insensitivity Toward Different Regulatory Axes. J Biol Chem. 2006 Feb 17;281(7):3810–20. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- 56.Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007 Jan 11;445(7124):219–23. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 57.Denechaud PD, Bossard P, Lobaccaro JMA, Millatt L, Staels B, Girard J, et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. [cited 2015 Nov 28];J Clin Invest [] 2008 Feb 1; doi: 10.1172/JCI34314. Internet. Available from: http://www.jci.org/articles/view/34314. [DOI] [PMC free article] [PubMed]
- 58.Bantubungi K, Prawitt J, Staels B. Control of metabolism by nutrient-regulated nuclear receptors acting in the brain. J Steroid Biochem Mol Biol. 2012 Jul;130(3–5):126–37. doi: 10.1016/j.jsbmb.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 59.Kruse MS, Rey M, Coirini H. Abstracts of the XXXII Annual Meeting - Sociedad De BiologÍA De Cuyo - XXXII Annual Meeting of the Cuyo Biology Society. 2015;39(2) [Google Scholar]
- 60.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JMA, Hammer RE, et al. Cholesterol and Bile Acid Metabolism Are Impaired in Mice Lacking the Nuclear Oxysterol Receptor LXRα. Cell. 1998 May 29;93(5):693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 61.Woodside B. Prolactin and the hyperphagia of lactation. Physiol Behav. 2007 Jul 24;91(4):375–82. doi: 10.1016/j.physbeh.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 62.García MC, López M, Gualillo O, Seoane LM, Diéguez C, Señarís RM. Hypothalamic levels of NPY, MCH, and prepro-orexin mRNA during pregnancy and lactation in the rat: role of prolactin. FASEB J. 2003 Jan 8;17(11):1392–400. doi: 10.1096/fj.02-0933com. [DOI] [PubMed] [Google Scholar]
- 63.Naef L, Woodside B. Prolactin/Leptin Interactions in the Control of Food Intake in Rats. Endocrinology. 2007 Dec 1;148(12):5977–83. doi: 10.1210/en.2007-0442. [DOI] [PubMed] [Google Scholar]
- 64.Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: Structure, Function, and Regulation of Secretion. Physiol Rev. 2000 Jan 10;80(4):1523–631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- 65.Walsh RJ, Slaby FJ, Posner BI. A Receptor-Mediated Mechanism for the Transport of Prolactin from Blood to Cerebrospinal Fluid. Endocrinology. 1987 May 1;120(5):1846–50. doi: 10.1210/endo-120-5-1846. [DOI] [PubMed] [Google Scholar]
- 66.Ben-Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR. Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab. 2006 Apr 1;17(3):110–6. doi: 10.1016/j.tem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 67.Byatt JC, Staten NR, Salsgiver WJ, Kostelc JG, Collier RJ. Stimulation of food intake and weight gain in mature female rats by bovine prolactin and bovine growth hormone. Am J Physiol - Endocrinol Metab. 1993 Jun 1;264(6):E986–92. doi: 10.1152/ajpendo.1993.264.6.E986. [DOI] [PubMed] [Google Scholar]
- 68.Gerardo-Gettens T, Moore BJ, Stern JS, Horwitz BA. Prolactin stimulates food intake in a dose-dependent manner. Am J Physiol - Regul Integr Comp Physiol. 1989 Jan 1;256(1):R276–80. doi: 10.1152/ajpregu.1989.256.1.R276. [DOI] [PubMed] [Google Scholar]
- 69.Freemark M, Fleenor D, Driscoll P, Binart N, Kelly PA. Body Weight and Fat Deposition in Prolactin Receptor-Deficient Mice. Endocrinology. 2001 Feb 1;142(2):532–7. doi: 10.1210/endo.142.2.7979. [DOI] [PubMed] [Google Scholar]
- 70.Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, et al. Pituitary Lactotroph Hyperplasia and Chronic Hyperprolactinemia in Dopamine D2 Receptor-Deficient Mice. Neuron. 1997 Jul 1;19(1):103–13. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 71.Cristina C, Díaz-Torga G, Baldi A, Góngora A, Rubinstein M, Low MJ, et al. Increased Pituitary Vascular Endothelial Growth Factor-A in Dopaminergic D2 Receptor Knockout Female Mice. Endocrinology. 2005 Jul 1;146(7):2952–62. doi: 10.1210/en.2004-1445. [DOI] [PubMed] [Google Scholar]
- 72.García-Tornadú I, Díaz-Torga G, Risso GS, Silveyra P, Cataldi N, Ramirez MC, et al. Hypothalamic orexin, OX1, αMSH, NPY and MCRs expression in dopaminergic D2R knockout mice. Neuropeptides. 2009 Aug 1;43(4):267–74. doi: 10.1016/j.npep.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 73.Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, et al. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011 Aug;14(8):1033–8. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010 May;13(5):635–41. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.García-Tornadu I, Risso G, Perez-Millan MI, Noain D, Diaz-Torga G, Low MJ, et al. Neurotransmitter Modulation of the GHRH-GH Axis. In: Arzt E, Bronstein M, Guitelman M, editors. Frontiers of Hormone Research. Basel: KARGER; 2010. pp. 59–69. [DOI] [PubMed] [Google Scholar]
- 76.Perez Millan MI, Luque GM, Ramirez MC, Noain D, Ornstein AM, Rubinstein M, et al. Selective Disruption of Dopamine D2 Receptors in Pituitary Lactotropes Increases Body Weight and Adiposity in Female Mice. Endocrinology. 2013 Jan 1;155(3):829–39. doi: 10.1210/en.2013-1707. [DOI] [PubMed] [Google Scholar]
- 77.Noaín D, Pérez-Millán MI, Bello EP, Luque GM, Cordero RC, Gelman DM, et al. Central Dopamine D2 Receptors Regulate Growth-Hormone-Dependent Body Growth and Pheromone Signaling to Conspecific Males. J Neurosci. 2013 Mar 27;33(13):5834–42. doi: 10.1523/JNEUROSCI.5673-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Augustine RA, Ladyman SR, Grattan DR. From feeding one to feeding many: hormone-induced changes in bodyweight homeostasis during pregnancy. J Physiol. 2008 Jan 15;586(2):387–97. doi: 10.1113/jphysiol.2007.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fleenor D, Arumugam R, Freemark M. Growth Hormone and Prolactin Receptors in Adipogenesis: STAT-5 Activation, Suppressors of Cytokine Signaling, and Regulation of Insulin-Like Growth Factor I. Horm Res. 2006;66(3):101–10. doi: 10.1159/000093667. [DOI] [PubMed] [Google Scholar]
- 80.Brandebourg TD, Bown JL, Ben-Jonathan N. Prolactin upregulates its receptors and inhibits lipolysis and leptin release in male rat adipose tissue. Biochem Biophys Res Commun. 2007 Jun 1;357(2):408–13. doi: 10.1016/j.bbrc.2007.03.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996 Mar;12(3):318–20. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- 82.Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996 Jul 1;137(7):3144–7. doi: 10.1210/endo.137.7.8770941. [DOI] [PubMed] [Google Scholar]
- 83.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996 Feb 15;379(6566):632–5. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 84.Yamaoka K, Saharinen P, Pesu M, Holt VE, Silvennoinen O, O'Shea JJ. The Janus kinases (Jaks) Genome Biol. 2004 Nov 30;5(12):253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Myers MG. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. 2004;59:287–304. doi: 10.1210/rp.59.1.287. [DOI] [PubMed] [Google Scholar]
- 86.Bjørbæk C, Uotani S, Silva B da, Flier JS. Divergent Signaling Capacities of the Long and Short Isoforms of the Leptin Receptor. J Biol Chem. 1997 Dec 19;272(51):32686–95. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 87.Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998 Mar 26;392(6674):398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 88.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003 Feb 20;421(6925):856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 89.Boulton TG, Zhong Z, Wen Z, Darnell JE, Stahl N, Yancopoulos GD. STAT3 activation by cytokines utilizing gp130 and related transducers involves a secondary modification requiring an H7-sensitive kinase. Proc Natl Acad Sci U S A. 1995 Jul 18;92(15):6915–9. doi: 10.1073/pnas.92.15.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic estrogen mimics leptin's effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007 Jan;13(1):89–94. doi: 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- 91.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase–phosphodiesterase 3B–cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002 Aug;5(8):727–8. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 92.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Schwartz MW. Intracellular signalling: Key enzyme in leptin-induced anorexia. Nature. 2001 Oct 25;413(6858):794–5. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 93.Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005 Apr 1;115(4):951–8. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Allison MB, Myers MG. 20 YEARS OF LEPTIN: Connecting leptin signaling to biological function. J Endocrinol. 2014 Jan 10;223(1):T25–35. doi: 10.1530/JOE-14-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000 Aug;3(8):757–8. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- 96.Patterson CM, Villanueva EC, Greenwald-Yarnell M, Rajala M, Gonzalez IE, Saini N, et al. Leptin action via LepR-b Tyr1077 contributes to the control of energy balance and female reproduction. Mol Metab. 2012 Jul 26;1(1-2):61–9. doi: 10.1016/j.molmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singireddy AV, Inglis MA, Zuure WA, Kim JS, Anderson GM. Neither Signal Transducer and Activator of Transcription 3 (STAT3) or STAT5 Signaling Pathways Are Required for Leptin's Effects on Fertility in Mice. Endocrinology. 2013 May 21;154(7):2434–45. doi: 10.1210/en.2013-1109. [DOI] [PubMed] [Google Scholar]
- 98.Björnholm M, Münzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, et al. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007 May 1;117(5):1354–60. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burgos-Ramos E, Chowen JA, Arilla-Ferreiro E, Canelles S, Argente J, Barrios V. Chronic central leptin infusion modifies the response to acute central insulin injection by reducing the interaction of the insulin receptor with IRS2 and increasing its association with SOCS3. J Neurochem. 2011 Apr 1;117(1):175–85. doi: 10.1111/j.1471-4159.2011.07191.x. [DOI] [PubMed] [Google Scholar]
- 100.White MF. IRS proteins and the common path to diabetes. Am J Physiol - Endocrinol Metab. 2002 Sep 1;283(3):E413–22. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 101.Pardini AW, Nguyen HT, Figlewicz DP, Baskin DG, Williams DL, Kim F, et al. Distribution of insulin receptor substrate-2 in brain areas involved in energy homeostasis. Brain Res. 2006 Sep 27;1112(1):169–78. doi: 10.1016/j.brainres.2006.06.109. [DOI] [PubMed] [Google Scholar]
- 102.Burks DJ, de Mora JF, Schubert M, Withers DJ, Myers MG, Towery HH, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000 Sep 21;407(6802):377–82. doi: 10.1038/35030105. [DOI] [PubMed] [Google Scholar]
- 103.Sadagurski M, Leshan RL, Patterson C, Rozzo A, Kuznetsova A, Skorupski J, et al. IRS2 Signaling in LepR-b Neurons Suppresses FoxO1 to Control Energy Balance Independently of Leptin Action. Cell Metab. 2012 May 2;15(5):703–12. doi: 10.1016/j.cmet.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002 May 31;296(5573):1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 105.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005 Apr 1;30(4):194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 106.Hörsch D, Kahn CR. Region-specific mRNA expression of phosphatidylinositol 3-kinase regulatory isoforms in the central nervous system of C57BL/6J mice. J Comp Neurol. 1999 Dec 6;415(1):105–20. [PubMed] [Google Scholar]
- 107.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome Off J Int Mamm Genome Soc. 2002 Mar;13(3):169–72. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 108.Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI, et al. Dominant Role of the p110β Isoform of PI3K over p110α in Energy Homeostasis Regulation by POMC and AgRP Neurons. Cell Metab. 2009 Nov 4;10(5):343–54. doi: 10.1016/j.cmet.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Geer EB, Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med. 2009;6(1):60–75. doi: 10.1016/j.genm.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roth SM, Ferrell RE, Peters DG, Metter EJ, Hurley BF, Rogers MA. Influence of age, sex, and strength training on human muscle gene expression determined by microarray. Physiol Genomics. 2002 Sep 3;10(3):181–90. doi: 10.1152/physiolgenomics.00028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Paula FJ, Pimenta WP, Saad MJ, Paccola GM, Piccinato CE, Foss MC. Sex-related differences in peripheral glucose metabolism in normal subjects. Diabète Métabolisme. 1990 Jun;16(3):234–9. [PubMed] [Google Scholar]
- 112.Arciero PJ, Goran MI, Poehlman ET. Resting metabolic rate is lower in women than in men. J Appl Physiol. 1993 Dec 1;75(6):2514–20. doi: 10.1152/jappl.1993.75.6.2514. [DOI] [PubMed] [Google Scholar]
- 113.Uranga AP, Levine J, Jensen M. Isotope tracer measures of meal fatty acid metabolism: reproducibility and effects of the menstrual cycle. Am J Physiol - Endocrinol Metab. 2005 Mar 1;288(3):E547–55. doi: 10.1152/ajpendo.00340.2004. [DOI] [PubMed] [Google Scholar]
- 114.De Maddalena C, Vodo S, Petroni A, Aloisi AM. Impact of testosterone on body fat composition. J Cell Physiol. 2012 Dec 1;227(12):3744–8. doi: 10.1002/jcp.24096. [DOI] [PubMed] [Google Scholar]
- 115.Corona G, Monami M, Rastrelli G, Aversa A, Sforza A, Lenzi A, et al. Type 2 diabetes mellitus and testosterone: a meta-analysis study. Int J Androl. 2011 Dec 1;34(6pt1):528–40. doi: 10.1111/j.1365-2605.2010.01117.x. [DOI] [PubMed] [Google Scholar]
- 116.Escobar-Morreale HF, Álvarez-Blasco F, Botella-Carretero JI, Luque-Ramírez M. The striking similarities in the metabolic associations of female androgen excess and male androgen deficiency. Hum Reprod. 2014 Oct 10;29(10):2083–91. doi: 10.1093/humrep/deu198. [DOI] [PubMed] [Google Scholar]
- 117.Pitteloud N, Mootha VK, Dwyer AA, Hardin M, Lee H, Eriksson KF, et al. Relationship Between Testosterone Levels, Insulin Sensitivity, and Mitochondrial Function in Men. Diabetes Care. 2005 Jan 7;28(7):1636–42. doi: 10.2337/diacare.28.7.1636. [DOI] [PubMed] [Google Scholar]
- 118.Law J, Bloor I, Budge H, Symonds ME. The influence of sex steroids on adipose tissue growth and function. Horm Mol Biol Clin Investig. 2014;19(1):13–24. doi: 10.1515/hmbci-2014-0015. [DOI] [PubMed] [Google Scholar]
- 119.Lund TD, Munson DJ, Haldy ME, Handa RJ. Dihydrotestosterone may inhibit hypothalamo–pituitary–adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett. 2004 Jul 15;365(1):43–7. doi: 10.1016/j.neulet.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 120.Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003 Jan 3;300(1):167–71. doi: 10.1016/s0006-291x(02)02774-2. [DOI] [PubMed] [Google Scholar]
- 121.Fan W, Yanase T, Nishi Y, Chiba S, Okabe T, Nomura M, et al. Functional Potentiation of Leptin-Signal Transducer and Activator of Transcription 3 Signaling by the Androgen Receptor. Endocrinology. 2008 Dec 1;149(12):6028–36. doi: 10.1210/en.2008-0431. [DOI] [PubMed] [Google Scholar]
- 122.Yu IC, Lin HY, Liu NC, Sparks JD, Yeh S, Fang LY, et al. Neuronal Androgen Receptor Regulates Insulin Sensitivity via Suppression of Hypothalamic NF-κB–Mediated PTP1B Expression. Diabetes. 2013 Jan 2;62(2):411–23. doi: 10.2337/db12-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nohara K, Laque A, Allard C, Münzberg H, Mauvais-Jarvis F. Central mechanisms of adiposity in adult female mice with androgen excess. Obesity. 2014 Jun 1;22(6):1477–84. doi: 10.1002/oby.20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lovejoy JC, Champagne CM, de Jonge L, Xie H, Smith SR. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int J Obes. 2008 Mar 11;32(6):949–58. doi: 10.1038/ijo.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maliqueo M, Sun M, Johansson J, Benrick A, Labrie F, Svensson H, et al. Continuous Administration of a P450 Aromatase Inhibitor Induces Polycystic Ovary Syndrome with a Metabolic and Endocrine Phenotype in Female Rats at Adult Age. Endocrinology. 2012 Nov 26;154(1):434–45. doi: 10.1210/en.2012-1693. [DOI] [PubMed] [Google Scholar]
- 126.Simpson ER, Jones ME. Of mice and men: the many guises of estrogens. Ernst Schering Found Symp Proc. 2006;(1):45–67. doi: 10.1007/2789_2006_016. [DOI] [PubMed] [Google Scholar]
- 127.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci. 2000 Jul 11;97(23):12729–34. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roesch DM. Effects of selective estrogen receptor agonists on food intake and body weight gain in rats. Physiol Behav. 2006 Jan 30;87(1):39–44. doi: 10.1016/j.physbeh.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 129.Yepuru M, Eswaraka J, Kearbey JD, Barrett CM, Raghow S, Veverka KA, et al. Estrogen Receptor-β-selective Ligands Alleviate High-fat Diet- and Ovariectomy-induced Obesity in Mice. J Biol Chem. 2010 Aug 10;285(41):31292–303. doi: 10.1074/jbc.M110.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, et al. Metabolic Actions of Estrogen Receptor Beta (ERβ) are Mediated by a Negative Cross-Talk with PPARγ. PLoS Genet. 2008 Jun 27;4(6):e1000108. doi: 10.1371/journal.pgen.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Barros RPA, Machado UF, Warner M, Gustafsson JÅ. Muscle GLUT4 regulation by estrogen receptors ERβ and ERα. Proc Natl Acad Sci. 2006 Jan 31;103(5):1605–8. doi: 10.1073/pnas.0510391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brock O, De Mees C, Bakker J. Hypothalamic Expression of Oestrogen Receptor α and Androgen Receptor is Sex-, Age- and Region-Dependent in Mice. J Neuroendocrinol. 2015 Apr 1;27(4):264–76. doi: 10.1111/jne.12258. [DOI] [PubMed] [Google Scholar]
- 133.Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, et al. Distinct Hypothalamic Neurons Mediate Estrogenic Effects on Energy Homeostasis and Reproduction. Cell Metab. 2011 Oct 5;14(4):453–65. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Asarian L, Geary N. Cyclic Estradiol Treatment Normalizes Body Weight and Restores Physiological Patterns of Spontaneous Feeding and Sexual Receptivity in Ovariectomized Rats. Horm Behav. 2002 Dec;42(4):461–71. doi: 10.1006/hbeh.2002.1835. [DOI] [PubMed] [Google Scholar]
- 135.Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci. 2007 Feb 13;104(7):2501–6. doi: 10.1073/pnas.0610787104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State. Endocrinology. 2013 Aug 22;154(11):4136–45. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Park CJ, Zhao Z, Glidewell-Kenney C, Lazic M, Chambon P, Krust A, et al. Genetic rescue of nonclassical ERα signaling normalizes energy balance in obese Erα-null mutant mice. J Clin Invest. 2011 Feb 1;121(2):604–12. doi: 10.1172/JCI41702. [DOI] [PMC free article] [PubMed] [Google Scholar]