

Abstract
Nod-like receptors (NLRs) have gained attention in recent years because of the ability of some family members to assemble into a multimeric protein complex known as the inflammasome. The role of NLRs and the inflammasome in regulating innate immunity against bacterial pathogens has been well studied. However, recent studies show that NLRs and inflammasomes also play a role during infections caused by protozoan parasites, which pose a significant global health burden. Herein, we review the diseases caused by the most common protozoan parasites in the world and discuss the roles of NLRs and inflammasomes in host immunity against these parasites.
Keywords: NLR, Inflammasome, Parasites, Protozoa, NOD, NLRP3
Introduction
Protozoa are unicellular eukaryotic microorganisms that can be free-living or parasitic. Although infection with most of the protozoa is harmless, some protozoan infections can be detrimental to human and animal health. In recent years, Plasmodium species, the causative agent of malaria, has garnered much attention because of the devastating effect of these protozoa in the infected host. Unfortunately, many other protozoan parasites that infect humans are still poorly understood and are categorized as neglected tropical disease by the World Health Organization (WHO). Although immune responses against some protozoan parasites have been relatively well studied, the roles of NLRs in regulating innate and adaptive immune responses against most of these parasites are only beginning to be understood. Here, we review immune responses and the roles of NLRs, inflammasomes and associated cytokines IL-1 and IL-18 in modulating adaptive immune responses during protozoan infections.
Nod-like receptors
Nucleotide-binding oligomerization domain receptors [Nod-like receptors (NLRs)] are cytoplasmic sensors that sense pathogen-associated molecular patterns (pathogens/foreign) or damage-associated molecular patterns (cells/self). To date, 22 NLRs in humans and 34 NLRs in mice have been characterized [1]. On the basis of the NLRs whose functions have been reported, NLRs can be classified into four major functional subgroups: (1) positive regulators of signaling pathways or inflammation, (2) negative regulators of signaling pathways or inflammation, (3) regulators involved in the formation of the inflammasome complex, and (4) regulators of transcription (Fig. 1). NLRC1 (NOD1) and NLRC2 (NOD2), which contain a caspase recruitment domain (CARD), were identified because of their ability to activate NFκB and mitogen-activated protein kinase (MAPK) in response to bacterial ligands [2–5]. NLRC3 [6], NLRC5 [7], NLRP6 [8], NLRP12 [9, 10], and NLRX1 (NOD5) [11, 12] negatively regulate the NFκB and MAPK signaling pathways. CIITA (NLRA) and NLRC5 are transcriptional activators of the major histocompatibility class II and class I molecules, respectively [13, 14]. Recent findings show that NLRP3 transcriptionally activates T-helper 2 (Th2) genes in T cells [15].
Fig. 1.
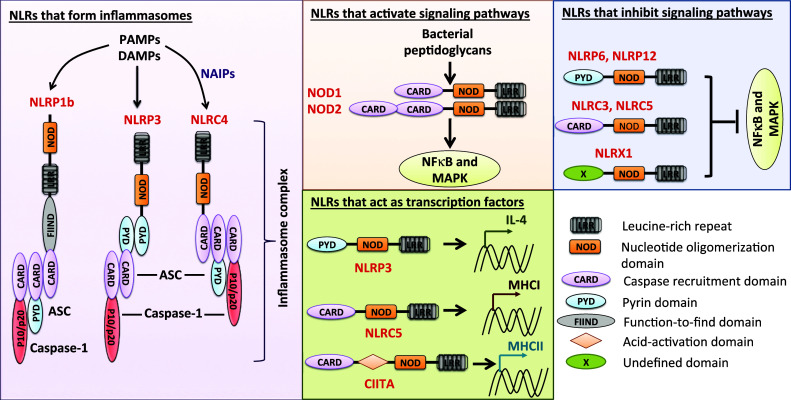
Diverse functions of NLR proteins. NLRs that form inflammasomes: NLRP1b, NLRP3 and NLRC4, upon sensing pathogen or damage signals, recruit ASC and caspase-1 to assemble the inflammasome complex. This inflammasome complex is crucial for activation of caspase-1 and ultimate cleavage of pro-IL-1β and pro-IL-18 into their mature forms. NLRs that activate signaling pathways: NOD1 (NLRC1) and NOD2 (NLRC2) recognize muramyl dipeptide and diaminopimelic acid from bacterial peptidoglycan and recruit downstream common adaptor receptor protein interacting kinase 2 to activate the NFκB and mitogen-activated protein kinase (MAPK) signaling pathways. NLRs that inhibit signaling pathways: NLRP6, NLRP12, NLRC3, NLRC5, and NLRX1 are negative regulators of NFκB and MAPK signaling pathways. NLRs that act as transcription factors: NLRP3 act as transcriptional regulator of IL-4. NLRC5 acts as a transcriptional activator of major histocompatibility complex (MHC) class I molecules, and CIITA has a well-established role as a transcriptional activator of MHC class II proteins
NLRP1b, NLRP3, and NLRC4 have well-established functions in forming the inflammasome, a multimeric protein complex that plays a central role in innate immunity [16–20]. More recently, NLRP6 and NLRP12 have also been implicated to form inflammasomes [21, 22], although additional independent and biochemical studies are needed to establish these findings. Upon sensing danger signals (self or foreign), these NLRs recruit caspase-1 and the apoptosis-associated speck-like protein containing a carboxy-terminal CARD (ASC) to form the inflammasome. Within the inflammasome complex, caspase-1 gets autocleaved and activated. Activated caspase-1 can then process pro-interleukin (IL)-1β and pro-IL-18 into their mature forms and induce pyroptotic cell death. Because of the known widespread functions of cytokines IL-1 and IL-18, NLRs that are associated with activation of the inflammasome complex are currently a major area of research.
Amebiasis
Amebiasis is a worldwide health concern caused by the enteric protozoan parasite Entamoeba histolytica and affects more than 100 million people, causing 100,000 deaths per year [23]. Most infected individuals are asymptomatic, but diarrhea, dysentery, colitis, and liver abscesses can occur [24, 25]. The disease is transmitted mainly through the ingestion of E. histolytica cysts under conditions of poor sanitation and contaminated water [25]. The results of a longitudinal study of a cohort of children in Bangladesh suggest that infant malnutrition is a major factor that predisposes young children to amebiasis [26, 27]. The treatment of amebiasis is usually limited to pan antibiotics such as metronidazole, which are often associated with severe side effects. A comprehensive understanding of the immune responses that are generated against E. histolytica is lacking [28].
Immune responses against E. histolytica
E. histolytica contains several virulence factors that allow it to persist and survive within the host. Although more than 90 % of E. histolytica infections are asymptomatic and remain as commensals within the host’s gut environment, under certain circumstances, the parasite invades the host epithelium and becomes pathogenic [27]. Some of the well-known key virulence factors of E. histolytica are Gal-lectin (the 170-kDa surface Gal/GalNAc lectin of E. histolytica), cysteine proteases, peroxiredoxin, and lipopeptidophosphoglycan (LPPG) [25, 29]. Although these virulence factors provide key survival advantages for E. histolytica in the infected host, they also activate the host’s immune responses.
The intestinal tract provides the first line of defense, where mucus prevents ameba penetrance into the intraepithelial cells. Gal-lectin of E. histolytica binds mucosal matrix and uses its cysteine proteases to cleave mucin to gain access to the intestinal epithelial cells (IECs) [30, 31]. Mice deficient in mucin (MUC2 knockout mice) are indeed highly susceptible to E. histolytica infection, further demonstrating the importance of the intestinal mucin coating [32]. The intestinal tract is also filled with antimicrobial compounds and secretory immunoglobulin A (sIgA) that provide additional protection against E. histolytica. As such, deficiency in antimicrobial protein REG1 or sIgA renders the host highly susceptible to E. histolytica infection [33, 34].
Once the initial luminal barrier is breached, the IECs respond to E. histolytica to eliminate infection. IECs recognize LPPG associated with E. histolytica and produce several chemokines and cytokines, including TNF-α, IL-6, and GM-CSF, to recruit neutrophils and monocytes [35]. In addition to actively secreting inflammatory cytokines, IEC death also promotes inflammation [35]. Neutrophils are rapidly recruited to the site of infection to combat infection once intestinal epithelial barrier integrity is breached [36, 37]. Thus, neutrophil-depleted mice have more intestinal lesions and higher susceptibility to E. histolytica than do wild-type (WT) mice [38]. Neutrophils are also important for establishing early resistance to hepatic amebiasis [39].
Similar to neutrophils, macrophages also play an important role during E. histolytica infection. Macrophages express Toll-like receptors (TLRs) that can recognize E. histolytica virulence factors to initiate amebicidal immune responses. TLR2 and TLR4 recognize E. histolytica LPPG, whereas TLR9 recognizes E. histolytica DNA [40, 41]. Cytokines such as interferon (IFN)-γ further activate macrophages to produce nitric oxide synthase (NOS) and prevent E. histolytica infection [42, 43]. LPPG also activates dendritic cells through TLRs, increasing their expression of costimulatory molecules such as CD80, CD86, and CD40 and promoting cytokine production [44]. NK and NKT cells are not important for intestinal immunity but are necessary for protection against E. histolytica liver colonization [45, 46]. These cells secrete IFN-γ and tumor necrosis factor (TNF)-α, factors that are critical for activating innate immune cells, including neutrophils and macrophages [45, 46]. Mice lacking NKT cells are highly susceptible to E. histolytica colonization in the liver and subsequent liver abscesses [45]. Deficiency of either IFN-γ or NOS results in severe liver infection with E. histolytica, demonstrating the importance of these critical cytokines [42, 47, 48].
With regard to adaptive immunity, protection from or resistance to E. histolytica also seem to depend on Th1 responses. Patients who were infected with Entamoeba but were asymptomatic had higher levels of IFN-γ, suggesting a protective role for Th1 responses [49]. In contrast, patients with invasive amebiasis had increased levels of the Th2-associated cytokine IL-4 [49]. Consistent with these correlative studies, the results of studies in mice showed that high IL-4 production by T cells plays an important role in pathogenicity and susceptibility during E. histolytica infection [50]. Conversely, production of IFN-γ by T cells is associated with protection against E. histolytica infection [50]. More importantly, the protection observed in susceptible mice immunized with Gal-lectin was due to the production of IFN-γ and IL-17 by CD4+ and CD8+ T cells [51]. Taken together, these results demonstrate that protection against E. histolytica requires a balance of Th1 and Th2 responses.
E. histolytica infection activates NLRP3 inflammasomes and IL-1 cytokine production
The role of NLRs, inflammasomes, and IL-1 and IL-18 during E. histolytica infection has not been thoroughly studied. The results of in vitro studies of cultured human epithelial and stromal cells as well as cell lines show that E. histolytica induces the robust production of IL-1α, which acts in a paracrine manner to induce strong cytokine production and inflammation [52]. The results of other studies show that the cysteine proteinase EhCP5 of E. histolytica can cleave and inactivate recombinant IL-1β and IL-18 [53, 54]. Furthermore, the monocyte locomotion inhibitory factor produced by E. histolytica downregulates Il1b expression [55]. Taken together, these results suggest a protective role for IL-1β and IL-18 in immunity against E. histolytica, which may be why this protozoan uses various strategies to neutralize these cytokines.
Although studies of NLRs are scarce, the results of studies by one group show that E. histolytica activates the inflammasome in vitro and in vivo (Fig. 2a). The Gal-lectin of E. histolytica induces robust caspase-1 activation and subsequent IL-1β and IL-18 production in mouse macrophages [56]. Similarly, the binding of E. histolytica to the integrin α5β1 recruits cysteine proteinase EhCP5, which activates the NLRP3 inflammasome and, subsequently, IL-1β and IL-18 in vitro and ex vivo [57]. Given that Gal-lectin induces inflammasome activation [56] and protective CD4 and CD8 T cell responses [51], the NLRP3 inflammasome may be involved in regulating adaptive immune responses. Further studies are needed to directly evaluate the effects of NLRP3, inflammasomes, and IL-1 and IL-18 on adaptive immune responses during E. histolytica infection.
Fig. 2.
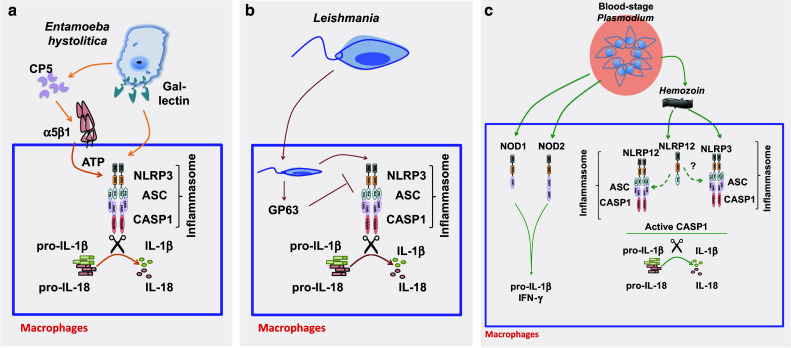
Role of the NLRP3 inflammasome during E. histolytica, Leishmania, and Plasmodium spp. infections. a The Entamoeba hystolitica Gal-lectin can directly activate the NLRP3 inflammasome through yet-unknown mechanisms. The Entamoeba protease CP5 can also engage the α5β1 integrin to modulate ATP and activate the NLRP3 inflammasome. b Leishmania activates the NLRP3 inflammasome to induce IL-1β and IL-18 production in vitro and in vivo. Leishmania-associated metalloprotease GP63 can inhibit NLRP3 inflammasomes by inhibiting ROS production and directly cleaving NLRP3. c Plasmodium spp. engage NOD1 and NOD2 to promote IL-1β and IFN-γ production. In addition, Plasmodium parasites can activate NLRP12 and NLRP3 inflammasomes through hemozoin. Whether NLRP12 is in complex with NLRP3 or forms independent inflammasomes is not known
Leishmaniasis
Leishmaniasis, a disease caused by Leishmania species, is a major health concern in tropical and subtropical regions around the world. More than 300 million people are at risk of this parasitic infection, including 12 million people currently infected and 1.5 million new cases per year [58–60]. Mouse models of Leishmania infections have been extremely useful in furthering our understanding of Th1 (hallmark cytokine, IFN-γ) and Th2 (hallmark cytokine, IL-4) responses [61]. L. major infection of C57BL/6 mice induces powerful Th1 responses that are dominated by IFN-γ production, leading to resistance in mice. However, L. major infection of BALB/c mice induces strong Th2 responses that are dominated by IL-4 production, making these mice highly susceptible to these infections. Thus, resistance or susceptibility to L. major infections is highly correlated with Th1 (IFN-γ) or Th2 (IL-4) responses, respectively [61].
Immune responses against Leishmania
The bite of an infected sandfly releases infectious Leishmania promastigotes in the skin, where they are taken up by phagocytic cells. The innate immune cells are critical for eliminating and clearing infections, but they also serve as a sanctuary for the obligate intracellular parasites. Neutrophils, macrophages, and dendritic cells play central roles in controlling and eliminating Leishmania. Neutrophils are one of the first cell types to be recruited to the site of infection and are constantly recruited to the parasitic lesions [62]. Depletion of neutrophils during L. braziliensis infection results in increased parasite load [63]. Similarly, infection of mice with Leishmania and neutrophils mixed together also leads to lower parasite burden both at the site of infection and within draining lymph nodes [63]. The neutrophil proteolytic enzyme elastase and neutrophil extracellular traps (NETs) have been proposed as mechanisms by which neutrophils kill and eliminate Leishmania [64, 65].
Similar to neutrophils, macrophages can phagocytize nascent Leishmania or Leishmania-infected dying cells. Macrophages are major effector cells that kill Leishmania parasites and, thus, are a major target for immune evasion by Leishmania. The inflammatory cytokines IFN-γ, IL-1, TNF-α, and type I IFNs activate macrophages to produce inducible nitric oxide synthase (iNOS) important for parasite killing. Indeed, mice deficient in iNOS are highly susceptible to Leishmania infection, even in a resistant C57BL/6 background [66].
Dendritic cells (DCs) are similar to macrophages in that they phagocytize Leishmania parasites and are critical antigen-presenting cells responsible for priming adaptive immune responses. One of the major cytokines that DCs produce upon activation is IL-12 [67], which is critical for developing protective immunity and resolving infection [68]. Activating these innate immune cells requires recognition of Leishmania-associated antigens via TLRs. Indeed, Myd88 −/− mice are highly susceptible to Leishmania infection and develop non-healing lesions, suggesting a strong role for TLR sensing [69–71]. Leishmania lipophosphoglycan (LPG), glycoproteins, and DNA activate TLR2, TLR4, and TLR9 signaling, respectively [72–76]. More importantly, activation of TLR2, TLR4, or TLR9 by their respective antigens results in significant protection from Leishmania pathogenesis and parasite burden [77]. Additionally, the results of recent studies show a surprising role for eosinophils and mast cells in inducing proper immune responses against Leishmania and in providing protection, mostly through the regulation of DCs and adaptive immunity [78, 79].
As described earlier, Leishmania studies have been instrumental in our current understanding of Th1/Th2 responses, with Th1 responses associated with IFN-γ production and protective immunity. Adaptive immunity that includes both humoral B-cell responses and T-cell-mediated immunity is critical in shaping immune responses during Leishmania infection. B cells and parasite-specific antibodies can be readily observed against Leishmania-induced skin lesions. Surprisingly, mice deficient in B cells are more resistant to Leishmania infections than are wild-type mice [80]. In contrast, T cell responses are critical in containing Leishmania infection, and infection of T cell-deficient nude or severe-combined immunodeficient (SCID) mice results in uncontrolled infection and death of the host [81, 82]. Both CD4+ and CD8+ T cells are important for protective immunity. Leishmania infection of either MHC class II-deficient (i.e., lacking CD4+ T cells) mice or CD4-depleted mice results in severe lesions, demonstrating the importance of CD4+ T cells [83, 84]. Interestingly, CD4-deficient mice clear infection similar to WT controls, owing to the generation of class II-restricted CD4−αβ+ T cells [85]. A similar supportive role of IFN-γ-producing CD8+ T cells in providing protection during L. major infection has also been demonstrated using CD8-deficient mice or CD8-depeletion strategies [86].
Roles of the IL-1 cytokine family in adaptive immunity against Leishmania
The roles of IL-1 and IL-18 in modulating adaptive immunity have been reported [87]. Both IL-1β and IL-18 promote Th1 and Th17 responses. Interestingly, during L. major infections, IL-1α, IL-1β, and IL-18 can be either protective or detrimental, depending on the genetic background of the host. Il1a −/− and Il1b −/− mice infected with L. major are slightly more resistant than are control BALB/c mice [88]. Consistent with this finding, a local IL-1β injection accelerates the progression of lesions in L. amazonensis-infected C57BL/6 mice [89]. Furthermore, IL-1 receptor antagonist-deficient (Il1ra −/−) BALB/c mice, which have heightened IL-1α and IL-1β signaling, are highly susceptible to leishmaniasis [88, 90]. The resistance in Il1a −/− and Il1b −/− mice is correlated with a subsequent increase in IFN-γ and a decrease in IL-4-producing T cells [88, 90]. Similar results were observed in T cells from susceptible Il1ra −/− mice during L. major infections. IL-18 increases the susceptibility of BALB/c mice to L. mexicana and L. major infections [91, 92]. Il18 −/− mice on a BALB/c background had slower lesion growth and a significantly lower parasite burden than did WT mice [91, 92]. Not surprisingly, the production of IL-4 by T cells was dramatically reduced and that of IFN-γ was increased in Il18 −/− BALB/c mice. Furthermore, the Th1-associated antibody IgG2a significantly increased whereas the Th2-associated antibody IgG1 significantly decreased in these mice [91]. Taken together, these results support the notion that the inflammasome-associated cytokines IL-1α, IL-1β, and IL-18 promote a Th2 milieu during leishmaniasis and render the BALB/c host susceptible to these parasites.
In contrast, in C57BL/6 mice, both IL-1α and IL-1β are dispensable for protection against Leishmania infection [93], and Il18 −/− mice are more susceptible to Leishmania infection than are WT mice, as demonstrated by a higher parasite burden and significantly increased lesion size [94–96]. The higher susceptibility of IL-18-deficient mice is associated with a concurrent decrease in IFN-γ production and an increase in IL-4 production by T cells. Taken together, these results suggest that IL-1 and IL-18 play a protective role in resistant C57BL/6 mice by promoting a Th1 environment.
NLRP3 inflammasomes regulate immune responses during Leishmania infections
Inflammasomes are the major protein complexes that process pro-IL-1β and pro-IL-18 to their mature bioactive forms. Despite several studies of IL-1 and IL-18, the roles of NLRs (the major regulators of IL-1 cytokine family) during Leishmania infections remain largely unknown. Thus far, only the NLRP3 inflammasome has been shown to be involved in modulating immune responses against Leishmania infections [97–100]. The results of these studies suggest that the NLRP3 inflammasome is directly involved in processing and releasing IL-1β and IL-18 during Leishmania infection in vitro and in vivo (Fig. 2b). The NLRP3 inflammasome is similarly required for IL-1β and IL-18 production during L. major infection in both C57BL/6 [98, 100] and BALB/c [97] mice. Leishmania-associated metalloprotease GP63 prevents NLRP3 inflammasome activation by inhibiting the production of reactive oxygen species and directly cleaving NLRP3 [99]. One of the first studies of NLRP3 inflammasomes showed that most Leishmania spp., including L. amazonensis, L. braziliensis, and L. mexicana, induce caspase-1 activation and IL-1β production upon infection of C57BL/6 macrophages [98]. Interestingly, IL-1β signaling promotes nitric oxide production, which promotes Leishmania killing in macrophages. As a result, C57BL/6 WT mice and mice deficient in components of the NLRP3 inflammasome (NLRP3, ASC, or caspase-1) have increased susceptibility to L. amazonensis infection and defects in clearance of the parasite. However, the authors of this study did not find any role for the NLRP3 inflammasome in clearing L. major, suggesting a species-specific role for the NLRP3 inflammasome. The results of a more recent study using a non-healing strain of L. major infection in C57BL/6 mice show that NLRP3 inflammasome-induced IL-1β promotes lesions through recruitment of neutrophils at the site of the infection [100]. Thus, mice deficient in the inflammasome components are significantly less protected from L. major infection in this model system. Similarly, the NLRP3 inflammasome promotes non-healing L. major infections in BALB/c mice, with NLRP3-dependent IL-18 directly promoting Th2 responses (IL-4 production) and inhibiting Th1 responses (IFN-γ production) [97]. Thus, the L. major-induced progression of disease or lesions in susceptible BALB/c WT mice can be partially rescued by neutralization of IL-18 in vivo. Mechanistically, IL-18 directly promotes the expression of GATA3 and cMAF, which are transcription factors for IL-4 [101, 102]. Whether increased IL-4 production results in a reduction in IFN-γ or whether IL-18 directly inhibits IFN-γ production by negatively regulating T-bet needs to be further examined. As reviewed elsewhere, these discrepancies in outcomes could be directly due to the differences in Leishmania strains or mouse genotypes tested [77].
Malaria
Malaria is a deadly parasitic disease that affects more than half of the world’s population. According to a WHO report in 2014, approximately 200 million cases of malaria were reported in 2013, resulting in approximately 0.5 million deaths [103]. Malaria is caused by protozoan parasites of the Plasmodium species, which are transmitted to humans and other mammals by mosquito bites. After its transmission into the host, the parasite undergoes two major stages to establish infection. First, the sporozoites (the infectious stage of Plasmodium) infect liver cells, where they divide and multiply and transform into merozoites. This stage is often referred to as the liver stage or pre-erythrocyte stage. After this cycle, the Plasmodium merozoites leave the liver to infect red blood cells (RBCs), and divide and multiply within these cells. This second stage of infection is known as the erythrocytic or blood stage. The periodic fever and symptoms of malaria are often associated with the cyclical bursting of merozoites from the infected RBCs [104].
Immune responses against Plasmodium spp.
Infection by Plasmodium parasites is often lethal if left untreated. Immune responses against Plasmodium start as soon as the sporozoites are released in the skin by mosquito bites. In the dermis, the sporozoites can come in contact with innate immune cells, including neutrophils, macrophages, dendritic cells, mast cells, NK and NKT cells, and γδ T cells. As in other protozoan infections described in this review, neutrophils are one of the early responders to sporozoites and can be found in the dermis as early as 20 min after infection [105]. However, neutrophil depletion during sporozoite infection does not affect parasite distribution and development in the liver, suggesting redundant compensatory roles for other cell types [106]. Once in the liver, Plasmodium sporozoites infect hepatocytes and transition into merozoites. Kupffer cells (liver resident macrophages) play important roles in containing and clearing the parasites, and depletion of Kupffer cells increases the sporozoite invasion of hepatocytes [107]. Furthermore, other innate cells, such as neutrophils, eosinophils, monocytes, and macrophages, all infiltrate the liver in response to Plasmodium sporozoites, and this infiltration is strongly correlated with resistance of BALB/c mice to P. berghei and P. yoelii infection [108, 109].
Once the infection develops into the erythrocytic stage, key innate mechanisms for clearance rely on the ability of monocytes and macrophages to quickly phagocytize and clear the infected erythrocytes. Indeed, CD36 is an important receptor for direct clearance of infected erythrocytes [110]. More importantly, human populations that are deficient in CD36 are more prone to developing severe malaria [111]. In addition to their roles in phagocytizing and killing Plasmodium-infected cells, innate immune cells secrete critical cytokines, such as IFN-γ and IL-12, in response to Plasmodium-associated pathogen-associated molecular patterns (PAMPs), including glycosylphosphatidylinositol (GPI), hemozoin, RNA, and DNA [112]. Both IFN-γ and IL-12 promote resistance against Plasmodium infection. Although recognition of Plasmodium RNA through the MDA5-MAVS pathway induces type I interferon responses that provide protection in some experimental settings, TLR-MyD88-mediated recognition of Plasmodium PAMPs (GPI, hemozoin, and DNA) promotes Plasmodium pathogenesis. Indeed, MyD88 −/− mice are highly resistant to P. chabaudi and P. berghei infection, and TLR antagonists protect mice from cerebral malaria [112].
Both CD8+ and CD4+ T cells are critical for protective immunity following Plasmodium infection [113]. CD8+ T cells are important for controlling and killing Plasmodium at the liver stage [114–117], whereas CD4+ T cells are important at both the liver and blood stages [118–121]. CD4+ T cells assist CD8+ T cells in mounting an efficient immune response against Plasmodium parasites [122, 123]. However, a study has shown that CD4+ T cells are critical in providing immunity whereas CD8+ T cells are expendable [124]. Thus, the coordinated orchestration between T cells is essential for protective immunity against Plasmodium parasites.
Role for NLRs during Plasmodium infection
Several cytoplasmic NLRs are involved during Plasmodium infection (Fig. 2c). The production of inflammatory cytokines, such as IL-1β and IFN-γ, is dramatically blunted in the absence of NOD1 and NOD2 in response to P. berghei ANKA infection, suggesting a role for these NLRs in sensing Plasmodium PAMPs [125]. However, mice deficient in NOD1 and NOD2 are comparable to WT mice and exhibit similar morbidity and mortality during P. berghei ANKA infection [125]. Future studies are needed to investigate the possible ligands from Plasmodium parasites that are recognized by NOD1 and NOD2 and their effects on protective immunity.
A major byproduct of the blood stage is hemozoin, a crystal of heme that is produced as a result of Plasmodium detoxifying the free heme present in the hemoglobin [126]. Hemozoin is perceived as a danger signal by the innate immune system and can directly activate inflammasomes in vitro and in vivo. When stimulated with hemozoin, WT macrophages induce caspase-1 activation and subsequent IL-1β and IL-18 production that are dependent on NLRP3 and ASC, suggesting the involvement of the NLRP3 inflammasome [127–129]. However, activation of the NLRP3 inflammasome and production of IL-1β during Plasmodium infection in vivo has detrimental effects. Mice deficient in NLRP3, ASC, caspase-1, or IL-1β demonstrate significant resistance to Plasmodium infection when compared to WT mice [127, 129]. The NLRP3 inflammasome-deficient mice have a similar parasitic burden, suggesting an immunopathologic role for the NLRP3 inflammasome and its associated cytokines. However, other studies have found no role for components of the inflammasome in Plasmodium disease pathology in vivo [130, 131]. In a separate model of Plasmodium-bacterial coinfection, both NLRP3 and NLRP12 promote immunopathology [132]. Specifically, WT mice infected with P. falciparum or P. vivax produce high levels of IL-1β in an NLRP3- and NLRP12-dependent manner. WT mice infected with P. faclipurum are highly sensitive to secondary bacterial infection or low-dose lipopolysaccharide injection (which mimics bacterial infection). In agreement with these results, mice deficient in NLRP3, NLRP12, or components of the inflammasome (ASC and caspase-1) have higher survival rates than do WT mice after Plasmodium-bacterial coinfection.
A recent publication analyzing single nucleotide polymorphisms in symptomatic human patients with P. vivax malaria demonstrated that polymorphisms associated with NLRP1 gene was associated with increased Plasmodium pathogenesis [133]. Although these studies are highly correlative at this time, it highlights a possible role for NLRP1 in modulating immune responses during Plasmodium infection.
Toxoplasmosis
Toxoplasmosis is caused by Toxoplasma gondii, an obligate intracellular protozoan that infects humans and animals. At least one third of the world’s population is infected with T. gondii, but only immunocompromised individuals are at high risk for developing complications such as pneumonia, organ failure, and encephalitis [134–137].
Immune responses against T. gondii
Both innate and adaptive components are important in providing immunity against T. gondii infections. Innate immune responses are critical for protection against T. gondii infections, as shown by the results of studies in mice lacking critical cytokines and molecules, including IFN-γ [138], IL-12 [139], and iNOS [140]. Mice lacking these innate effector molecules are highly susceptible to T. gondii infections. Innate immune cells that produce and respond to these cytokines include neutrophils, macrophages, DCs, and NK cells. T. gondii causes increased morbidity and mortality in mouse depleted of these innate cell populations [141, 142].
TLRs play an integral role in innate recognition of T. gondii-associated PAMPs and in initiating cytokine responses by the innate immune cells. As such, MyD88 −/− mice are highly susceptible to T. gondii infection. Several TLRs, including TLR2, TLR4, TLR7, TLR9, and TLR11, are involved in innate recognition of T. gondii and promote production of IL-12 and IFN-γ [143]. More recently, the T. gondii-secreted protein profilin was shown to be the specific ligand for TLR11, suggesting that TLR11 is the principal innate sensor [144]. However, deficiency in individual TLRs does not render mice more susceptible to T. gondii infection than WT controls are [143]. These results suggest a possible redundancy between TLRs in recognizing and clearing T. gondii infection in vivo.
T cells are essential for providing complete protection against T. gondii, which is confirmed by the finding that mice deficient in T cells are highly susceptible and die as a result of uncontrollable proliferation of the parasite in various organs, including the brain [145, 146]. Both CD8+ and CD4+ T cells are important for controlling T. gondii infection, and IFN-γ production by these cells is critical for protection [147–149]. T. gondii infection of IFN-γ-deficient mice or WT mice with neutralized IFN-γ results in severe acute inflammation and development of necrotic lesions in the brain [150, 151]. Unlike its role in other protozoan infections, IL-4 is protective against T. gondii infection. IL-4-deficient mice are highly susceptible to T. gondii, and all mice succumbed to the infection [152]. IL-4-deficient mice have significantly fewer IFN-γ-producing T cells than did WT mice, supporting the role for IL-4 in promoting Th1 cell differentiation.
Role for NLRs during T. gondii infection
Several NLR molecules are involved in providing protective immunity against T. gondii (Fig. 3a). NOD2 was one of the first NLRs shown to be important in providing protective immunity against T. gondii., with all WT mice infected with T. gondii surviving and all NOD2-deficient mice succumbing to infection by day 21 [150]. Interestingly, NOD2-mediated protection against T. gondii is independent of its adaptor protein RIPK2, as RIPK2-deficient mice are completely protected [150]. Although NOD2 deficiency in antigen-presenting cells is not important for immunity, NOD2 expression is critical in T cells and required for optimal IFN-γ production. Mechanistically, NOD2 in T cells is required for optimal IL-2 production and proliferation. In T cells, NOD2 binds to c-Rel to promote T-cell activation and IFN-γ production. As a result, c-Rel translocation in NOD2-deficient T cells is severely blunted. Thus, NOD2 has a T-cell-intrinsic role in promoting proliferation and the production of effector cytokines during T. gondii infection [150]. However, these findings have yet to be substantiated by independent groups. Indeed, one study has found no significant role for NOD2 in T cells and shown that NOD2 is dispensable for protection during T. gondii infection of mice [153]. This discrepancy in findings from two independent studies might be due to the differences in mouse genetic backgrounds or differences in microbiota.
Fig. 3.

Role of NLR in modulating immune responses during Toxoplasma gondii and Trypanosoma cruzi infection. a Toxoplasma gondii activates both NLRP1b and NLRP3 inflammasomes to induce the robust production of IL-1β and IL-18. In addition, NOD2 in T cells directly interacts with c-Rel to induce the production of IFN-γ and IL-2 during T. gondii infection and is required for resistance. b NOD1 promotes T. cruzi killing within the macrophages. T. cruzi also activates the NLRP3 inflammasome through mechanisms that involve K+ efflux and reactive oxygen species
In addition to NOD2, both NLRP1b and NLRP3 are also involved in protection against T. gondii. Single-nucleotide polymorphisms in the NLRP1 gene are associated with increased susceptibility to toxoplasmosis in humans [154]. These findings were further confirmed via RNA interference-mediated knockdown of NLRP1 in human monocytic cell lines. Activation of the inflammasome during T. gondii infection in human monocytes was determined via short hairpin RNA-mediated knockdown of ASC and caspase-1 [155]. The involvement of the NLRP1 inflammasome during T. gondii infection has also been confirmed in both murine and rat models [156]. These studies’ results show that the activation of caspase-1 and subsequent production of IL-1β and IL-18 in human cells in response to T. gondii infection are mediated by the NLRP1 inflammasome. More recent in vitro and in vivo studies of T. gondii infection in mice suggest a dual role for NLRP3 and NLRP1 inflammasomes [157]. NLRP3-, NLRP1b-, ASC-, caspase-1-, or caspase-11-deficient mice are much more susceptible to T. gondii infection than are WT mice. Although IL-1β production is detectable during in vitro stimulation of macrophages with T. gondii, only IL-18 is measurable in the serum of T. gondii-infected mice in vivo. Interestingly, both IL-1 and IL-18 signaling are required to protect mice during T. gondii infection because IL-1R- and IL-18-deficient mice are much more susceptible than are WT mice.
Altogether, the results of these studies show that several NLRs function in both innate and adaptive immune cells to recognize and combat T. gondii infection. Whether other NLRs are also involved in various immune cell types should be an active research area for future studies.
Trypanosomiasis or Chagas disease
Trypanosomiasis or Chagas disease is caused primarily by the obligate intracellular parasite Trypanosoma cruzi. More than 10 million people worldwide are infected with T. cruzi, of which approximately 30 % develop cardiac diseases and complications [158–160]. The precise molecular mechanisms involved in providing protection against T. cruzi are largely unknown because of the paucity of studies in this field. However, both innate and adaptive immune responses are necessary for protective immunity and resistance to T. cruzi in vivo [161–163].
Immune responses against T. cruzi
Innate immune cells, including neutrophils, monocyte/macrophages, DCs, and NK cells, play an important role in controlling T. cruzi infection in the host. Recognition of T. cruzi by the innate immune cells is important for production of protective factors such as IL-12, TNF-α, IFN-γ, and nitric oxide. Indeed, mice deficient in IL-12, IFN-γ, and iNOS are highly susceptible to T. cruzi infection [164–166]. Depletion of neutrophils and macrophages in BALB/c mice is detrimental during T. cruzi infection, demonstrating these molecules’ importance, and similar important roles for NK cells have also been shown by depletion studies [167, 168]. NK cells are not only directly cytotoxic but are also the major source of IFN-γ at acute time points. The IFN-γ signaling is critical for macrophages to effectively kill intracellular T. cruzi. The TLR signaling axis is critical in priming these immune responses by innate immune cells. Mice lacking MyD88 are highly susceptible to T cruzi infections as a result of failure to produce proinflammatory cytokines such as IL-12 and IFN-γ. Attempts to identify upstream TLRs that recognize T. cruzi ligands have identified TLR2, TLR4, TLR7, and TLR9 as possible innate receptors [169]. TLR2 recognizes T. cruzi glycophosphatidylinositol, and TLR4 recognizes glycoinositolphospholipid. TLR7 and TLR9 recognize T. cruzi-derived RNA and DNA fragments respectively. Although deficiency of any individual TLR results in increased susceptibility to T. cruzi, the disease in such deficient mice is less severe than that in Myd88 −/− mice, suggesting partial redundancy between the TLRs [169].
Adaptive T cells play an extremely important role in providing protection against T. cruzi infection. Mice deficient in either CD4+ or CD8+ T cells are more susceptible to T. cruzi infection than are WT mice and quickly succumb to infection [170, 171]. Upon activation, CD4+ and CD8+ T cells produce IFN-γ that induces further activation of phagocytic cells, including macrophages, to promote parasite killing [172–174]. CD8+ T cells, in particular, can directly kill infected cells. Unfortunately, the adaptive immune response is not sufficient to achieve sterilizing immunity, and the parasite can establish chronic infections in most individuals.
Roles of NLRs during T. cruzi infection
Evidence for the roles of NLRs during T. cruzi infection is limited, and the few studies conducted on the roles of NLRs during T. cruzi infection are discussed here (Fig. 3b). NOD1 and NLRP3 play a protective role during T. cruzi infections. The results of one study showed that NOD1, but not NOD2, is important in providing protection against T. cruzi in mice in vivo [175]. Mice deficient in NOD1 bear a significantly increased parasite load and have higher mortality rates than NOD2-deficient or WT mice. Interestingly, NOD1 does not seem to affect overall serum cytokine levels during T. cruzi infection but is required for IFN-γ sensitivity in macrophages to clear T. cruzi.
The results of two independent studies suggest a role for the NLRP3 inflammasome in response to T. cruzi infection [176, 177]. T. cruzi infected-macrophages induce caspase-1 activation and subsequent IL-1β and IL-18 production. The activation of the NLRP3 inflammasome in response to T. cruzi is dependent on established activators, such as potassium efflux, ROS production, and lysosomal damage. T. cruzi infection of mice deficient in NLRP3, ASC, and caspase-1 results in severe illness, high parasitic burden, and death. It is unclear whether IL-1β and IL-18 are involved in these protective effects in vivo. Of note, the in vitro protective effects of WT macrophages (killing of T. cruzi) are not dependent on IL-1β or IL-18 [176]. However, the in vivo protection in mice could be due to IL-1β- and IL-18-mediated effects on priming the adaptive immune responses, especially CD8+ T-cell responses.
NLRs in modulating adaptive immune responses during protozoan infection: a perspective
NLRs are novel players in the regulation of innate immunity during protozoan infections. As discussed earlier in the review, NLRs can function directly as either positive or negative regulators of pro-inflammatory signaling in various innate immune cell types in response to pathogenic insults. Overall, protozoan PAMP sensing by NLRs results in the production of pro-inflammatory cytokines such as TNF-α, IFN-γ, IL-12, IL-1β, and IL-18; the production of reactive oxygen and nitrogen species; and inflammatory cell death. Although our understanding of NLRs in regulating innate immunity against protozoan infections is beginning to unfold, several important findings suggest that NLRs can also modulate adaptive immune responses. Here, we have presented a perspective on how these NLRs are involved in modulating adaptive immune responses against protozoan infection, with specific examples wherever applicable.
NLRs could use one or more of the following mechanisms to regulate T cell immune responses during protozoan infections: (1) NLR-mediated production of pro-inflammatory cytokines by innate immune cells may regulate T cell responses, (2) cell-intrinsic roles of NLRs in innate immune cells may ultimately affect T cells, (3) inflammasome-activation-induced IL-1β and IL-18 and inflammatory cell death may affect adaptive immunity, and (4) NLRs may have cell-intrinsic roles in T cells.
NOD1 and NOD2 are activated in response to bacterial peptidoglycans. The result of this recognition is activation of NFκB and MAPK signaling pathways and subsequent production of pro-inflammatory cytokines. In the context of protozoan infections, both NOD1 and NOD2 are involved in the recognition of an unknown Plasmodium ligand and promote production of pro-inflammatory cytokines such as IL-1β and IFN-γ. Given that IL-1β drives T cell proliferation and survival [87] and that IFN-γ promotes Th1 differentiation [178], it could be posited that NOD1 and NOD2 play an important role in regulating adaptive immunity during Plasmodium infections.
Similarly, both NOD1 and NOD2 are required for activation of NFκB and MAPK signaling induced by T. cruzi infection in macrophages. As a result, production of cytokines such as IL-12, IFN-γ, and TNF-α are blunted in T. cruzi-stimulated Nod1- and Nod2-deficient macrophages. Interestingly, only Nod1 −/− mice are susceptible to T. cruzi infection. Although the Nod1 −/− mice had similar levels of IL-12 and IFN-γ as WT mice, they had lower NOS levels. NOS-induced production of nitric oxide by macrophages is known to promote Th1 immune responses and, thus, could be a mechanism of NOD1-mediated protection against T. cruzi [179]. More interestingly, Nod1 −/− mice die 15 days post infection, strengthening the argument that NOD1 has a role in regulating adaptive immune responses during T. cruzi infection.
Cell-intrinsic roles for NLRs in macrophages and DCs have been reported, but this area needs further investigation. NOD2 has been shown to regulate DC function and survival. Specifically, Nod2 −/− DCs exhibit reduced expression of CD80 and CD86 costimulatory molecules and defects in survival compared to WT DCs [180]. As a result, Nod2 −/− DCs are less efficient in priming T cell responses during influenza virus infection. A similar intrinsic function for NLR molecules has not been observed for macrophages or DCs during protozoan infections. However, NOD1 is required for IFN-γ-induced killing of T. cruzi in macrophages, suggesting a possible macrophage intrinsic role for NOD1 in protozoa killing [175]. Although there are no current studies on the roles of CIITA and NLRC5 during protozoan infection, these two molecules regulate MHCI and MHCII transcription, both of which are important for antigen presentation and activation of T cell responses [181].
The NLRP3 inflammasome is activated in response to all protozoan parasites discussed in this review. In addition to the NLRP3 inflammasome, Plasmodium hemozoin also activates the putative NLRP12 inflammasome, and Toxoplasma gondii activates the NLRP1b inflammasome. The functional consequence of inflammasome activation in the cell is release of cytokines IL-1β and IL-18, leading to pyroptosis, an inflammatory form of cell death. Both IL-1β and IL-18 promote T cell responses. In the context of Leishmania infection, NLRP3 inflammasome-induced IL-18 promotes Th2 immune responses, rendering mice susceptible to infection [97]. Of interest, inflammasome activation is also detrimental for Plasmodium infection [127, 129]. Whether IL-1β and IL-18 prime Th2 responses in these settings is not known, and future studies are needed to understand these molecular and cellular underpinnings. In contrast, inflammasome activation during E. histolytica, T. gondii, and T. cruzi infection are all protective [56, 57, 157, 176]; thus, it could be posited that the IL-1β and IL-18 cytokines may prime a protective Th1 immune response in these infectious settings.
A major consequence of inflammasome activation is pyroptotic cell death, which is often overlooked when studying immune responses. The results of previous studies show that DCs that phagocytize activated dead cells or necrotic dead cells upregulate CD80/CD86 and produce higher levels of IL-12 cytokines [182]. Pyroptosis is an inflammatory cell death and thus it could be hypothesized that macrophages and DCs that uptake these pyroptotic dead cells will have upregulated expression of co-stimulatory molecules and increased IL-12 production that could ultimately affect T cell activation. In contrast, higher rates of pyroptosis in macrophages and DCs could directly limit the source of antigen presenting cells available for T cell activation.
Although NLRs can regulate T cell responses through their role in macrophages and DCs as discussed in this review, NLRs also have T cell-intrinsic functions. NLRP3 acts as a transcriptional factor for IL-4 (in transactivation with Interferon regulatory factor 4 (IRF4)) in T cells and drives Th2 responses [15]. More recently, NLRP12 was shown to negatively regulate NFκB and ERK signaling in T cells and to promote T cell hyperproliferation and IL-4 production during experimental autoimmune encephalitis in mice [183]. Additionally, inflammasome-induced pyroptosis of CD4+ T cells is suggested to be the major reason for T cell loss in HIV infected patients [184]. With regards to protozoan infections, NOD2 has T cell-intrinsic functions and drives T cell proliferation and IFN-γ production during T. gondii infection [150]. Mechanistically, NOD2 directly interacts with the transcription factor c-Rel to drive T cell proliferation and IFN-γ production [150]. Future studies should reveal more information on the potential role of NLRs in T cells during several protozoan infections.
Conclusion
Despite the high morbidity and mortality associated with the protozoan infections discussed in this review, viable treatment options for these diseases are largely lacking. Most of the treatments for these protozoan parasites are non-specific and often associated with severe side effects. With the exception of malaria, the research, attention, and funding required to develop effective therapies against these protozoan infections are severely lacking. As a result, the WHO continues to categorize most of these diseases as neglected tropical diseases.
Studies of the roles of NLRs during protozoan infections are still in their infancy. Moreover, even for the NLRs that have been shown to be involved in these protozoan infections, their exact roles in different cell types and whether they affect adaptive immunity remains unknown. With only a few of the known NLRs being tested thus far, there remains a large knowledge gap in our understanding of how various NLRs are involved in providing or modulating immunity during these protozoan infections. Thus, immediate research on the role of NLRs during protozoan infections is warranted to design new immunotherapies and vaccines.
Acknowledgments
We thank Drs. Vani J. Shanker and Cherise M. Guess of St. Jude Children’s Research Hospital’s Department of Scientific Editing for her help with critical editing of the manuscript. We also thank Drs. Farrah Phillips, Si Ming Man and Ankit Malik for their critical reading of the manuscript. PG is a postdoctoral fellow supported by the Paul Barrett Endowed Fellowship from St. Jude Children’s Research Hospital. This work was supported in part by grants from the National Institute of Health (Grants AR056296, CA163507, and AI101935) and American Lebanese Syrian Associated Charities to T-D.K.
Compliance with ethical standards
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Harton JA, Linhoff MW, Zhang J, Ting JP. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol. 2002;169(8):4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- 2.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, Valvano MA, Foster SJ, Mak TW, Nunez G, Inohara N. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 3.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 4.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278(8):5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 5.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 6.Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, Davis BK, Allen IC, Holl EK, Ye Z, Rahman AH, Conti BJ, Eitas TK, Koller BH, Ting JP. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol. 2012;13(9):823–831. doi: 10.1038/ni.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang RF. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141(3):483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti TD. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488(7411):389–393. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RM, Davis BK, Uronis JM, Herfarth HH, Jobin C, Rogers AB, Ting JP. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. 2012;36(5):742–754. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, Kanneganti TD. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. 2011;20(5):649–660. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JP. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34(6):854–865. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, Wang RF. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34(6):843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol. 2000;20(17):6185–6194. doi: 10.1128/MCB.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA. 2010;107(31):13794–13799. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruchard M, Rebe C, Derangere V, Togbe D, Ryffel B, Boidot R, Humblin E, Hamman A, Chalmin F, Berger H, Chevriaux A, Limagne E, Apetoh L, Vegran F, Ghiringhelli F. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol. 2015;16(8):859–870. doi: 10.1038/ni.3202. [DOI] [PubMed] [Google Scholar]
- 16.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nunez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 17.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 19.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 20.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 21.Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, Pevsner-Fischer M, Shapiro H, Christ A, Harmelin A, Halpern Z, Latz E, Flavell RA, Amit I, Segal E, Elinav E. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163(6):1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA, Lien E. The NLRP12 inflammasome recognizes Yersinia pestis . Immunity. 2012;37(1):96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanley SL., Jr Amoebiasis. Lancet. 2003;361(9362):1025–1034. doi: 10.1016/S0140-6736(03)12830-9. [DOI] [PubMed] [Google Scholar]
- 24.Haque R, Huston CD, Hughes M, Houpt E, Petri WA., Jr Amebiasis. N Engl J Med. 2003;348(16):1565–1573. doi: 10.1056/NEJMra022710. [DOI] [PubMed] [Google Scholar]
- 25.Mortimer L, Chadee K. The immunopathogenesis of Entamoeba histolytica . Exp Parasitol. 2010;126(3):366–380. doi: 10.1016/j.exppara.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Mondal D, Haque R, Sack RB, Kirkpatrick BD, Petri WA., Jr Attribution of malnutrition to cause-specific diarrheal illness: evidence from a prospective study of preschool children in Mirpur, Dhaka, Bangladesh. Am J Trop Med Hyg. 2009;80(5):824–826. [PMC free article] [PubMed] [Google Scholar]
- 27.Verkerke HP, Petri WA, Jr, Marie CS. The dynamic interdependence of amebiasis, innate immunity, and undernutrition. Semin Immunopathol. 2012;34(6):771–785. doi: 10.1007/s00281-012-0349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassily S, Farid Z, el-Masry NA, Mikhail EM. Treatment of intestinal E. histolytica and G. lamblia with metronidazole, tinidazole and ornidazole: a comparative study. J Trop Med Hyg. 1987;90(1):9–12. [PubMed] [Google Scholar]
- 29.Guo X, Houpt E, Petri WA., Jr Crosstalk at the initial encounter: interplay between host defense and ameba survival strategies. Curr Opin Immunol. 2007;19(4):376–384. doi: 10.1016/j.coi.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chadee K, Petri WA, Jr, Innes DJ, Ravdin JI. Rat and human colonic mucins bind to and inhibit adherence lectin of Entamoeba histolytica . J Clin Investig. 1987;80(5):1245–1254. doi: 10.1172/JCI113199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moncada DM, Kammanadiminti SJ, Chadee K. Mucin and Toll-like receptors in host defense against intestinal parasites. Trends Parasitol. 2003;19(7):305–311. doi: 10.1016/S1471-4922(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 32.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6(5):e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haque R, Ali IM, Sack RB, Farr BM, Ramakrishnan G, Petri WA., Jr Amebiasis and mucosal IgA antibody against the Entamoeba histolytica adherence lectin in Bangladeshi children. J Infect Dis. 2001;183(12):1787–1793. doi: 10.1086/320740. [DOI] [PubMed] [Google Scholar]
- 34.Peterson KM, Guo X, Elkahloun AG, Mondal D, Bardhan PK, Sugawara A, Duggal P, Haque R, Petri WA., Jr The expression of REG 1A and REG 1B is increased during acute amebic colitis. Parasitol Int. 2011;60(3):296–300. doi: 10.1016/j.parint.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Becker SM, Cho KN, Guo X, Fendig K, Oosman MN, Whitehead R, Cohn SM, Houpt ER. Epithelial cell apoptosis facilitates Entamoeba histolytica infection in the gut. Am J Pathol. 2010;176(3):1316–1322. doi: 10.2353/ajpath.2010.090740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chadee K, Moreau F, Meerovitch E. Entamoeba histolytica: chemoattractant activity for gerbil neutrophils in vivo and in vitro. Exp Parasitol. 1987;64(1):12–23. doi: 10.1016/0014-4894(87)90003-8. [DOI] [PubMed] [Google Scholar]
- 37.Salata RA, Ahmed P, Ravdin JI. Chemoattractant activity of Entamoeba histolytica for human polymorphonuclear neutrophils. J Parasitol. 1989;75(4):644–646. doi: 10.2307/3282920. [DOI] [PubMed] [Google Scholar]
- 38.Rivero-Nava L, Aguirre-Garcia J, Shibayama-Salas M, Hernandez-Pando R, Tsutsumi V, Calderon J. Entamoeba histolytica: acute granulomatous intestinal lesions in normal and neutrophil-depleted mice. Exp Parasitol. 2002;101(4):183–192. doi: 10.1016/S0014-4894(02)00106-6. [DOI] [PubMed] [Google Scholar]
- 39.Seydel KB, Zhang T, Stanley SL., Jr Neutrophils play a critical role in early resistance to amebic liver abscesses in severe combined immunodeficient mice. Infect Immun. 1997;65(9):3951–3953. doi: 10.1128/iai.65.9.3951-3953.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivory CP, Prystajecky M, Jobin C, Chadee K. Toll-like receptor 9-dependent macrophage activation by Entamoeba histolytica DNA. Infect Immun. 2008;76(1):289–297. doi: 10.1128/IAI.01217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maldonado-Bernal C, Kirschning CJ, Rosenstein Y, Rocha LM, Rios-Sarabia N, Espinosa-Cantellano M, Becker I, Estrada I, Salazar-Gonzalez RM, Lopez-Macias C, Wagner H, Sanchez J, Isibasi A. The innate immune response to Entamoeba histolytica lipopeptidophosphoglycan is mediated by toll-like receptors 2 and 4. Parasite Immunol. 2005;27(4):127–137. doi: 10.1111/j.1365-3024.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- 42.Seydel KB, Smith SJ, Stanley SL., Jr Innate immunity to amebic liver abscess is dependent on gamma interferon and nitric oxide in a murine model of disease. Infect Immun. 2000;68(1):400–402. doi: 10.1128/IAI.68.1.400-402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siman-Tov R, Ankri S. Nitric oxide inhibits cysteine proteinases and alcohol dehydrogenase 2 of Entamoeba histolytica . Parasitol Res. 2003;89(2):146–149. doi: 10.1007/s00436-002-0716-2. [DOI] [PubMed] [Google Scholar]
- 44.Vivanco-Cid H, Alpuche-Aranda C, Wong-Baeza I, Rocha-Ramirez LM, Rios-Sarabia N, Estrada-Garcia I, Villasis-Keever MA, Lopez-Macias C, Isibasi A. Lipopopeptidephosphoglycan from Entamoeba histolytica activates human macrophages and dendritic cells and reaches their late endosomes. Parasite Immunol. 2007;29(9):467–474. doi: 10.1111/j.1365-3024.2007.00963.x. [DOI] [PubMed] [Google Scholar]
- 45.Lotter H, Gonzalez-Roldan N, Lindner B, Winau F, Isibasi A, Moreno-Lafont M, Ulmer AJ, Holst O, Tannich E, Jacobs T. Natural killer T cells activated by a lipopeptidophosphoglycan from Entamoeba histolytica are critically important to control amebic liver abscess. PLoS Pathog. 2009;5(5):e1000434. doi: 10.1371/journal.ppat.1000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsutsumi V, Mena-Lopez R, Anaya-Velazquez F, Martinez-Palomo A. Cellular bases of experimental amebic liver abscess formation. Am J Pathol. 1984;117(1):81–91. [PMC free article] [PubMed] [Google Scholar]
- 47.Guo X, Barroso L, Becker SM, Lyerly DM, Vedvick TS, Reed SG, Petri WA, Jr, Houpt ER. Protection against intestinal amebiasis by a recombinant vaccine is transferable by T cells and mediated by gamma interferon. Infect Immun. 2009;77(9):3909–3918. doi: 10.1128/IAI.00487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lotter H, Jacobs T, Gaworski I, Tannich E. Sexual dimorphism in the control of amebic liver abscess in a mouse model of disease. Infect Immun. 2006;74(1):118–124. doi: 10.1128/IAI.74.1.118-124.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanchez-Guillen Mdel C, Perez-Fuentes R, Salgado-Rosas H, Ruiz-Arguelles A, Ackers J, Shire A, Talamas-Rohana P. Differentiation of Entamoeba histolytica/Entamoeba dispar by PCR and their correlation with humoral and cellular immunity in individuals with clinical variants of amoebiasis. Am J Trop Med Hyg. 2002;66(6):731–737. doi: 10.4269/ajtmh.2002.66.731. [DOI] [PubMed] [Google Scholar]
- 50.Guo X, Stroup SE, Houpt ER. Persistence of Entamoeba histolytica infection in CBA mice owes to intestinal IL-4 production and inhibition of protective IFN-gamma. Mucosal Immunol. 2008;1(2):139–146. doi: 10.1038/mi.2007.18. [DOI] [PubMed] [Google Scholar]
- 51.Guo X, Barroso L, Lyerly DM, Petri WA, Jr, Houpt ER. CD4+ and CD8+ T cell- and IL-17-mediated protection against Entamoeba histolytica induced by a recombinant vaccine. Vaccine. 2011;29(4):772–777. doi: 10.1016/j.vaccine.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eckmann L, Reed SL, Smith JR, Kagnoff MF. Entamoeba histolytica trophozoites induce an inflammatory cytokine response by cultured human cells through the paracrine action of cytolytically released interleukin-1 alpha. J Clin Investig. 1995;96(3):1269–1279. doi: 10.1172/JCI118161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Que X, Kim SH, Sajid M, Eckmann L, Dinarello CA, McKerrow JH, Reed SL. A surface amebic cysteine proteinase inactivates interleukin-18. Infect Immun. 2003;71(3):1274–1280. doi: 10.1128/IAI.71.3.1274-1280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Wang L, Seydel KB, Li E, Ankri S, Mirelman D, Stanley SL., Jr Entamoeba histolytica cysteine proteinases with interleukin-1 beta converting enzyme (ICE) activity cause intestinal inflammation and tissue damage in amoebiasis. Mol Microbiol. 2000;37(3):542–548. doi: 10.1046/j.1365-2958.2000.02037.x. [DOI] [PubMed] [Google Scholar]
- 55.Velazquez JR, Garibay-Martinez L, Martinez-Tejada P, Leal YA. An amebic anti-inflammatory peptide down-regulates ex vivo IL-1beta expression in patients with rheumatoid arthritis. Reumatol Clin. 2012;8(6):315–320. doi: 10.1016/j.reuma.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 56.Mortimer L, Moreau F, Cornick S, Chadee K. Gal-lectin-dependent contact activates the inflammasome by invasive Entamoeba histolytica . Mucosal Immunol. 2014;7(4):829–841. doi: 10.1038/mi.2013.100. [DOI] [PubMed] [Google Scholar]
- 57.Mortimer L, Moreau F, Cornick S, Chadee K. The NLRP3 inflammasome is a pathogen sensor for invasive Entamoeba histolytica via activation of alpha5beta1 integrin at the macrophage-amebae intercellular junction. PLoS Pathog. 2015;11(5):e1004887. doi: 10.1371/journal.ppat.1004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M, Team WHOLC. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.CDC (2013) Center for Disease Control and Prevention. Parasites-Leishmaniasis. Global Health—Division of Parasitic Diseases and Malaria. http://www.cdc.gov/parasites/leishmaniasis/epi.html
- 60.Kedzierski L. Leishmaniasis vaccine: where are we today? J Glob Infect Dis. 2010;2(2):177–185. doi: 10.4103/0974-777X.62881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. 2002;2(11):845–858. doi: 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- 62.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321(5891):970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Novais FO, Santiago RC, Bafica A, Khouri R, Afonso L, Borges VM, Brodskyn C, Barral-Netto M, Barral A, de Oliveira CI. Neutrophils and macrophages cooperate in host resistance against Leishmania braziliensis infection. J Immunol. 2009;183(12):8088–8098. doi: 10.4049/jimmunol.0803720. [DOI] [PubMed] [Google Scholar]
- 64.Afonso L, Borges VM, Cruz H, Ribeiro-Gomes FL, DosReis GA, Dutra AN, Clarencio J, de Oliveira CI, Barral A, Barral-Netto M, Brodskyn CI. Interactions with apoptotic but not with necrotic neutrophils increase parasite burden in human macrophages infected with Leishmania amazonensis . J Leukoc Biol. 2008;84(2):389–396. doi: 10.1189/jlb.0108018. [DOI] [PubMed] [Google Scholar]
- 65.Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, Saraiva EM. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA. 2009;106(16):6748–6753. doi: 10.1073/pnas.0900226106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 67.von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J Exp Med. 1998;188(8):1547–1552. doi: 10.1084/jem.188.8.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinzel FP, Rerko RM, Ahmed F, Hujer AM. IFN-gamma-independent production of IL-12 during murine endotoxemia. J Immunol. 1996;157(10):4521–4528. [PubMed] [Google Scholar]
- 69.de Veer MJ, Curtis JM, Baldwin TM, DiDonato JA, Sexton A, McConville MJ, Handman E, Schofield L. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. 2003;33(10):2822–2831. doi: 10.1002/eji.200324128. [DOI] [PubMed] [Google Scholar]
- 70.Debus A, Glasner J, Rollinghoff M, Gessner A. High levels of susceptibility and T helper 2 response in MyD88-deficient mice infected with Leishmania major are interleukin-4 dependent. Infect Immun. 2003;71(12):7215–7218. doi: 10.1128/IAI.71.12.7215-7218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muraille E, De Trez C, Brait M, De Baetselier P, Leo O, Carlier Y. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J Immunol. 2003;170(8):4237–4241. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 72.Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, Ruiz A, Cervantes R, Torres AP, Cabrera N, Gonzalez A, Maldonado C, Isibasi A. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasitol. 2003;130(2):65–74. doi: 10.1016/S0166-6851(03)00160-9. [DOI] [PubMed] [Google Scholar]
- 73.Karmakar S, Bhaumik SK, Paul J, De T. TLR4 and NKT cell synergy in immunotherapy against visceral leishmaniasis. PLoS Pathog. 2012;8(4):e1002646. doi: 10.1371/journal.ppat.1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paul J, Karmakar S, De T. TLR-mediated distinct IFN-gamma/IL-10 pattern induces protective immunity against murine visceral leishmaniasis. Eur J Immunol. 2012;42(8):2087–2099. doi: 10.1002/eji.201242428. [DOI] [PubMed] [Google Scholar]
- 75.Liese J, Schleicher U, Bogdan C. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol. 2007;37(12):3424–3434. doi: 10.1002/eji.200737182. [DOI] [PubMed] [Google Scholar]
- 76.Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, Fischer JA, Weiss S, Kalinke U, Kunz S, Bogdan C. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med. 2007;204(4):893–906. doi: 10.1084/jem.20061293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gurung P, Kanneganti TD. Innate immunity against Leishmania infections. Cell Microbiol. 2015 doi: 10.1111/cmi.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kautz-Neu K, Schwonberg K, Fischer MR, Schermann AI, von Stebut E. Dendritic cells in Leishmania major infections: mechanisms of parasite uptake, cell activation and evidence for physiological relevance. Med Microbiol Immunol. 2012;201(4):581–592. doi: 10.1007/s00430-012-0261-2. [DOI] [PubMed] [Google Scholar]
- 79.Rodriguez NE, Wilson ME. Eosinophils and mast cells in leishmaniasis. Immunol Res. 2014;59(1–3):129–141. doi: 10.1007/s12026-014-8536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smelt SC, Cotterell SE, Engwerda CR, Kaye PM. B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J Immunol. 2000;164(7):3681–3688. doi: 10.4049/jimmunol.164.7.3681. [DOI] [PubMed] [Google Scholar]
- 81.Holaday BJ, Sadick MD, Wang ZE, Reiner SL, Heinzel FP, Parslow TG, Locksley RM. Reconstitution of Leishmania immunity in severe combined immunodeficient mice using Th1- and Th2-like cell lines. J Immunol. 1991;147(5):1653–1658. [PubMed] [Google Scholar]
- 82.Mitchell GF. Murine cutaneous leishmaniasis: resistance in reconstituted nude mice and several F1 hybrids infected with Leishmania tropica major. J Immunogenet. 1983;10(5):395–412. doi: 10.1111/j.1744-313X.1983.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 83.Erb K, Blank C, Ritter U, Bluethmann H, Moll H. Leishmania major infection in major histocompatibility complex class II-deficient mice: CD8+ T cells do not mediate a protective immune response. Immunobiology. 1996;195(2):243–260. doi: 10.1016/S0171-2985(96)80043-X. [DOI] [PubMed] [Google Scholar]
- 84.Titus RG, Milon G, Marchal G, Vassalli P, Cerottini JC, Louis JA. Involvement of specific Lyt-2+ T cells in the immunological control of experimentally induced murine cutaneous leishmaniasis. Eur J Immunol. 1987;17(10):1429–1433. doi: 10.1002/eji.1830171007. [DOI] [PubMed] [Google Scholar]
- 85.Locksley RM, Reiner SL, Hatam F, Littman DR, Killeen N. Helper T cells without CD4: control of leishmaniasis in CD4-deficient mice. Science. 1993;261(5127):1448–1451. doi: 10.1126/science.8367726. [DOI] [PubMed] [Google Scholar]
- 86.Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, Udey MC, Sacks D. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major . J Immunol. 2002;168(8):3992–4000. doi: 10.4049/jimmunol.168.8.3992. [DOI] [PubMed] [Google Scholar]
- 87.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10(2):89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 88.Voronov E, Dotan S, Gayvoronsky L, White RM, Cohen I, Krelin Y, Benchetrit F, Elkabets M, Huszar M, El-On J, Apte RN. IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. Int Immunol. 2010;22(4):245–257. doi: 10.1093/intimm/dxq006. [DOI] [PubMed] [Google Scholar]
- 89.Xin L, Li Y, Soong L. Role of interleukin-1beta in activating the CD11c(high) CD45RB- dendritic cell subset and priming Leishmania amazonensis-specific CD4+ T cells in vitro and in vivo. Infect Immun. 2007;75(10):5018–5026. doi: 10.1128/IAI.00499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kautz-Neu K, Kostka SL, Dinges S, Iwakura Y, Udey MC, von Stebut E. A role for leukocyte-derived IL-1RA in DC homeostasis revealed by increased susceptibility of IL-1RA-deficient mice to cutaneous leishmaniasis. J Invest Dermatol. 2011;131(8):1650–1659. doi: 10.1038/jid.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bryson KJ, Wei XQ, Alexander J. Interleukin-18 enhances a Th2 biased response and susceptibility to Leishmania mexicana in BALB/c mice. Microbes Infect/Institut Pasteur. 2008;10(7):834–839. doi: 10.1016/j.micinf.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 92.Wei XQ, Niedbala W, Xu D, Luo ZX, Pollock KG, Brewer JM. Host genetic background determines whether IL-18 deficiency results in increased susceptibility or resistance to murine Leishmania major infection. Immunol Lett. 2004;94(1–2):35–37. doi: 10.1016/j.imlet.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 93.Kautz-Neu K, Kostka SL, Dinges S, Iwakura Y, Udey MC, von Stebut E. IL-1 signalling is dispensable for protective immunity in Leishmania-resistant mice. Exp Dermatol. 2011;20(1):76–78. doi: 10.1111/j.1600-0625.2010.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Monteforte GM, Takeda K, Rodriguez-Sosa M, Akira S, David JR, Satoskar AR. Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J Immunol. 2000;164(11):5890–5893. doi: 10.4049/jimmunol.164.11.5890. [DOI] [PubMed] [Google Scholar]
- 95.Wei XQ, Leung BP, Niedbala W, Piedrafita D, Feng GJ, Sweet M, Dobbie L, Smith AJ, Liew FY. Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J Immunol. 1999;163(5):2821–2828. [PubMed] [Google Scholar]
- 96.Ohkusu K, Yoshimoto T, Takeda K, Ogura T, Kashiwamura S, Iwakura Y, Akira S, Okamura H, Nakanishi K. Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infect Immun. 2000;68(5):2449–2456. doi: 10.1128/IAI.68.5.2449-2456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, Kanneganti TD. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J Clin Investig. 2015;125(3):1329–1338. doi: 10.1172/JCI79526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, Gutierrez FR, Bellio M, Bortoluci KR, Flavell RA, Bozza MT, Silva JS, Zamboni DS. Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nat Med. 2013;19(7):909–915. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 99.Shio MT, Christian JG, Jung JY, Chang KP, Olivier M. PKC/ROS-mediated NLRP3 inflammasome activation is attenuated by leishmania zinc-metalloprotease during infection. PLoS Negl Trop Dis. 2015;9(6):e0003868. doi: 10.1371/journal.pntd.0003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, Mayer-Barber K, Tacchini-Cottier F, Sacks DL. The Nlrp3 inflammasome, IL-1beta, and neutrophil recruitment are required for susceptibility to a non-healing strain of Leishmania major in C57BL/6 mice. Eur J Immunol. 2015 doi: 10.1002/eji.201546015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoover DL, Berger M, Hammer CH, Meltzer MS. Complement-mediated serum cytotoxicity for Leishmania major amastigotes: killing by serum deficient in early components of the membrane attack complex. J Immunol. 1985;135(1):570–574. [PubMed] [Google Scholar]
- 102.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89(4):587–596. doi: 10.1016/S0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 103.WHO (2014) World Health Organization. Malaria. World Malaria Report 2014. http://www.whoint/malaria/media/world_malaria_report_2014/en/
- 104.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19(2):156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Amino R, Giovannini D, Thiberge S, Gueirard P, Boisson B, Dubremetz JF, Prevost MC, Ishino T, Yuda M, Menard R. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe. 2008;3(2):88–96. doi: 10.1016/j.chom.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 106.Mac-Daniel L, Buckwalter MR, Berthet M, Virk Y, Yui K, Albert ML, Gueirard P, Menard R. Local immune response to injection of Plasmodium sporozoites into the skin. J Immunol. 2014;193(3):1246–1257. doi: 10.4049/jimmunol.1302669. [DOI] [PubMed] [Google Scholar]
- 107.Vreden SG. The role of Kupffer cells in the clearance of malaria sporozoites from the circulation. Parasitol Today. 1994;10(8):304–308. doi: 10.1016/0169-4758(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 108.Scheller LF, Wirtz RA, Azad AF. Susceptibility of different strains of mice to hepatic infection with Plasmodium berghei . Infect Immun. 1994;62(11):4844–4847. doi: 10.1128/iai.62.11.4844-4847.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vanderberg JP, Khan ZM, Stewart MJ. Induction of hepatic inflammatory response by Plasmodium berghei sporozoites protects BALB/c mice against challenge with Plasmodium yoelii sporozoites. J Parasitol. 1993;79(5):763–767. doi: 10.2307/3283617. [DOI] [PubMed] [Google Scholar]
- 110.Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW, Pravenec M, Kain KC. CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis. 2004;189(2):204–213. doi: 10.1086/380764. [DOI] [PubMed] [Google Scholar]
- 111.Aitman TJ, Cooper LD, Norsworthy PJ, Wahid FN, Gray JK, Curtis BR, McKeigue PM, Kwiatkowski D, Greenwood BM, Snow RW, Hill AV, Scott J. Malaria susceptibility and CD36 mutation. Nature. 2000;405(6790):1015–1016. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 112.Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. 2014;14(11):744–757. doi: 10.1038/nri3742. [DOI] [PubMed] [Google Scholar]
- 113.Tsuji M. A retrospective evaluation of the role of T cells in the development of malaria vaccine. Exp Parasitol. 2010;126(3):421–425. doi: 10.1016/j.exppara.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodrigues MM, Cordey AS, Arreaza G, Corradin G, Romero P, Maryanski JL, Nussenzweig RS, Zavala F. CD8+ cytolytic T cell clones derived against the Plasmodium yoelii circumsporozoite protein protect against malaria. Int Immunol. 1991;3(6):579–585. doi: 10.1093/intimm/3.6.579. [DOI] [PubMed] [Google Scholar]
- 115.Romero P, Maryanski JL, Corradin G, Nussenzweig RS, Nussenzweig V, Zavala F. Cloned cytotoxic T cells recognize an epitope in the circumsporozoite protein and protect against malaria. Nature. 1989;341(6240):323–326. doi: 10.1038/341323a0. [DOI] [PubMed] [Google Scholar]
- 116.Sano G, Hafalla JC, Morrot A, Abe R, Lafaille JJ, Zavala F. Swift development of protective effector functions in naive CD8(+) T cells against malaria liver stages. J Exp Med. 2001;194(2):173–180. doi: 10.1084/jem.194.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zavala F, Rodrigues M, Rodriguez D, Rodriguez JR, Nussenzweig RS, Esteban M. A striking property of recombinant poxviruses: efficient inducers of in vivo expansion of primed CD8(+) T cells. Virology. 2001;280(2):155–159. doi: 10.1006/viro.2000.0792. [DOI] [PubMed] [Google Scholar]
- 118.Perez-Mazliah D, Langhorne J. CD4 T-cell subsets in malaria: TH1/TH2 revisited. Front Immunol. 2014;5:671. doi: 10.3389/fimmu.2014.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tsuji M, Romero P, Nussenzweig RS, Zavala F. CD4+ cytolytic T cell clone confers protection against murine malaria. J Exp Med. 1990;172(5):1353–1357. doi: 10.1084/jem.172.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang R, Charoenvit Y, Corradin G, De La Vega P, Franke ED, Hoffman SL. Protection against malaria by Plasmodium yoelii sporozoite surface protein 2 linear peptide induction of CD4+ T cell- and IFN-gamma-dependent elimination of infected hepatocytes. J Immunol. 1996;157(9):4061–4067. [PubMed] [Google Scholar]
- 121.Weiss WR, Mellouk S, Houghten RA, Sedegah M, Kumar S, Good MF, Berzofsky JA, Miller LH, Hoffman SL. Cytotoxic T cells recognize a peptide from the circumsporozoite protein on malaria-infected hepatocytes. J Exp Med. 1990;171(3):763–773. doi: 10.1084/jem.171.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13(2):188–195. doi: 10.1038/ni.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carvalho LH, Sano G, Hafalla JC, Morrot A, Curotto de Lafaille MA, Zavala F. IL-4-secreting CD4+ T cells are crucial to the development of CD8+ T-cell responses against malaria liver stages. Nat Med. 2002;8(2):166–170. doi: 10.1038/nm0202-166. [DOI] [PubMed] [Google Scholar]
- 124.Oliveira GA, Kumar KA, Calvo-Calle JM, Othoro C, Altszuler D, Nussenzweig V, Nardin EH. Class II-restricted protective immunity induced by malaria sporozoites. Infect Immun. 2008;76(3):1200–1206. doi: 10.1128/IAI.00566-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Finney CA, Lu Z, LeBourhis L, Philpott DJ, Kain KC. Disruption of Nod-like receptors alters inflammatory response to infection but does not confer protection in experimental cerebral malaria. Am J Trop Med Hyg. 2009;80(5):718–722. [PubMed] [Google Scholar]
- 126.Egan TJ. Haemozoin formation. Mol Biochem Parasitol. 2008;157(2):127–136. doi: 10.1016/j.molbiopara.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 127.Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. 2009;4(8):e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. 2009;183(8):5208–5220. doi: 10.4049/jimmunol.0713552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, Sutterwala FS, Bohle DS, Descoteaux A, Flavell RA, Olivier M. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5(8):e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kordes M, Matuschewski K, Hafalla JC. Caspase-1 activation of interleukin-1beta (IL-1beta) and IL-18 is dispensable for induction of experimental cerebral malaria. Infect Immun. 2011;79(9):3633–3641. doi: 10.1128/IAI.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reimer T, Shaw MH, Franchi L, Coban C, Ishii KJ, Akira S, Horii T, Rodriguez A, Nunez G. Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur J Immunol. 2010;40(3):764–769. doi: 10.1002/eji.200939996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, Elian S, Martins FS, Pereira D, Reed G, Fitzgerald KA, Golenbock DT, Gazzinelli RT. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. 2014;10(1):e1003885. doi: 10.1371/journal.ppat.1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Santos ML, Reis EC, Bricher PN, Sousa TN, Brito CF, Lacerda MV, Fontes CJ, Carvalho LH, Pontillo A. Contribution of inflammasome genetics in Plasmodium vivax malaria. Infect Genet Evol. 2016 doi: 10.1016/j.meegid.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 134.Flegr J, Prandota J, Sovickova M, Israili ZH. Toxoplasmosis—a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS One. 2014;9(3):e90203. doi: 10.1371/journal.pone.0090203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363(9425):1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 136.Robert-Gangneux F, Darde ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. 2012;25(2):264–296. doi: 10.1128/CMR.05013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol. 2009;39(8):895–901. doi: 10.1016/j.ijpara.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, Hieny S, Gazzinelli RT, Sher A. In the absence of endogenous IFN-gamma, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J Immunol. 1996;157(9):4045–4054. [PubMed] [Google Scholar]
- 139.Gazzinelli RT, Hieny S, Wynn TA, Wolf S, Sher A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci USA. 1993;90(13):6115–6119. doi: 10.1073/pnas.90.13.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii . J Exp Med. 1997;185(7):1261–1273. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bliss SK, Gavrilescu LC, Alcaraz A, Denkers EY. Neutrophil depletion during Toxoplasma gondii infection leads to impaired immunity and lethal systemic pathology. Infect Immun. 2001;69(8):4898–4905. doi: 10.1128/IAI.69.8.4898-4905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS, Aliberti J. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J Immunol. 2006;177(1):31–35. doi: 10.4049/jimmunol.177.1.31. [DOI] [PubMed] [Google Scholar]
- 143.Yarovinsky F. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol. 2014;14(2):109–121. doi: 10.1038/nri3598. [DOI] [PubMed] [Google Scholar]
- 144.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308(5728):1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 145.Lindberg RE, Frenkel JK. Toxoplasmosis in nude mice. J Parasitol. 1977;63(2):219–221. doi: 10.2307/3280044. [DOI] [PubMed] [Google Scholar]
- 146.Suzuki Y, Joh K, Kobayashi A. Tumor necrosis factor-independent protective effect of recombinant IFN-gamma against acute toxoplasmosis in T cell-deficient mice. J Immunol. 1991;147(8):2728–2733. [PubMed] [Google Scholar]
- 147.Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii . J Immunol. 1992;149(1):175–180. [PubMed] [Google Scholar]
- 148.Hunter CA, Litton MJ, Remington JS, Abrams JS. Immunocytochemical detection of cytokines in the lymph nodes and brains of mice resistant or susceptible to toxoplasmic encephalitis. J Infect Dis. 1994;170(4):939–945. doi: 10.1093/infdis/170.4.939. [DOI] [PubMed] [Google Scholar]
- 149.Schluter D, Hein A, Dorries R, Deckert-Schluter M. Different subsets of T cells in conjunction with natural killer cells, macrophages, and activated microglia participate in the intracerebral immune response to Toxoplasma gondii in athymic nude and immunocompetent rats. Am J Pathol. 1995;146(4):999–1007. [PMC free article] [PubMed] [Google Scholar]
- 150.Shaw MH, Reimer T, Sanchez-Valdepenas C, Warner N, Kim YG, Fresno M, Nunez G. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii . Nat Immunol. 2009;10(12):1267–1274. doi: 10.1038/ni.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suzuki Y, Conley FK, Remington JS. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J Immunol. 1989;143(6):2045–2050. [PubMed] [Google Scholar]
- 152.Suzuki Y, Yang Q, Yang S, Nguyen N, Lim S, Liesenfeld O, Kojima T, Remington JS. IL-4 is protective against development of toxoplasmic encephalitis. J Immunol. 1996;157(6):2564–2569. [PubMed] [Google Scholar]
- 153.Caetano BC, Biswas A, Lima DS, Jr, Benevides L, Mineo TW, Horta CV, Lee KH, Silva JS, Gazzinelli RT, Zamboni DS, Kobayashi KS. Intrinsic expression of Nod2 in CD4+ T lymphocytes is not necessary for the development of cell-mediated immunity and host resistance to Toxoplasma gondii . Eur J Immunol. 2011;41(12):3627–3631. doi: 10.1002/eji.201141876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, Cavailles P, Bisanz C, Cesbron-Delauw MF, Fournie GJ, McLeod R. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79(2):756–766. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gov L, Karimzadeh A, Ueno N, Lodoen MB (2013) Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio 4(4). doi:10.1128/mBio.00255-13 [DOI] [PMC free article] [PubMed]
- 156.Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii . Infect Immun. 2014;82(1):460–468. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, Masters S, Sher A, Leppla SH, Moayeri M, Saeij JP, Grigg ME (2014) Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5(1). doi:10.1128/mBio.01117-13 [DOI] [PMC free article] [PubMed]
- 158.Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl Trop Dis. 2008;2(9):e300. doi: 10.1371/journal.pntd.0000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lescure FX, Le Loup G, Freilij H, Develoux M, Paris L, Brutus L, Pialoux G. Chagas disease: changes in knowledge and management. Lancet Infect Dis. 2010;10(8):556–570. doi: 10.1016/S1473-3099(10)70098-0. [DOI] [PubMed] [Google Scholar]
- 160.Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, Factor SM, Kirchhoff LV, Weiss LM. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease) Prog Cardiovasc Dis. 2009;51(6):524–539. doi: 10.1016/j.pcad.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, Ropert C, Gazzinelli RT. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. 2004;172(3):1711–1718. doi: 10.4049/jimmunol.172.3.1711. [DOI] [PubMed] [Google Scholar]
- 162.Roggero E, Wildmann J, Passerini MO, del Rey A, Besedovsky HO. Different peripheral neuroendocrine responses to Trypanosoma cruzi infection in mice lacking adaptive immunity. Ann N Y Acad Sci. 2012;1262:37–44. doi: 10.1111/j.1749-6632.2012.06645.x. [DOI] [PubMed] [Google Scholar]
- 163.Teixeira MM, Gazzinelli RT, Silva JS. Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol. 2002;18(6):262–265. doi: 10.1016/S1471-4922(02)02283-3. [DOI] [PubMed] [Google Scholar]
- 164.Graefe SE, Jacobs T, Gaworski I, Klauenberg U, Steeg C, Fleischer B. Interleukin-12 but not interleukin-18 is required for immunity to Trypanosoma cruzi in mice. Microbes Infect. 2003;5(10):833–839. doi: 10.1016/S1286-4579(03)00176-X. [DOI] [PubMed] [Google Scholar]
- 165.Holscher C, Kohler G, Muller U, Mossmann H, Schaub GA, Brombacher F. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect Immun. 1998;66(3):1208–1215. doi: 10.1128/iai.66.3.1208-1215.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Torrico F, Heremans H, Rivera MT, Van Marck E, Billiau A, Carlier Y. Endogenous IFN-gamma is required for resistance to acute Trypanosoma cruzi infection in mice. J Immunol. 1991;146(10):3626–3632. [PubMed] [Google Scholar]
- 167.Chen L, Watanabe T, Watanabe H, Sendo F. Neutrophil depletion exacerbates experimental Chagas’ disease in BALB/c, but protects C57BL/6 mice through modulating the Th1/Th2 dichotomy in different directions. Eur J Immunol. 2001;31(1):265–275. doi: 10.1002/1521-4141(200101)31:1<265::AID-IMMU265>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 168.Lieke T, Graefe SE, Klauenberg U, Fleischer B, Jacobs T. NK cells contribute to the control of Trypanosoma cruzi infection by killing free parasites by perforin-independent mechanisms. Infect Immun. 2004;72(12):6817–6825. doi: 10.1128/IAI.72.12.6817-6825.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rodrigues MM, Oliveira AC, Bellio M. The immune response to Trypanosoma cruzi: role of Toll-like receptors and perspectives for vaccine development. J Parasitol Res. 2012;2012:507874. doi: 10.1155/2012/507874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tarleton RL, Grusby MJ, Postan M, Glimcher LH. Trypanosoma cruzi infection in MHC-deficient mice: further evidence for the role of both class I- and class II-restricted T cells in immune resistance and disease. Int Immunol. 1996;8(1):13–22. doi: 10.1093/intimm/8.1.13. [DOI] [PubMed] [Google Scholar]
- 171.Tarleton RL, Koller BH, Latour A, Postan M. Susceptibility of beta 2-microglobulin-deficient mice to Trypanosoma cruzi infection. Nature. 1992;356(6367):338–340. doi: 10.1038/356338a0. [DOI] [PubMed] [Google Scholar]
- 172.Antunez MI, Cardoni RL. Early IFN-gamma production is related to the presence of interleukin (IL)-18 and the absence of IL-13 in experimental Trypanosoma cruzi infections. Immunol Lett. 2001;79(3):189–196. doi: 10.1016/S0165-2478(01)00283-8. [DOI] [PubMed] [Google Scholar]
- 173.Silva JS, Morrissey PJ, Grabstein KH, Mohler KM, Anderson D, Reed SG. Interleukin 10 and interferon gamma regulation of experimental Trypanosoma cruzi infection. J Exp Med. 1992;175(1):169–174. doi: 10.1084/jem.175.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tarleton RL. CD8+ T cells in Trypanosoma cruzi infection. Semin Immunopathol. 2015;37(3):233–238. doi: 10.1007/s00281-015-0481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Silva GK, Gutierrez FR, Guedes PM, Horta CV, Cunha LD, Mineo TW, Santiago-Silva J, Kobayashi KS, Flavell RA, Silva JS, Zamboni DS. Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol. 2010;184(3):1148–1152. doi: 10.4049/jimmunol.0902254. [DOI] [PubMed] [Google Scholar]
- 176.Goncalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, Rodrigues MM, Alvarez JM, Bortoluci KR. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PLoS Negl Trop Dis. 2013;7(10):e2469. doi: 10.1371/journal.pntd.0002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FR, Guedes PM, Andrade WA, De Niz M, Gazzinelli RT, Zamboni DS, Silva JS. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1beta response and host resistance to Trypanosoma cruzi infection. J Immunol. 2013;191(6):3373–3383. doi: 10.4049/jimmunol.1203293. [DOI] [PubMed] [Google Scholar]
- 178.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2(12):933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 179.Niedbala W, Cai B, Liew FY. Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis. 2006;65(Suppl 3):iii37–iii40. doi: 10.1136/ard.2006.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lupfer C, Thomas PG, Kanneganti TD. Nucleotide oligomerization and binding domain 2-dependent dendritic cell activation is necessary for innate immunity and optimal CD8+ T Cell responses to influenza A virus infection. J Virol. 2014;88(16):8946–8955. doi: 10.1128/JVI.01110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12(12):813–820. doi: 10.1038/nri3339. [DOI] [PubMed] [Google Scholar]
- 182.Griffith TS, Ferguson TA. Cell death in the maintenance and abrogation of tolerance: the five Ws of dying cells. Immunity. 2011;35(4):456–466. doi: 10.1016/j.immuni.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, Vogel P, Chi H, Kanneganti TD. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity. 2015;42(4):654–664. doi: 10.1016/j.immuni.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505(7484):509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]