

Activation of innate immune related pathways including Toll-like receptor (TLR) signaling play a key role in chronic liver diseases. Apart from the finding that TLR9 is involved in the development of nonalcoholic steatohepatitis (NASH), very little is known about the identity of TLR9 ligands and how the TLR9 pathway is activated in NASH. In the recent Journal Clinical Investigation article, Garcia-Martinez et al1 uncovered a mitochondrial DNA (mtDNA) – TLR9 axis as an important signaling pathway by which hepatocytes communicate with leukocytes via extracellular vesicles to mount an inflammatory response.
Nonalcoholic Fatty Liver Disease (NAFLD) is currently the most common form of chronic liver disease affecting both adults and children in the United States and many other parts of the world. The spectrum of NAFLD includes isolated steatosis and NASH. While patients with isolated steatosis appear to have a benign non-progressive clinical course, those with NASH, characterized by steatosis along with hepatocellular injury, inflammation and varying degrees of fibrosis, may have a potentially serious condition.2 Among these patients, those with liver fibrosis appear to be the ones at higher risk of overall and liver-related morbidity and mortality.3 The clinical importance of NAFLD and the current lack of effective medications to stop or reverse disease progression in patients with NASH have sparked great interest and intense investigation to identify relevant pathophysiologic mechanisms that can be target for development of novel therapies. Lipotoxicity, a process by which accumulation of certain toxic lipids such as saturated free fatty acids (SFA), free cholesterol, or ceramides among others in hepatocytes triggers various molecular pathways of cell stress in particular mitochondrial dysfunction and eventually cell death have evolved as a key event during NASH development.4 Indeed, studies in experimental models of NASH, as well as in humans with NASH have demonstrated that liver cells have both structural and functional mitochondrial abnormalities.5 Structural abnormalities include mitochondrial enlargement and development of crystalline inclusions, whereas functional mitochondrial abnormalities are characterized by enhanced production of reactive oxygen species, accumulation of lipid peroxides, and release of cytochrome c into the cytoplasm. More recently, release of extracellular vesicles, a heterogeneous population of small membrane-bound structures that include exosomes and microparticles released by cells in the extracellular environment as well as in the bloodstream have been identified as a consequence of hepatocyte lipotoxicity.6 These vesicles are effective communicators that are generated by a cell of origin and can act on a number of target cells in a paracrine manner in the microenvironment where they are released as well as in an endocrine manner, acting as long-range signals. Extracellular vesicles released by lipotoxic hepatocytes have been shown to activate immune cells as well as hepatic stellate cells and thus may link excess lipid deposition in hepatocytes to innate immune activation, inflammation, and fibrosis.7 The molecular mechanisms involved in these effects are just now starting to be understood.
First, Garcia-Martinez et al showed that plasma from both mice and patients with NASH have increased levels of mitochondrial DNA and oxidized DNA in microparticles that have the ability to activate the endosomal pattern recognition receptor TLR9. For initial studies, mice were fed a high fat diet (HFD) for 12-week, a model associated with the early stages of NASH including steatosis, mild inflammation and ballooning of hepatocytes but without fibrosis. Plasma mtDNA was increased in these mice when compared to mice fed a control diet. Using FACS analysis combining size gating and dual staining with Mitotracker Red, for mitochondria, and PKH67 for microparticles, they found that most of the circulating mitochondrial and mtDNA were present inside microparticles of hepatocyte origin. Depletion of microparticles from plasma of HFD fed mice resulted in loss of the ability to activate a TLR9 reporter cell line. To assess whether these changes were also present in humans they used plasma from three groups of patients: (1) lean subjects without liver disease and normal ALT levels, (2) obese subjects with normal ALT levels, and (3) obese subjects with elevated ALT levels. They found that the human data mirrored the findings on mice on the high fat diet.
Previous studies by this group showed that global TLR9 deficiency results in protection from the development of NASH in experimental models. To address the cell type specificity that is responsible for the TLR9 signaling, they used a Lysozyme (Lysm)-Cre approach to delete TLR9 in myeloid-derived cells. Those mice were placed on a HFD for 12-weeks as before. Both the global Tlr9-KO and the Lysm-Cre Tlr9fl/fl mice showed protection from hepatic steatosis, as well as ballooning, and inflammation induced by this diet. Levels of total liver inflammatory cytokine gene expression were also significantly reduced in Tlr9-KO and Lysm-Cre Tlr9fl/fl mice when compared to WT mice fed a HFD.
Authors concluded that the findings of high plasma TLR9 ligand activity in NASH and the requirement of TLR9 in the development of HFD-induced NASH might have direct therapeutic implications. In order to investigate this hypothesis, they used a TLR7/9 antagonist IRS954. WT mice were placed on a HFD and concurrently administered IRS954 subcutaneously (5 mg/kg weekly). This resulted in a significant reduction in steatosis, ballooning and inflammation, serum transaminases, and inflammatory cytokine transcript levels. A therapeutic protocol calling for initiation of IRS954 after 8 weeks of a HFD, up to a total of 12 weeks on a HFD, also resulted in a significant reduction in hepatic steatosis, ballooning and inflammation, and serum transaminases, identifying TLR9 inhibition as a potential novel therapeutic approach for NASH.
Some of the limitations of the study included the use of an experimental model that recapitulates only the very early stages of NASH and thus did not allow to assess the relationship with more severe liver injury and in particular with the presence of liver fibrosis. Also, the group of patients tested did not have liver biopsy available to address the association of circulating microparticles containing mitochondrial DNA with histological features of disease severity in these patients. As mentioned before, IRS954 has inhibitory effects on both TLR9 and TLR7 signaling. Here, a word of caution should be brought with regards to inhibition of TLR7 in NASH and in particular in fibrotic-NASH. Recent studies have shown that stimulation of TLR7 in dendritic cells has anti-fibrotic effects, suggesting that TLR7 activation could be used as a therapeutic strategy in fibrosis.8
In summary, the findings by Garcia-Martinez et al uncover a novel role for TLR9 in the association between mitochondrial dysfunction and oxidized mtDNA, two key changes related to lipotoxicity, and the development of a proinflammatory response. Moreover, the data reveals new therapeutic targets, including mtDNA oxidation, microparticles release and uptake, as well as TLR9 ligand binding. Further studies will be necessary to elucidate the role of this novel signaling pathway in more advance stages in particular those associated with liver fibrosis that represent the subgroup at more risk for liver related complications. Moreover, the possibility that hepatocyte-derived mtDNA activate immune cells, especially macrophages, systemically or in extrahepatic sites should be explored.
Fig. 1.
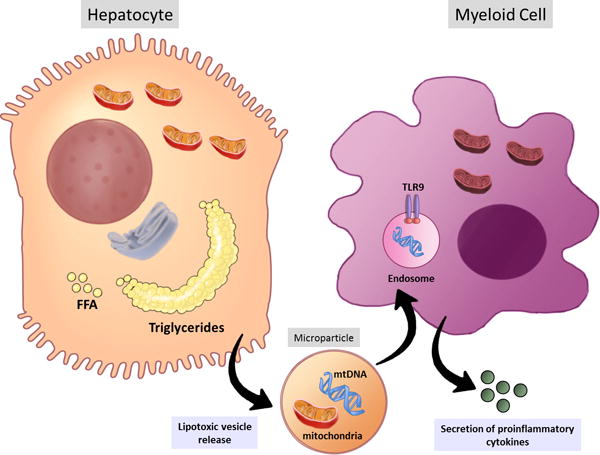
Lipotoxic hepatocytes release microparticles containing mtDNA and oxidized DNA as well as intact mitochondria. mtDNA is recognized by TLR9 expressed in the endosomes of the myeloid cells triggering the secretion of proinflammatory cytokines, such as TNF-alpha and IL1b, which can further enhance hepatic damage. FFA. Free fatty acids; mtDNA: mitochondrial DNA; TLR9: Toll like receptor 9.
Acknowledgments
Funding: This work was funded by NIH grants R01 DK082451, U01 AA022489 to AEF. AW is support by the START-Program of the Faculty of Medicine, RWTH Aachen, and DFG grant WR173/3-1.
Footnotes
Conflict of Interest: The authors state no conflict of interest.
References
- 1.Garcia-Martinez I, Santoro N, Chen Y, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. The Journal of Clinical Investigation. 126:859–864. doi: 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Machado MV, Diehl AM. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rinella ME, Sanyal AJ. Management of NAFLD: a stage-based approach. Nat Rev Gastroenterol Hepatol. 2016 doi: 10.1038/nrgastro.2016.3. [DOI] [PubMed] [Google Scholar]
- 4.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–79. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Begriche K, Massart J, Robin MA, et al. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497–507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- 6.Povero D, Feldstein AE. Novel Molecular Mechanisms in the Development of Non-Alcoholic Steatohepatitis. Diabetes Metab J. 2016;40:1–11. doi: 10.4093/dmj.2016.40.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Povero D, Panera N, Eguchi A, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol Gastroenterol Hepatol. 2015;1:646–663 e4. doi: 10.1016/j.jcmgh.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roh YS, Park S, Kim JW, et al. Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. Hepatology. 2014;60:237–49. doi: 10.1002/hep.26981. [DOI] [PMC free article] [PubMed] [Google Scholar]