

Supplemental Digital Content is available in the text.
Keywords: collagen, fibrosis, heart failure, hypertension, right ventricular dysfunction
Abstract
Background—
The purpose of this study was to determine the relative contribution of fibrosis-mediated and myofibril-mediated stiffness in rats with mild and severe right ventricular (RV) dysfunction.
Methods and Results—
By performing pulmonary artery banding of different diameters for 7 weeks, mild RV dysfunction (Ø=0.6 mm) and severe RV dysfunction (Ø=0.5 mm) were induced in rats. The relative contribution of fibrosis- and myofibril-mediated RV stiffness was determined in RV trabecular strips. Total myocardial stiffness was increased in trabeculae from both mild and severe RV dysfunction in comparison to controls. In severe RV dysfunction, increased RV myocardial stiffness was explained by both increased fibrosis-mediated stiffness and increased myofibril-mediated stiffness, whereas in mild RV dysfunction, only myofibril-mediated stiffness was increased in comparison to control. Histological analyses revealed that RV fibrosis gradually increased with severity of RV dysfunction, whereas the ratio of collagen I/III expression was only elevated in severe RV dysfunction. Stiffness measurements in single membrane-permeabilized RV cardiomyocytes demonstrated a gradual increase in RV myofibril stiffness, which was partially restored by protein kinase A in both mild and severe RV dysfunction. Increased expression of compliant titin isoforms was observed only in mild RV dysfunction, whereas titin phosphorylation was reduced in both mild and severe RV dysfunction.
Conclusions—
RV myocardial stiffness is increased in rats with mild and severe RV dysfunction. In mild RV dysfunction, stiffness is mainly determined by increased myofibril stiffness. In severe RV dysfunction, both myofibril- and fibrosis-mediated stiffness contribute to increased RV myocardial stiffness.
Patients with pulmonary arterial hypertension (PAH) develop right heart failure (RHF) because of a progressive increase in right ventricular (RV) pressure overload.1 Although it is known for some years that RV systolic adaptation is of clinical importance, it has just recently became clear that RV diastolic stiffness increases and may contribute to disease progression in PAH.2,3 In addition, we have previously shown that RV diastolic stiffness was closely associated with a doubling in sarcomere-derived cardiomyocyte stiffness and increased myocardial fibrosis in end-stage PAH patients.2,4
See Clinical Perspective
Changes in myofibril stiffness are closely regulated by the giant elastic protein titin.5 Titin stiffness can be regulated via both post-transcriptional and post-translational modifications. Post-transcriptional modification includes a shift from the compliant N2BA isoform to the stiffer N2B isoform.6 Post-translational modification is mainly regulated via phosphorylation of titin by the protein kinases A (PKA), G, and C. Titin phosphorylation by PKA7 and protein kinase G8 reduce myofibril stiffness, whereas protein kinase C–mediated titin phosphorylation results in increased RV diastolic stiffness.9–11 We have previously demonstrated that in end-stage PAH, no alterations in titin isoform composition are observed,2 whereas PKA-mediated titin phosphorylation was significantly reduced.4
Changes in fibrosis could also contribute to diastolic stiffness and involve differences in collagen fiber type secretion, collagen type I/III ratio, cross-linking, or degradation.12–14 Previous studies found a positive correlation between markers of collagen degradation measured in the serum of patients with PAH and the severity of the disease.15 In addition, late gadolinium enhancement studies in patients with PAH further indicate a positive association between RV fibrosis and worsening of RV function.16,17
However, the functional relevance of increased fibrosis and myofibril stiffness to RV diastolic stiffness remains to be elucidated. Moreover, it is currently unclear whether diastolic stiffening of the RV already occurs at earlier stages of PAH-induced RHF, because obtaining human RV tissue is only limited to end-stage PAH because of the risks of performing RV biopsies in vivo.
We propose that there are stage-specific changes in the structure of the RV, which influence its diastolic function.
To mimic disease severity observed in patients with PAH, a novel rat model of pressure overload–induced RV remodeling was developed.18 By performing pulmonary artery banding of different diameters, the following 2 phenotypes were created: (1) mild RV dysfunction, induced by a moderate increase in RV afterload/RV systolic pressure resulting in reduced RV ejection fraction (RVEF) and cardiac output but without extracardiac signs of RHF (ascites or signs of liver failure) and (2) severe RV dysfunction, induced by a further increase in RV afterload resulting in a severe reduction in RVEF and cardiac output.18
Thus, the aim of this study was to determine the relative contribution of fibrosis-mediated and myofibril-mediated RV myocardial stiffness in rats with mild and severe RV dysfunction.
Methods
Study Design
We used rat RV trabecular tissue obtained from a previous study protocol. Institutional review board approval was obtained in accordance with institutional guidelines, and rats were treated according to Danish national guidelines. All experiments were conducted in accordance with institutional guidelines and the Danish law for animal research – authorization number: 2012-15-2934-00384 Danish Ministry of Justice.18 Fifteen male Wistar Galas rats were used for this study (M&B Taconic, Ry, Denmark), which were all treatment naive. Control rats were sham operated (n=5). The 0.6-mm clip led to mild RV dysfunction (n=5), and the 0.5-mm clip led to severe RV dysfunction (n=5). After 7 weeks of pulmonary artery banding, all animals underwent hemodynamic assessment as described previously,18 and RV tissue was harvested for further analyses.
Force Measurements on Skinned Muscle Strips
Thin muscle strips with an average length of 1 mm and diameter of ≈ 0.2 mm were dissected respecting the longitudinal orientation of the fibers.19–22 The integrity of the trabecular muscle strip was checked before the stiffness determination by activating the preparation. Thereafter, the trabecular strip was transferred to a relaxing solution where it was stretched by 20% from the initial slack length with a stretch speed of 10% preparation length per second. Passive force was recorded at the end of stretch and divided by the corresponding strip cross-sectional area to normalize for variation in trabecular strip diameters (passive tension [kN/m2]=total RV myocardial stiffness). To determine the relative contribution of fibrosis and myofibrils to total RV myocardial stiffness, thick and thin filaments were extracted by immersing the muscle strips in relaxing solution containing 0.6 mol/L KCl (60 minutes at 20 °C) followed by a relaxing solution containing 1 mol/L KI (60 minutes at 20 °C).19–22 Subsequently, the muscle strips were transferred to fresh relaxing solutions, and passive force development was measured again at the end of the 20% stretch and was assumed to represent fibrosis-mediated stiffness. Myofibril-mediated stiffness was determined as total RV myocardial stiffness minus fibrosis-mediated stiffness.19–22
RV Fibrosis
Absolute RV myocardial fibrosis and collagen isoform composition were assessed as described in the Data Supplement.
Force Measurements on Skinned Cardiomyocyte
Small RV myocardial pieces were defrosted in relaxing solution, and single cardiac cells were isolated mechanically as previously described (control n=5, mild RV dysfunction n=5, and severe RV dysfunction n=5 samples).2 A detailed description of the force measurements on skinned cardiomyocytes can be found in the Data Supplement.
Titin Isoform and Phosphorylation
To determine titin isoform expression and phosphorylation, frozen RV free-wall tissue samples of control rats, rats with mild RV dysfunction, and rats with severe RV dysfunction were weighed and pulverized in liquid nitrogen using a mortar and a pestle. See Data Supplement for detailed description.
Statistical Analyses
Statistical analyses were performed using Prism 5 for Windows (GraphPad Software, Inc, San Diego, CA). P values <0.05 were considered significant. All data are presented as mean±SEM.
Because the assumption of equal variances across the groups failed, we have transformed the data by log transformation before the analyses. All analyses were performed using Kruskal–Wallis test with Dunn multiple comparison tests between control, mild, and severe RV dysfunction, unless stated otherwise. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test was performed to assess the following: (1) the difference in RV cardiomyocyte stiffness at sarcomere lengths of 1.8 to 2.4 μm between control, mild, and severe RV dysfunction (sarcomere length×group); (2) the difference in RV cardiomyocyte stiffness after PKA incubation at sarcomere lengths of 1.8 to 2.4 μm between control, mild, and severe RV dysfunction (sarcomere length×group); and (3) the effects of PKA incubation on RV cardiomyocyte stiffness of rats with mild RV dysfunction and severe RV dysfunction at a sarcomere length of 2.2 μm (PKA×group).
Results
Relative Contribution of Fibrosis- and Myofibril-Mediated Stiffness
RV diastolic stiffness was measured on small RV muscle strips of control rats (n=5; RV systolic pressure: 26±6 mm Hg; RVEF: 72±1%), rats with mild RV dysfunction (n=5; RV systolic pressure: 84±8 mm Hg; RVEF: 55±2%; both P<0.05 versus control), and rats with severe RV dysfunction (n=5; RV systolic pressure: 115±8 mm Hg; RVEF: 45±3%; both P<0.001 versus control). The hemodynamic characteristics of the rats are presented in the Table.
Table.
General Characteristics
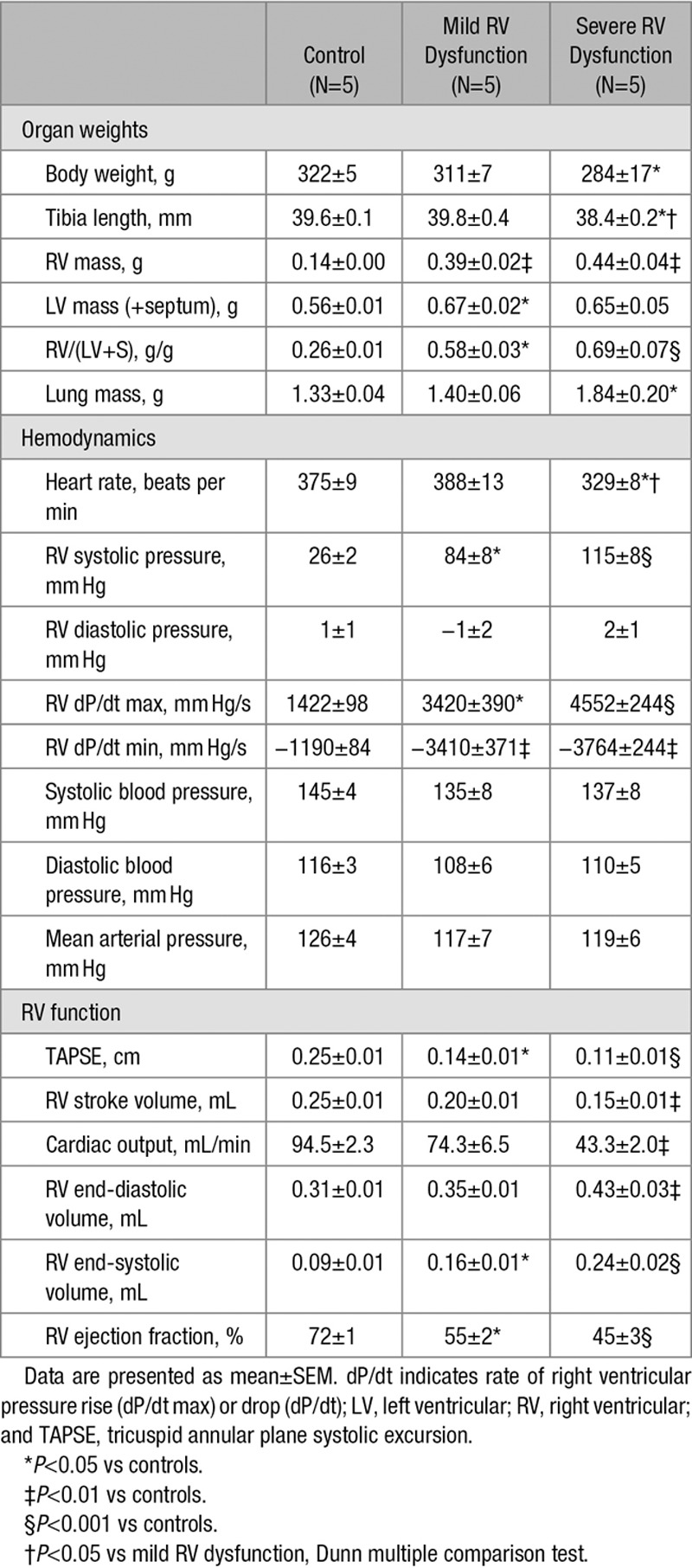
As can be observed in Figure 1, RV myocardial stiffness was significantly increased in both rats with mild and rats with severe RV dysfunction in comparison to controls. To determine the separate contribution of fibrosis on RV myocardial stiffness, the sarcomeric component of the tissue was extracted by KI/KCl treatment. This treatment disrupts titin anchoring to the thick filament and thereby eliminates the contribution of sarcomeric stiffness to the overall muscle strip stiffness, the residual stiffness being attributed to the fibrotic component. We found a stepwise increase in fibrosis-mediated stiffness with a moderate increase in fibrosis-mediated stiffness in rats with mild RV dysfunction and a further significant increase in fibrosis-mediated stiffness in rats with severe RV dysfunction. Subsequently, myofibril-derived stiffness was calculated by subtracting fibrosis stiffness from total stiffness. RV myofibril-derived stiffness was increased in all rats with RV dysfunction independent of the severity of the RV dysfunction, but only statistically significant in rats with mild RV dysfunction in comparison to control.
Figure 1.
Right ventricular (RV) myocardial stiffness in skinned trabecular strips. A, Schematic representation of skinned trabecular strip measurements. B, Representative examples of RV myocardial stiffness measurements in skinned trabecular strips of control, mild RV dysfunction, and severe RV dysfunction before (solid line) and after incubation with KCl/KI. C, Total RV myocardial stiffness was significantly increased in both mild RV dysfunction (blue bars) and severe RV dysfunction (red bars) in comparison to controls (white bars). In severe RV dysfunction, increased RV myocardial stiffness could be explained by both increased fibrosis- and myofibril-mediated stiffness, whereas in mild RV dysfunction only myofibril-mediated stiffness was increased in comparison to control. Data are presented as mean±SEM. Controls: n=5, mild RV dysfunction: n=5, and severe RV dysfunction: n=5. **P<0.01 vs control.
These data suggest that RV myocardial stiffness is closely associated with RV dysfunction. RV myofibril stiffness contributes to RV myocardial stiffness in both mild and severe RV dysfunction, whereas fibrosis-mediated stiffness led to a further increase in RV myocardial stiffness in rats with severe RV dysfunction only.
RV Fibrosis
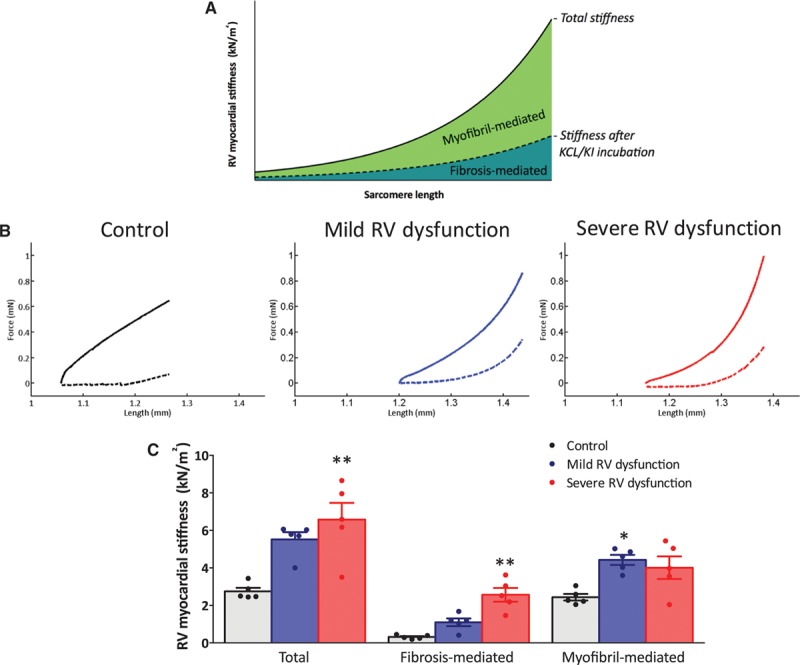
To explain the increasing contribution of fibrosis-mediated stiffness with the severity of RV dysfunction, we further investigated the presence of fibrotic areas in the RV free wall. As observed previously,18 the percentage of fibrosis gradually increased with the severity of RV dysfunction (Figure 2A). Fibrosis-mediated RV myocardial stiffness can further be influenced by the predominant type of collagen fibers expressed, where increased collagen I/III ratio is known to be associated with increased myocardial stiffness.13 Interestingly, RV collagen I/III ratio was only increased in rats with severe RV dysfunction (Figure 2B). These data suggest that the increased fibrosis-mediated stiffness is a consequence of increased fibrosis and increased collagen I/III expression in the right ventricle of rats with severe RV dysfunction.
Figure 2.
Right ventricular (RV) fibrosis and collagen I/III ratio. A, RV histology sections were stained for collagen using a Picrosirius red staining and analyzed under double-polarized light, results from Andersen et al.18 A gradual increase in RV fibrosis was found in mild and severe RV dysfunction in comparison to control. Data presented as percentage of controls. B, Collagen type I and III mRNA expression was determined by qPCR, and the collagen I/III ratio was calculated. Collagen I/III expression was significantly increased in severe RV dysfunction. Data presented as mean±SEM. Controls: n=13, mild RV dysfunction: n=5, and severe RV dysfunction: n=5. *P<0.05, #P<0.05.
RV Myofibril-Mediated Stiffness
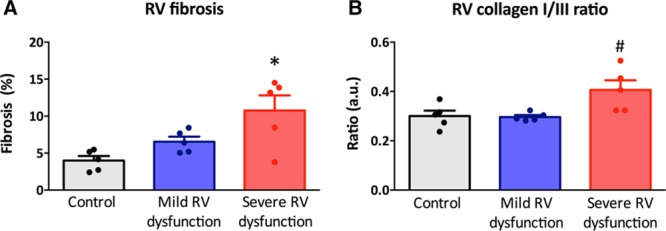
To investigate in more detail the myofibril-derived stiffness, we subsequently determined myofibril stiffness in single membrane-permeabilized RV cardiomyocytes from control rats, rats with mild RV dysfunction, and rats with severe RV dysfunction. As can be observed in Figure 3A, RV myofibril-mediated stiffness gradually increased with the severity of RV dysfunction. Especially at sarcomere length of 2.2 and 2.4 μm, RV myofibril stiffness was increased in rats with mild and severe RV dysfunction in comparison to controls (Figure 3A and 3B). To investigate whether reduced PKA-mediated phosphorylation of the giant elastic filament titin may contribute to the observed increased RV myofibril stiffness, measurements of RV myofibril stiffness was also assessed after PKA incubation. As can be observed in Figure 3C, PKA incubation resulted in a reduction in RV myofibril stiffness, but it remained elevated especially in rats with severe RV dysfunction in comparison to control. In addition, the effect of PKA incubation in rats with severe RV dysfunction was larger than in that in rats with mild RV dysfunction or controls (Pinteraction<0.001; Figure 3D), suggesting that PKA-mediated phosphorylation of sarcomeric proteins is more hampered in RV cardiomyocytes of rats with severe RV dysfunction.
Figure 3.
Skinned cardiomyocytes. A and B, Right ventricular (RV) cardiomyocyte stiffness was gradually increased in mild and severe RV dysfunction in comparison to control. C and D, Protein kinase A (PKA) incubation significantly decreased cardiomyocyte stiffness in mild and severe RV dysfunction but remained significantly elevated in comparison to controls. Data are presented as mean±SEM. Controls: n=5, mild RV dysfunction: n=5, and severe RV dysfunction: n=5. **P<0.01, ***P<0.001 vs control. The Pinteraction value represents the response of passive tension on increase in sarcomere length or PKA incubation.
Titin Isoform and Phosphorylation
To further explain the findings of RV myofibril stiffness, we investigated whether titin isoform composition or titin phosphorylation was altered in RV dysfunction. We observed an increase in expression of the compliant titin isoform (N2BA) in rats with mild RV dysfunction, whereas titin isoform composition was unaltered in rats with severe RV dysfunction (Figure 4A). This was also observed when expressed as N2BA/N2B ratio: control 3.4±1.4% versus mild RV dysfunction 12.4±1.3% versus severe RV dysfunction 4.8±0.89%. Subsequently, we determined overall phosphorylation levels of titin (ProQ Diamond staining) normalized to total protein content (Sypro Ruby staining). Titin phosphorylation was lower in both rats with mild or severe RV dysfunction, but only reached significance in rats with mild RV dysfunction (Figure 4B and 4C). These data suggest that although more compliant titin isoform was expressed in rats with mild RV dysfunction, this compensatory mechanism was insufficient to prevent an increase in RV myofibril stiffness, probably because of a reduced phosphorylation of titin.
Figure 4.
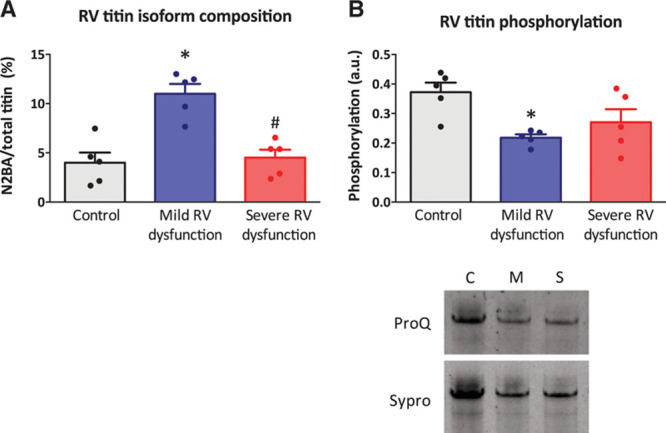
Right ventricular (RV) titin isoform composition and titin phosphorylation. A, Titin isoform ratio determined on 1% agarose gels stained with Coomassie Blue by dividing the N2BA isoform content to the more abundant N2B isoform and expressed in percentages. N2BA/total titin ratio was similar in control and severe RV dysfunction, whereas elevated in mild RV dysfunction. B, Total titin phosphorylation was determined by ProQ staining for phosphorylation divided by Sypro staining for total protein content. Titin phosphorylation was lower both in mild and severe RV dysfunction. C, Example of ProQ and Sypro staining of titin. Data are presented as mean±SEM. Controls: n=5, mild RV dysfunction: n=5, and severe RV dysfunction: n=5. *P<0.05. A.u. indicates arbitrary units.
Discussion
By combining RV mechanics with protein and histological analyses, we demonstrated that:
1. RV myocardial stiffness is increased in both mild and severe RV dysfunction; myofibril-derived stiffness contributes to both mild and severe RV dysfunction, whereas fibrosis-mediated stiffness plays an additional role in severe RV dysfunction.
2. RV fibrosis-mediated stiffness is associated with gradually increased fibrosis deposition in mild and severe RV dysfunction and increased collagen I/III ratio in rats with severe RV dysfunction.
3. RV myofibril-mediated stiffness gradually increases with severity of RV dysfunction. Probably explained by the finding that phosphorylation of titin was reduced in both mild and severe RV dysfunction, whereas titin isoform composition was only changed in mild RV dysfunction toward more compliant titin.
Fibrosis-Mediated Stiffness
The role of fibrosis in RV remodeling in PAH is inconclusive. With imaging techniques, fibrotic areas are only observed in the insertion points between the septum and RV wall,16,17 whereas in tissue of PAH patients with end-stage RHF and in several PAH animal models RV fibrosis is either absent23 or mildly increased in the RV free wall.2,24–29 However, until now, it was unclear whether the observed increase in RV fibrosis had any functional consequence. Here, we demonstrate for the first time that increased RV fibrosis, especially in rats with severe RV dysfunction, significantly impairs RV diastolic function. Besides histologically observed increase in RV fibrosis, we could also demonstrate a shift in collagen isoform expression. This suggests that even though the increase in RV fibrosis is small, when this occurs in combination with increased expression of the stiff collagen isoform, increased fibrosis can have important functional consequences for RV relaxation in clinical PAH.
Because increased fibrosis-mediated stiffness, increased fibrosis, and collagen I/III isoform shift occurred mainly in rats with severe RV dysfunction, the presence of RV fibrosis may be used as a tool to predict deterioration of RV function in PAH patients. This suggestion is further supported by the recent finding by Safdar et al15 that a biological marker of collagen metabolism (N-terminal propeptide of type III procollagen) could be used to predict prognosis and disease progression in patients with PAH. In addition, the development of more advanced imaging modalities may further improve the sensitivity to detect RV fibrosis in patients with PAH, which may be used to predict RV dysfunction in future.30
The underlying mechanism of increased RV fibrosis in RV dysfunction remains elusive. One may speculate that increased neurohormonal activity could play a role.31 Previously, we have shown increased activation of the renin-angiotensin-aldosterone system, which was closely associated with disease progression.32 Increased levels of angiotensin II could lead to the activation of transforming growth factor-β and increased collagen production by RV fibroblasts.14,33,34 Furthermore, overactive renin-angiotensin-aldosterone system could also increase RV fibrosis via aldosterone signaling pathways via the activation of mitogen-activated protein kinases, including extracellular signal-regulated kinases with a net effect of increased mRNA levels of types I, III, and IV collagen.14,34 But further studies are needed to determine the exact underlying mechanism of RV fibrosis in PAH, which could further be used as therapeutic target.
Myofibril-Mediated Stiffness
In this study, we determined the contribution of myofibril-derived stiffness in 2 ways: (1) by subtracting fibrosis-mediated stiffness from total RV myocardial stiffness of RV trabecular muscle strips and (2) by measuring myofibril stiffness in single demembranated single RV cardiomyocytes. Interestingly, no difference in myofibril-derived RV myocardial stiffness was observed in trabecular muscle strips of mild versus severe RV dysfunction, whereas on single cardiomyocyte level, we observed a further increase in RV myofibril stiffness in severe RV dysfunction compared with mild RV dysfunction. The myofibril stiffness derived from the trabecular stiffness may be underestimated because of the normalization of the stiffness to the trabecular cross-sectional area, which includes not only cardiomyocytes but also the fibrotic component. Because the collagen fraction is increased in severe RV dysfunction, we expect that the area of the total cross-sectional area occupied by cardiomyocytes is smaller, leading to an underestimation of the overall myofibril stiffness (Figure 5).
Figure 5.
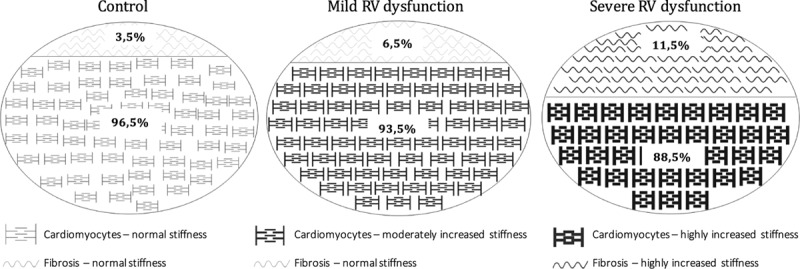
Components of trabecular stiffness. Schematic representation of the relative contribution of fibrosis and cardiomyocytes to trabecular stiffness. RV indicates right ventricular.
The giant sarcomeric protein titin is an important regulator of myofibril-mediated stiffness.5 Changes in titin phosphorylation or titin isoform composition contribute closely to stiffening of cardiomyocytes. In this study, we observed that overall titin phosphorylation was reduced in both mild and severe RV dysfunction, although the differences were only statistically significant in rats with mild RV dysfunction. The reduction of global titin phosphorylation in severe RV dysfunction relative to controls (0.74±0.12) was comparable to previous observations in RV tissue of PAH patients with severe RV dysfunction (0.82±0.05) relative to controls.2
Although PKA-mediated titin phosphorylation could not be specifically measured in rat RV tissue, the reduction observed in RV diastolic stiffness after PKA incubation suggests that the reduced titin phosphorylation is partly mediated by reduced PKA activity in both mild and severe RV dysfunction. A decreased intracellular PKA-mediated phosphorylation of titin could be a direct consequence of the downregulation and desensitization of the β-adrenergic receptor pathway.7,35 However, PKA incubation could not fully restore RV myocardial stiffness, indicating that other kinases and post-translational modifications may play an additional role.
Although titin phosphorylation was reduced in both mild and severe RV dysfunction, increased expression of the compliant titin isoform was observed only in rats with mild RV dysfunction and unaltered in rats with severe RV dysfunction. This is in line with our previous data from tissue of patients with end-stage RHF, in which we did not observe any change in titin isoform expression compared with control samples.2 This may suggest that titin isoform switch is a dynamic process, which in mild RV dysfunction may play a protective role in maintaining myofibril stiffness relatively low by enhancing the expression of the compliant N2BA isoform, whereas in later stages of RV dysfunction titin isoform composition reverses toward a N2BA/N2B ratios that are similar to those in controls.
In addition to a role in myofibril stiffness, important mechanosensing properties and hypertrophy-inducing signals have been associated with titin.36,37 Therefore, changes in titin isoform and phosphorylation may not only increase myofibril stiffness but also alter the capacity of the cardiomyocytes to correctly sense the afterload and stop the hypertrophic signaling triggered by the increase in RV wall stress. At this point, the transition from a hypertrophic compensated RV phenotype to a dilative failing RV phenotype may occur.
RV Versus Left Ventricular Pressure Overload
In patients with PAH, the right ventricle is exposed to a ≤4-fold increase in pressure.35 This magnitude of pressure overload in PAH is much higher than that observed in left ventricular (LV) pressure overload induced by, for example, hypertension or aortic stenosis. This may explain why some of our findings are not in line with previously published results on LV pressure overload. First of all, RV myocardial stiffness is increased in all severities of PAH, whereas in LV pressure overload, increased myocardial stiffness is only observed in patients with hypertension and heart failure.38,39 Second, increased expression of the more compliant titin isoform (N2BA) is frequently observed in patients with decompensated LV pressure overload,38,40,41 whereas our study only observed a shift to the more compliant titin isoform in mild RV dysfunction and not in end-stage disease.
Besides the magnitude of pressure overload, also embryological differences between the RV and LV may underlie the differential response to pressure overload.
Limitations
In this study, we have used the pulmonary artery banding model to investigate the contribution of fibrosis and myofibrillar stiffness in mild and severe RV dysfunction. No animal model exists that fully recapitulates the human disease, but the advantage of this model is that we could induce 2 stages of RV dysfunction without using higher doses of toxins (ie, monocrotaline), which may have their own secondary (cardiac) effects. After fully characterizing the rats with right heart catheterization and magnetic resonance imaging, we are of the opinion that this animal model features the most essential clinical characteristics of mild and severe RV dysfunction such as a gradual decrease in RVEF and increase in end-diastolic volume (Table).
To determine the relative contributions of myofibril stiffness and fibrosis to RV myocardial stiffness, we isolated RV trabecular tissue, which we considered more representative for the overall RV free-wall morphological and molecular changes than papillary muscle strips. However, in contrast to strips of papillary muscles, trabecular muscle strips have a more heterogeneous fiber orientation with unevenly distributed sarcomeres, limiting accurate sarcomere length determination.21 Therefore, we performed our experiments at a 20% increase in slack length. As a consequence, it is possible that the sarcomere length may have been unevenly distributed between the experiments, with variable influence on the myofibril stiffness. Therefore, to accurately determine myofibril stiffness in relation to sarcomere length, we also isolated cardiomyocytes from the free wall and measured stiffness at increasing sarcomere lengths (from 1.8 to 2.4 µm).
High cardiomyocyte stiffness may contribute to impaired diastolic function, which at the cellular level may be caused by increased titin-based passive stiffness of the sarcomeres. There are, however, multiple other cellular protein modifications, which may underlie impaired cardiomyocyte relaxation. Changes in intracellular calcium handling and high myofilament calcium sensitivity may impair proper relaxation of cardiomyocytes. In a previous study, we found that sarcoendoplasmic reticulum Ca2+-ATPase expression and phospholamban phosphorylation were altered in idiopathic pulmonary arterial hypertension, whereas calcium sensitivity of the myofilaments was increased. These findings suggest that in addition to stiffer myofibrils, calcium dysregulation and high myofilament calcium sensitivity might also play an important role in altering RV diastolic function.
This study does not compare RV to LV myocardial stiffness or the relative contributions of fibrosis- and myofibril-mediated stiffness between the ventricles. Although of interest, the LV cannot be used as the same animal control for the RV, because the LV cardiomyocytes are affected in PAH as well.42 Probably, because of a decreased RV stroke volume, the LV in PAH is exposed to reduced filling volumes and pressures, which induces LV atrophy and reduced contractile function.42 Therefore, we used disease-free sham RV tissues as control.
Finally, the sample size of this study was small, but sufficient to observe differences between the groups. Therefore, we have analyzed all data with exact methods.
Clinical Implications
Little is known about the presence and impact of RV diastolic dysfunction in idiopathic pulmonary arterial hypertension patients. In a previous clinical study, we show a marked increase in RV diastolic stiffness in patients with idiopathic PAH. Furthermore, we showed a significant correlation between increased RV diastolic stiffness and disease severity (characterized by reduced stroke volume, N-terminal pro–B-type natriuretic peptide levels, and 6-minute walk distance).2 More recently, we demonstrated that increased RV diastolic stiffness is associated with worse outcome in patients with PAH.3 Therefore, diastolic dysfunction is likely to be of clinical relevance. Whether RV diastolic stiffness is restricted to idiopathic pulmonary arterial hypertension or is also present in other disorders that are associated with elevated RV pressure is currently unclear. However, because we observed clear RV diastolic stiffness in this animal model with only RV pressure overload, it is likely that increased RV myocardial stiffness also occurs in other conditions where RV pressures are elevated.
Although an abnormally high fibrotic response in severe RV dysfunction may imply that treatment should be directed toward reducing fibrosis, it is important to point out that myofibril stiffness is already increased in rats with mild RV dysfunction. Therefore, efforts should be directed toward improving lusitropy by targeting both the fibrotic component and myofibril stiffness. Restoring the neurohormonal-dependent cellular and extracellular signaling pathways by, for instance, β-blocker therapy or renin-angiotensin-aldosterone system inhibitors has already been shown to be effective in reducing overall RV diastolic stiffness in PAH animal models.25,32 Whether this effect is mediated by reduction of both fibrosis and myofibril stiffness should be further investigated.
Conclusions
RV myocardial stiffness is increased in rats with mild and severe RV dysfunction. However, the underlying mechanism differs between the groups. In mild RV dysfunction, RV myocardial stiffness is mainly contributed to myofibril-mediated stiffness, as a consequence of hypophosphorylation of the giant elastic titin filament. In contrast, in severe RV dysfunction, RV myocardial stiffness is mediated by both myofibril- and fibrosis-mediated stiffness, as a consequence of hypophosphorylation of titin as well as increased ratio of collagen I/III expression.
Acknowledgments
We would like to thank Dr Emmy Manders for her help pertaining to the figures of the article.
Sources of Funding
Drs de Man, Ottenheijm, van der Velden, and Vonk-Noordegraaf were supported by a VENI (916.14.099) and VIDI (917.12.319; 917.11.344; 917.96.306) grant from the Dutch Foundation for Scientific Research. Drs Rain and Vonk-Noordegraaf were supported by the Netherlands Organization for Health Research and Development (ZonMw 95110079). Dr Bogaard was supported by the National Institute of Health (NIH HL062881). Drs de Man, Bogaard, and Vonk-Noordegraaf were supported by the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development, and the Royal Netherlands Academy of Sciences (Cardiovascular onderzoek Nederland 2012-08). D. da Silva Gonçalves Bós and Dr Handoko were supported by Coordinator for the Development of Graduate Human Resources – Science Without Borders grant, National Council for Scientific and Technological Development Brazil (245849). Drs Schultz, S. Andersen, and A. Andersen were supported by the Danish Research Council (11–108354). Dr de Man is further supported by L’Oreal/Unesco for Women in Science and Netherlands Institute for Advanced Studies, the American Thoracic Society (Jerry Wojciechowski Memorial Pulmonary Hypertension Research Grant), and the European Respiratory Society.
Disclosures
None.
Footnotes
The Data Supplement is available at http://circheartfailure.ahajournals.org/lookup/suppl/doi:10.1161/CIRCHEARTFAILURE.115.002636/-/DC1.
Clinical Perspective
Patients with pulmonary arterial hypertension develop right heart failure because of a progressive increase in right ventricular (RV) pressure overload. Although it has been known for some years that RV systolic adaptation is of clinical importance, it just recently became clear that RV diastolic stiffness increases and may contribute to disease progression in pulmonary arterial hypertension. Increased sarcomere-derived cardiomyocyte stiffness and myocardial fibrosis were identified as possible contributing factors to RV diastolic stiffness in patients with pulmonary arterial hypertension. However, the relative contribution of fibrosis-mediated and myofibril-mediated stiffness remained elusive. In this study, we observed that although RV myocardial stiffness is increased in both rats with mild RV dysfunction as well as in rats with severe RV dysfunction, the underlying mechanism differs between the groups. In mild RV dysfunction, RV myocardial stiffness is mainly attributed to myofibril-mediated stiffness, as a consequence of hypophosphorylation of the giant elastic titin filament. In contrast, in severe RV dysfunction, RV myocardial stiffness is mediated by both myofibril- and fibrosis-mediated stiffness, as a consequence of hypophosphorylation of titin and increased ratio of collagen I/III expression. Although an abnormally high fibrotic response in severe RV dysfunction may imply that treatment should be directed toward reducing fibrosis, it is important to point out that myofibril stiffness is already increased in rats with mild RV dysfunction. Therefore, efforts should be directed toward improving lusitropy by targeting both the fibrotic component and myofibril stiffness.
References
- 1.Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62(suppl 25):D22–D33. doi: 10.1016/j.jacc.2013.10.027. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 2.Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128:2016–25, 1. doi: 10.1161/CIRCULATIONAHA.113.001873. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 3.Trip P, Rain S, Handoko ML, van der Bruggen C, Bogaard HJ, Marcus JT, Boonstra A, Westerhof N, Vonk-Noordegraaf A, de Man FS. Clinical relevance of right ventricular diastolic stiffness in pulmonary hypertension. Eur Respir J. 2015;45:1603–1612. doi: 10.1183/09031936.00156714. doi: 10.1183/09031936.00156714. [DOI] [PubMed] [Google Scholar]
- 4.Rain S, Bos Dda S, Handoko ML, Westerhof N, Stienen G, Ottenheijm C, Goebel M, Dorfmüller P, Guignabert C, Humbert M, Bogaard HJ, Remedios CD, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc. 2014;3:e000716. doi: 10.1161/JAHA.113.000716. doi: 10.1161/JAHA.113.000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trombitás K, Wu Y, Labeit D, Labeit S, Granzier H. Cardiac titin isoforms are coexpressed in the half-sarcomere and extend independently. Am J Physiol Heart Circ Physiol. 2001;281:H1793–H1799. doi: 10.1152/ajpheart.2001.281.4.H1793. [DOI] [PubMed] [Google Scholar]
- 7.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 8.Krüger M, Kötter S, Grützner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 9.Anderson BR, Bogomolovas J, Labeit S, Granzier H. The effects of PKCalpha phosphorylation on the extensibility of titin’s PEVK element. J Struct Biol. 2010;170:270–277. doi: 10.1016/j.jsb.2010.02.002. doi: 10.1016/j.jsb.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–8, 17 p following 638. doi: 10.1161/CIRCRESAHA.109.198465. doi: 10.1161/CIRCRESAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson BD, Hidalgo CG, Gotthardt M, Granzier HL. Excision of titin’s cardiac PEVK spring element abolishes PKCalpha-induced increases in myocardial stiffness. J Mol Cell Cardiol. 2010;48:972–978. doi: 10.1016/j.yjmcc.2009.12.006. doi: 10.1016/j.yjmcc.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman D, Weber KT, Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res. 1990;67:787–794. doi: 10.1161/01.res.67.4.787. [DOI] [PubMed] [Google Scholar]
- 13.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 14.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 15.Safdar Z, Tamez E, Chan W, Arya B, Ge Y, Deswal A, Bozkurt B, Frost A, Entman M. Circulating collagen biomarkers as indicators of disease severity in pulmonary arterial hypertension. J AM COLL CARDIOL. Heart Fail. 2014;2:412–421. doi: 10.1016/j.jchf.2014.03.013. doi: 10.1016/j.jchf.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blyth KG, Groenning BA, Martin TN, Foster JE, Mark PB, Dargie HJ, Peacock AJ. Contrast enhanced-cardiovascular magnetic resonance imaging in patients with pulmonary hypertension. Eur Heart J. 2005;26:1993–1999. doi: 10.1093/eurheartj/ehi328. doi: 10.1093/eurheartj/ehi328. [DOI] [PubMed] [Google Scholar]
- 17.Freed BH, Gomberg-Maitland M, Chandra S, Mor-Avi V, Rich S, Archer SL, Jamison EB, Jr, Lang RM, Patel AR. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson. 2012;14:11. doi: 10.1186/1532-429X-14-11. doi: 10.1186/1532-429X-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen S, Schultz JG, Andersen A, Ringgaard S, Nielsen JM, Holmboe S, Vildbrad MD, de Man FS, Bogaard HJ, Vonk-Noordegraaf A, Nielsen-Kudsk JE. Effects of bisoprolol and losartan treatment in the hypertrophic and failing right heart. J Card Fail. 2014;20:864–873. doi: 10.1016/j.cardfail.2014.08.003. doi: 10.1016/j.cardfail.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circ Res. 2011;109:858–866. doi: 10.1161/CIRCRESAHA.111.246819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H. Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol. 2000;32:2151–2162. doi: 10.1006/jmcc.2000.1281. doi: 10.1006/jmcc.2000.1281. [DOI] [PubMed] [Google Scholar]
- 21.Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–1044. doi: 10.1016/S0006-3495(95)80278-X. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM, González A, Tschöpe C, Díez J, Linke WA, Leite-Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. doi: 10.1161/CIRCHEARTFAILURE.113.000539. doi: 10.1161/CIRCHEARTFAILURE.113.000539. [DOI] [PubMed] [Google Scholar]
- 23.Overbeek MJ, Mouchaers KT, Niessen HM, Hadi AM, Kupreishvili K, Boonstra A, Voskuyl AE, Belien JA, Smit EF, Dijkmans BC, Vonk-Noordegraaf A, Grünberg K. Characteristics of interstitial fibrosis and inflammatory cell infiltration in right ventricles of systemic sclerosis-associated pulmonary arterial hypertension. Int J Rheumatol. 2010;2010 doi: 10.1155/2010/604615. doi: 10.1155/2010/604615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Handoko ML, de Man FS, Happé CM, Schalij I, Musters RJ, Westerhof N, Postmus PE, Paulus WJ, van der Laarse WJ, Vonk-Noordegraaf A. Opposite effects of training in rats with stable and progressive pulmonary hypertension. Circulation. 2009;120:42–49. doi: 10.1161/CIRCULATIONAHA.108.829713. doi: 10.1161/CIRCULATIONAHA.108.829713. [DOI] [PubMed] [Google Scholar]
- 25.de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105. doi: 10.1161/CIRCHEARTFAILURE.111.964494. doi: 10.1161/CIRCHEARTFAILURE.111.964494. [DOI] [PubMed] [Google Scholar]
- 26.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. doi: 10.1164/rccm.201003-0335OC. doi: 10.1164/rccm.201003-0335OC. [DOI] [PubMed] [Google Scholar]
- 27.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med. 2011;183:1402–1410. doi: 10.1164/rccm.201007-1106OC. doi: 10.1164/rccm.201007-1106OC. [DOI] [PubMed] [Google Scholar]
- 28.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:1239–1247. doi: 10.1165/rcmb.2010-0412OC. doi: 10.1165/rcmb.2010-0412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 30.García-Álvarez A, García-Lunar I, Pereda D, Fernández-Jimenez R, Sánchez-González J, Mirelis JG, Nuño-Ayala M, Sánchez-Quintana D, Fernández-Friera L, García-Ruiz JM, Pizarro G, Agüero J, Campelos P, Castellá M, Sabaté M, Fuster V, Sanz J, Ibañez B. Association of myocardial T1-mapping CMR with hemodynamics and RV performance in pulmonary hypertension. JACC Cardiovasc Imaging. 2015;8:76–82. doi: 10.1016/j.jcmg.2014.08.012. doi: 10.1016/j.jcmg.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 31.de Man FS, Handoko ML, Guignabert C, Bogaard HJ, Vonk-Noordegraaf A. Neurohormonal axis in patients with pulmonary arterial hypertension: friend or foe? Am J Respir Crit Care Med. 2013;187:14–19. doi: 10.1164/rccm.201209-1663PP. doi: 10.1164/rccm.201209-1663PP. [DOI] [PubMed] [Google Scholar]
- 32.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:780–789. doi: 10.1164/rccm.201203-0411OC. doi: 10.1164/rccm.201203-0411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horiguchi M, Ota M, Rifkin DB. Matrix control of transforming growth factor-β function. J Biochem. 2012;152:321–329. doi: 10.1093/jb/mvs089. doi: 10.1093/jb/mvs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lijnen PJ, van Pelt JF, Fagard RH. Stimulation of reactive oxygen species and collagen synthesis by angiotensin II in cardiac fibroblasts. Cardiovasc Ther. 2012;30:e1–e8. doi: 10.1111/j.1755-5922.2010.00205.x. doi: 10.1111/j.1755-5922.2010.00205.x. [DOI] [PubMed] [Google Scholar]
- 35.Rain S, Handoko ML, Vonk Noordegraaf A, Bogaard HJ, van der Velden J, de Man FS. Pressure-overload-induced right heart failure. Pflugers Arch. 2014;466:1055–1063. doi: 10.1007/s00424-014-1450-1. doi: 10.1007/s00424-014-1450-1. [DOI] [PubMed] [Google Scholar]
- 36.Linke WA, Krüger M. The giant protein titin as an integrator of myocyte signaling pathways. Physiology (Bethesda) 2010;25:186–198. doi: 10.1152/physiol.00005.2010. doi: 10.1152/physiol.00005.2010. [DOI] [PubMed] [Google Scholar]
- 37.Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, Bull MM, Saripalli C, Pappas CT, Gregorio CC, Smith JE., 3rd Deleting titin’s I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci USA. 2014;111:14589–14594. doi: 10.1073/pnas.1411493111. doi: 10.1073/pnas.1411493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, van Heerebeek L, Stienen GJ, Niessen HW, Leite-Moreira AF, Paulus WJ. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. 2011;124:1151–1159. doi: 10.1161/CIRCULATIONAHA.111.025270. doi: 10.1161/CIRCULATIONAHA.111.025270. [DOI] [PubMed] [Google Scholar]
- 40.Borbély A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- 41.van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 42.Manders E, Bogaard HJ, Handoko ML, van de Veerdonk MC, Keogh A, Westerhof N, Stienen GJ, Dos Remedios CG, Humbert M, Dorfmüller P, Fadel E, Guignabert C, van der Velden J, Vonk-Noordegraaf A, de Man FS, Ottenheijm CA. Contractile dysfunction of left ventricular cardiomyocytes in patients with pulmonary arterial hypertension. J Am Coll Cardiol. 2014;64:28–37. doi: 10.1016/j.jacc.2014.04.031. doi: 10.1016/j.jacc.2014.04.031. [DOI] [PubMed] [Google Scholar]