

Supplemental Digital Content is available in the text
Keywords: Gastrointestinal tract, Pathology, Perivascular epithelioid cell tumor, Rare tumor
Abstract
Perivascular epithelioid cell tumor (PEComa) is a rare entity with distinctive morphology and of expressing myomelanocytic markers. Gastrointestinal tract (GI) is one of the most common anatomic sites of origin and counts for 20% to 25% of all reported cases of perivascular epithelioid cell tumors not otherwise specified (PEComas-NOS). However, the biologic behavior of perivascular epithelioid cell tumors of gastrointestinal tract (GI PEComas-NOS) is still unclear. The aim of conducting this systematic review is to sum up what is known so far of the epidemiology, natural history, management and prognosis of GI PEComas-NOS.
A systematic research was performed on PubMed and EMBASE using the following terms: (“perivascular epithelioid cell tumor” or “PEComa”) and (“gastrointestinal tract” or “GI” or “oral ” or “mouth” or “esophagus” or “gullet” or “gastric” or “stomach” or “duodenum” or “jejunum” or “ileum” or “cecum” or “colon” or “colorectal” or “sigmoid” or “rectum” or “anus” or “mesentery”) up to December 1, 2015. Retrieved GI PEComas-NOS publications, which included these terms, contains case reports, case series to case characteristic researches.
A total of 168 articles were reviewed, 41 GI PEComa-NOS English studies among which were retrieved for analysis. We reviewed epidemiology, natural history, management and prognosis of GI PEComa-NOS. Generally GI PEComa-NOS is believed to have women predomination. The most frequently involved location is colon with non-specific clinical signs. Pathologically, GI PEComas-NOS shows epithelioid predominance (70%), meanwhile coexpresses melanocytic and muscle markers characteristically, while immunohistochemistry is a useful tool for identify, which indicates that HMB-45 is regarded as the most sensitive reagent. Complete resection served as mainstay of treatment, while chemotherapy should be unanimously considered to apply in malignant cases. Eventually, it is necessary for closed and long-term follow-up with endoscope and imaging for ruling out local recurrence or distant metastasis of this tumor.
GI PEComas-NOS lives with unclear behavior. There are still many unverified clinicopathological issues of GI PEComas-NOS that needs to be clarified. Further studies and analyses concerning this rare entity should be brought out. Thus, the randomized clinical researches (RCTs) are required to be conducted.
1. Introduction
Perivascular epithelioid cell tumors (PEComas) is rare mesenchymal neoplasm with distinctive morphology and immunohistochemical characteristics. In 1992, Bonetti et al[1] firstly initiatived the term of perivascular epithelioid cell (PEC) to portray some epitheliod type cells with a perivascular distribution and immunoreactive for melanocyte markers. In 2002, the World Health Organization took the definition of “mesenchymal tumors composed of histologically and immunohistochemically distinctive perivascular epithelioid cells” as perivascular epithelioid cell tumors (PEComas).[2] Lately, the family of PEComas had grown to include angiomyolipoma (AML), clear-cell “sugar” tumor (CCST), lymphangioleiomyomatosis (LAM), clear-cell myomelanocytic tumor of the falciform ligament/ligamentum teres and rare clear cell tumors of other anatomic locations such as uterus, colon, and soft tissues. The term perivascular epithelioid cell tumors–not otherwise specified (PEComas-NOS) was proposed to describe the latter subgroup.[3]
PEComas-NOS arises at diverse visceral and soft tissue sites. Perivascular epithelioid cell tumors of gastrointestinal tract (GI PEComas-NOS) takes a proportion of about 20% to 25% of PEComas-NOS.[4] There are 50 cases of GI PEComas-NOS recorded in the English literature approximately and most of them are case report or small-scale case series research. For the rarity of such a distinctive neoplasm, the epidemiology, clinical presentation, optimal management, and prognosis of it are still unascertainable. Aiming to better know of this entity, a systematic review was made by us, focusing on what have been known so far with concerning of the epidemiology, natural history, management, and prognosis of GI PEComas-NOS.
2. Methods
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines were followed to complete the searches and review. Database of PubMed and EMBASE was searched for all related literature, using combinations of the following search terms: (“perivascular epithelioid cell tumor” or “PEComa”) and (“gastrointestinal tract” or “GI” or “oral ” or “mouth” or “esophagus” or “gullet” or “gastric” or “stomach” or “duodenum” or “jejunum” or “ileum” or “cecum” or “colon” or “colorectal” or “sigmoid” or “rectum” or “anus” or “mesentery”). Figure 1 is a flowchart summarizing study identification and selection according to PRISMA.
Figure 1.
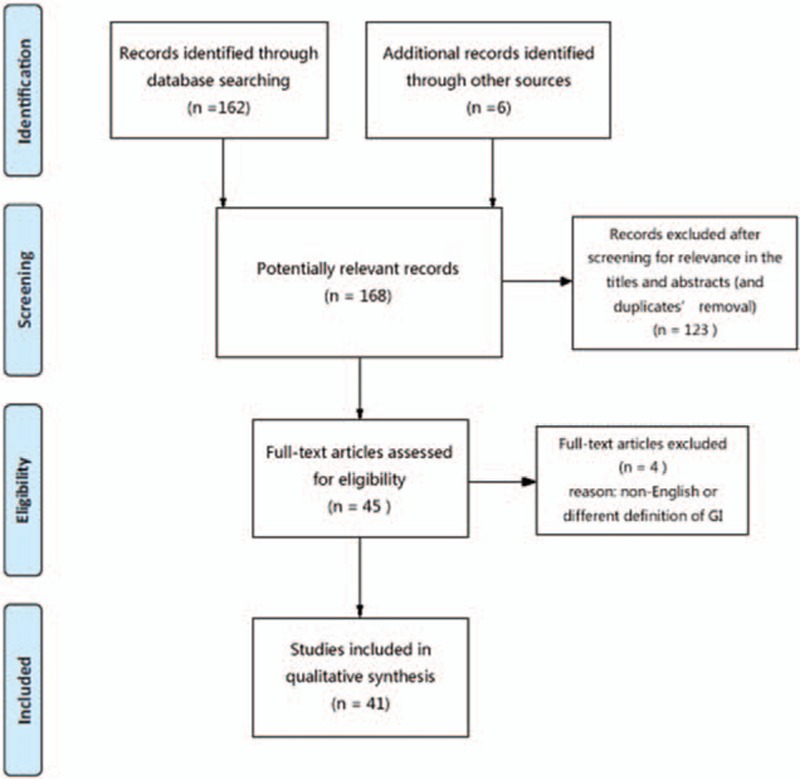
PRISMA flow chart of literature search. PRISMA = Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Ethics committee approval was not necessary for systematic reviews.
2.1. Studies selection
Inclusion criteria for the present systematic review included: case reports, case series, and case characteristic researches published before December 1, 2015, concerning patients admitted with a diagnosis of GI PEComas-NOS. We excluded the studies that did not accord with our definition of gastrointestinal tract. Only English-language articles were evaluated. Moreover, reference lists of relevant articles were also searched to identify additional studies.
2.2. Data abstraction
We take the definition of GI as the tract includes all structures between the mouth and the anus as well as mesentery. Two independent investigators scanned the potential literatures and screened out identified literatures based on the title and abstract. The full text would be reviewed if the former content was not satisfied for evaluation. Any discrepancies between the independent searchers would be solved in consensus with another two authors. The following information of the literatures were took down: name of the first author, the year of publication, sample size of cases, location of lesion, pathological characteristics, therapeutic method, duration of follow-up, and result. All the results were reviewed by another investigator.
3. Result
The initial systematic search produced 168 potentially relevant records. Our screening strategy filtrated out 45 eligible articles for further assessment. Subsequently, among these literatures we ruled out 3 articles that were written in German, French, and Korean language, respectively. One research was excluded since their definition of GI is different from the meaning that we will state in this review. The included and reviewed studies ranges from case reports, case series to case characteristic researches. After the data of the included studies were analyzed, we were able to summed up the conclusions of all aspects of GI PEComas-NOS.
3.1. Etiology
The exact cause of GI PEComas-NOS is not clear. It is believed to originate from PEC but the natural histological counterpart and how it can be a potential neoplastic originator are out of clarity.[5]
PEC is characterized by perivascular location, often arranging around the vascular lumen radically. It expresses myogenic and melanocytic markers immunohistochemically and can modulate its morphology and immunophenotype: PEC can be spindle shape of muscular features and be positive of actin than HMB-45 or it can be epithelioid feature and positive of HMB-45 and a mild, if any, reaction for actin.
There are some investigators advancing hypothesis of the derivational source of PEC. Fernandez-Flores proposed PEC derives from neural crest for their expressing many melanocytic and muscular markers. Apart from melanocytic or muscular markers, there are other markers that link PEComas to the neural crest such as neural stem cell markers NG2 and L1 are both found in AML. Another evidence is that PEComas-NOS seems so ubiquitous that they had been described in almost every visceral and somatic locations. An origin from the neural crest would explain such ubiquity since derivatives from neural crest are found in somatic as well as in visceral locations.[6]
Stone et al[7] presented PEC originates from smooth muscle with possible molecular alteration that brings to expression of melanogenesis and melanocytic markers. They applied electron microscopic examination of spindle cells in renal AML and revealed ultrastructural characteristics of smooth muscle cells. The spindle cells contained more microfilaments with occasional dense bodies, subplasmalemmal dense plaques and pinocytotic vesicles, the characteristics of smooth muscle cells. Moreover, the positiveness of calponin and muscle-specific actin immunoreactivity in renal AML further characterizes the smooth muscle cell nature.
Ardeleanu et al[8] advanced that the telocyte might be the cell of origin of PEComas because markers described in telocytes (CD34, S-100, smooth muscle actin, and vascular endothelial growth factor) have been reported as positive in cases of PEComas. Telocytes described in many anatomic sites could display a wide spectrum of differentiation.
These hypotheses are concluded by phenomenon and personal observations. It is still insufficiently convincing. Therefore, more convictive evidence supporting the origin for PEC might be found from molecular and genetic proof.
3.2. Epidemiology
There are 50 cases of GI PEComas-NOS according to the included literatures. GI is the second most frequent site of PEComas, only behind gynecological tract.[9]
Table 1 summarizes the clinicopathologic features. The age of patients who were diagnosed with GI PEComas-NOS ranging from 5.5 to 97 years with an average age of 38.9. In keeping with research in other system, GI PEComas-NOS is more likely to affect people in the fourth and fifth decade of life.[10] Thirty two women and 18 men were recorded in the literatures so that it is believed to have women predomination. The most frequently involved location is colon (N = 20),[11–26] followed by mesentery (N = 10),[27–33] stomach (N = 4),[34–37] duodenum (N = 4),[38–41] ileum (N = 4),[4,42,43] cecum (N = 3),[12,44,45] oral cavity (N = 1),[46] esophagus (N = 1),[47] jejunum (N = 1),[48] ileocecal junction (N = 1)[49] and one case without exact site of origin (N = 1, multiple parts of GI involved).[9] Refined to GI PEComas-NOS of colon, rectum (N = 7) seems to be the most common involved region.
Table 1.
Clinicopathologic features of gastrointestinal perivascular epithelioid cell tumors-not otherwise specified (GI PEComas-NOS).
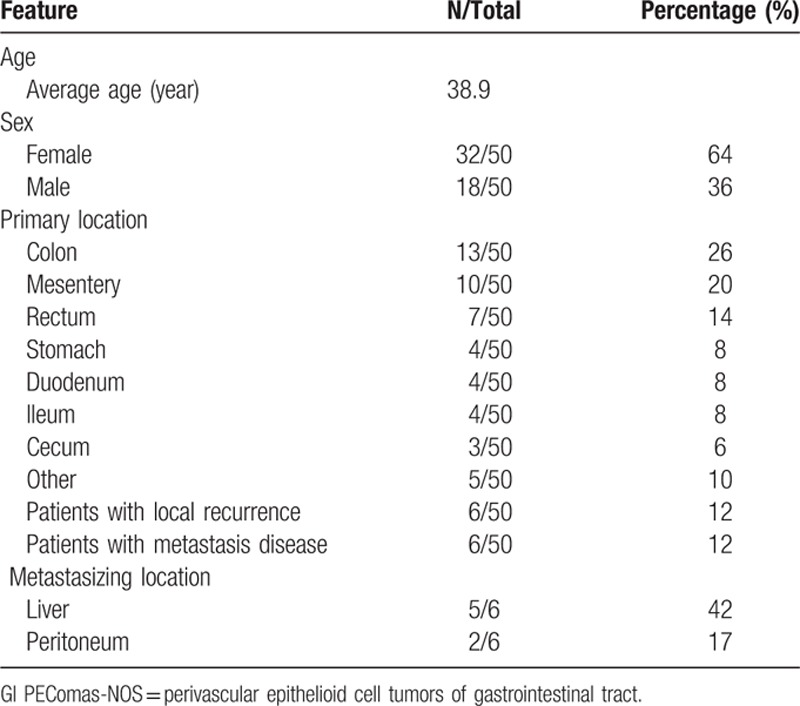
Tuberous sclerosis is an autosomal dominant genetic disease as a result of heterozygous mutations in either the TSC1 (9q34) or TSC2 (16p13.3) gene and it is strongly associated with AML, LAM, and CCST. There are also some cases of PEComas-NOS of gynecological tract showed concurrence with tuberous sclerosis. There are limited genomic studies on PEComas of other sites. Pan et al[50] showed multiple chromosomal imbalances, with 16p loss in 6 cases and X-chromosomal gains in 6 cases using comparative genomic hybridization on 9 PEComas. Kenerson et al[51] demonstrated mTOR pathway activation in sporadic AMLs and extrarenal PEComas by immunohistochemistry (increased levels of phosphor-p70S6K, reduced levels of phospho-AKT and loss of tuberin).
Unlike the other site of PEComa-NOS, there is no case of GI PEComas-NOS showing tuber sclerosis concurrence with information to date. We are, therefore, of the opinion that GI PEComas-NOS seems irrelevant to tuberous sclerosis but this need further verification by more cases and genetic studies.
3.3. Clinical manifestations
GI PEComa-NOS presents with non-specific clinical signs. With symptoms relying on the different organ involved, the size of neoplasm and tumor volume, principal clinical presentations include abdominal pain, melaena, rectal bleeding, obstruction, weight loss, anemia, and some even asymptomatic. Abdominal pain is most frequent (35%) that may result from oppression, impaction, and hemorrhage. Medical imageology, in accordance with clinical presentations, has no specific signs. Most GI PEComas-NOS manifests as well-demarcated mass with homogeneous density in plain computed tomography (CT) and shows heterogeneous or homogenous enhancement in contrast-enhancement CT. Hogemann et al[52] proposed arteriovenous hypervascularity in contrast-enhanced CT is a feature. Usually, the lesions are hypointense to isointense on T1-weighted imaging and heterogeneously hyperintense on T2-weighted imaging when applying magnetic resonance imaging (MRI). Ultrasonography may represent a highly vascularized heterogeneous mass. However, abdominal CT, MRI, and ultrasonography are not sufficiently sensitive to enable the diagnosis of PEComas because of their nonspecific imaging characteristics. There is no clear distinction between the benign and malignant counterparts but the aforementioned imaging modalities can help to detect suspicious lymphovascular invasion and metastatic lesion.[53] As a useful tool of gastroenterologist, endoscopy can help to detect the lesion, which includes polypoid tumor or fungating mass protruding into the lumen with ulcerated or smooth surface, however, also has no specific sign.
3.4. Pathology
Macroscopically, GI PEComa-NOS is always well circumscribed and no capsule formation, while some can show ill-defined border or infiltrative growth. It can be a solid or soft polypoid lesion locating in the mucosa or submucosa. The neoplasm also can be a round or ovoid mass which originates from any layer and even invades all the wall of the tract, protruding into mucosal and serous surfaces. The cut surface color varies from grayish-white, dark red, or pinkish-grey. Central ulcer, hemorrhage, and necrosis are also frequently detected.[3]
Microscopically, GI PEComa-NOS grows in a biphasic pattern, which shows a mixture of epithelioid and spindle cell components with clear-to-eosinophilic granular cytoplasm. Proportion of epithelioid and spindled cell varies in different anatomic site while GI PEComa-NOS shows epithelioid predominance (70%). Perivascular cells are nearly all epithelioid, which are arranged in sheets or nest pattern. Spindled components resembling smooth muscle are usually away from vessels, which are arranged in cordal or fascicular pattern with rich vascularity. The cytoplasm of epithelioid cell is usually clear to pale eosinophilic. The nuclei are oval to polygonal and the nucleoli is prominent and centrally located. Spindled cell usually shows slightly eosinophilic or granular cytoplasm, oval nuclei, and small distinct nucleoli.[3,27]
Immunohistochemical findings of included articles are summed up in Table 2. We listed all the markers used by the literatures (supplemental Table 1). The PECs coexpress melanocytic and muscle markers characteristically and immunohistochemistry is a useful tool for indentify. With all data available, each case of GI PEComas-NOS expressed one melanocytic marker at least; HMB-45 was regarded as the most sensitive reagent that being positive in 44 of 46 tumors (96%), followed by Melan-A in 22 of 34 tumors (65%), and MiTF in 5 of 9 tumors (55%). Smooth muscle actin (SMA) was positive in 21 of 33 lesions (64%). Desmin and S-100 were less often positive. CD 117 was expressed only in one single case and no case of CD34. Melan-A + SMA in 15/25 cases (60%) was the dominant patterns of immunehistochemistry, followed by HMB-45 + SMA in 19/32 cases (59.3%) and Melan-A + Vimentin in 8/20 cases (40%).
Table 2.
Immunohistochemical findings of gastrointestinal perivascular epithelioid cell tumors-not otherwise specified (GI PEComas-NOS).
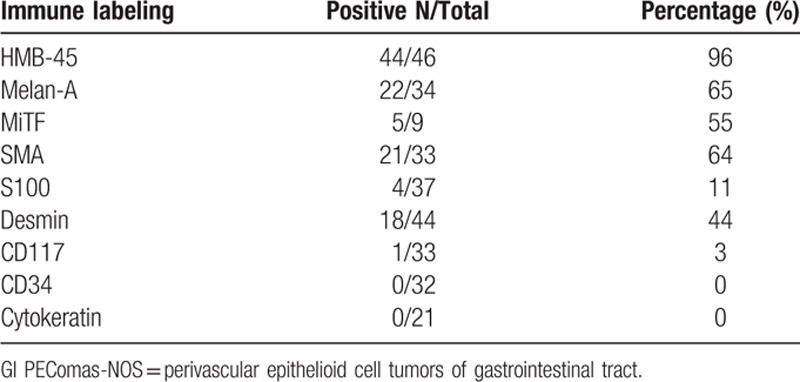
TFE3, which was proposed by Cho et al[20] might have potential role in the acquisition of the distinct histopathologic appearance, being reactive in 6 of 10 tumors (60%). Tanaka et al[21] presented an interesting phenomena that in TFE3+ cases they always show negative for Melan-A and SMA. There are 6 TFE3+ cases included in our study and in accordance with their findings, among which 4 TFE3+ cases are negative for Melan-A and SMA, the other 2 TFE3+ cases did not detect Melan-A and SMA. The phenomenon of TFE3+ cases is always negative for Melan-A and SMA is interesting, which needs further study. Furthermore, there are 3 cases exploring the genetic status by FISH(Fluorescence in situ hybridization)/PCR(Polymerase Chain Reaction)/Sequencing of TFE3. The authors used FISH assay, which indicates a rearrangement of TFE3 in a child of 14 years old. Further molecular cloning by 5′ rapid amplification of complementary DNA ends and subsequent RT-PCR(Reverse Transcription-Polymerase Chain Reaction) revealed an SFPQ/PSF-TFE3 gene fusion, thus suggests at least a subset of PEComas constitutes a new member of the microphthalmia transcription factor family of tumors. Lee et al[24] performed FISH assay to assess Ewing sarcoma (EWS) gene breakage but showed no evidence of EWS rearrangement in a 62-year-old TFE3+ patient. Lu and colleagues[4] performed RT-PCR assay to detect related gene fusions, including PSF-TFE3, PRCC-TFE3, and EWS-ATF-1 in a 29-year-old TFE3+ patient, which is finally confirmed the PSF-TFE3 gene fusion. Sequencing of the amplified DNA, analyses the expression levels of MiTF and its downstream genes showed that MiTF, TYR, CDK2, TBX2, and C-MET were upregulated in the tumor.[4] Recently, other studies proposed the subset of tumors shows TFE3 positive, sharing distinctive clinicopathological features such as relatively young age, more-feminine, epithelioid cells with eosinophilic cytoplasm.[27] TFE3 seems to play an important role in the tumor of PEComas, and deserves further research.
Cathepsin-K was recently reported to be a more powerful marker in diagnosing PEComas,[54] however, it was only detected in 2 cases and both showed positiveness, which needs further verification.
3.5. Classification
GI PEComa-NOS manifests a broad biological behavior spectrum from indolent growth to aggressive progression. Most GI PEComas-NOS behave in a benign manner while some extend as malignant. Local recurrence or distant metastasis was observed in some cases. Clinical follow-up data (43 of 50 patients, 86%; range, 2.5–180, months) of included literatures revealed 10 local recurrences and/or distant metastases. Five patients died of tumor (12%) and 32 patients were alive without disease (74%). Given the paucity of such a distinctive disease and no systematic evaluation, there are still no firm criteria to help classify benign or malignant.
The 2002 WHO Soft Tissue and Bone book wrote: “PEComas displaying any combination of infiltrative growth, marked hypercellularity, nuclear enlargement and hyperchromasia, high mitotic activity, atypical mitotic figures, and coagulative necrosis should be regarded as malignant,” however, no exact guideline was presented.[2] By studying 35 cases of PEComa with evaluation of prognostic parameters, Doyle et al[55] concluded that there was a strong association between marked nuclear atypia, diffuse pleomorphism, >2 mitoses per 10 high power field (HPF) and metastases, and suggested these factors should be strongest predictors of malignant behavior. Apart from Folpe's criteria, Im et al[15] presented that large tumor size, infiltrative growth pattern, high nuclear grade, and high proliferative activity are possible predictors of aggressive behavior. After correlated the clinicopathologic of intestinal PEComas with tumor recurrence and/or metastasis, Lee et al[24] put forward that infiltrative growth, tumor location, and tumor size were important prognostic factors.
Even not unanimous, the most widely used method of classification at present was proposed by Folpe et al[27] (Table 3). They suggested a provisional classification strategy that stratifies PEComas as “benign,” “uncertain malignant potential” and “malignant”. Tumor size >5 cm, infiltrative growth pattern, high nuclear grade, necrosis, and mitotic activity >1/50 HPF were identified for predicting aggressive clinical behavior. “Benign” is free of the above features. “Malignant” refers to hold 2 or more that features. “Uncertain malignant potential” means that the tumor has only one of the following characteristics: nuclear pleomorphism/multinucleated giant cells or a size >5 cm.[27]
Table 3.
Classification of gastrointestinal perivascular epithelioid cell tumors-not otherwise specified (GI PEComas-NOS) proposed by Folpe et al[27].
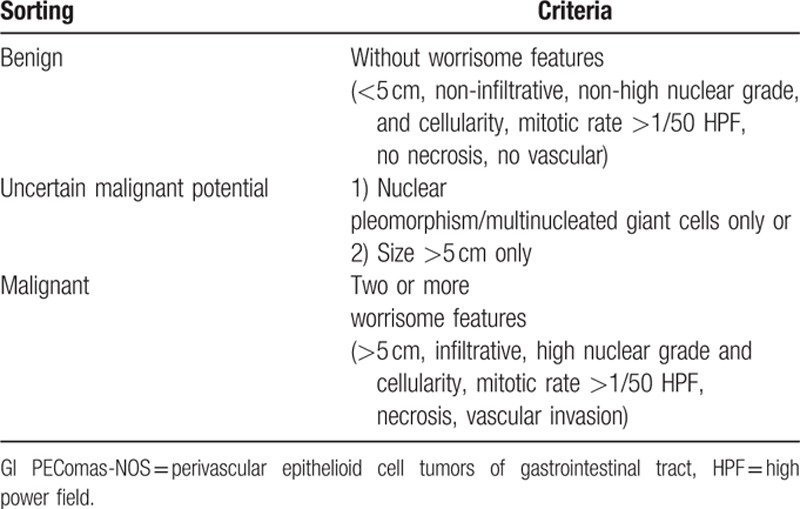
According to Folpe's criterion, there are 26 (52%) malignant cases. With 23 cases follow-up data available (mean: 16.5 months; range, 3–180 months) and 8 in that 23 cases developed local recurrences and/or distant metastases. Folpe's criterion is somewhat accurate for it included 8 of 10 recurrences and/or metastases cases as malignant. It is considered as the best available method now but needs more cases with long follow-up data to verify.
Hence, the firm criteria for classification have not been well established and the proposals need further validation.
3.6. Differential diagnosis
GI PEComa-NOS is difficult to diagnose because it is easy to be misdiagnosed as gastrointestinal stromal tumor (GIST), malignant melanoma (MM, primary or metastatic), smooth muscle tumor, renal cell carcinoma (RCC), paraganglioma, clear cell sarcoma, alveolar soft part sarcoma (ASPS) as well as other carcinomas (adrenocortical or hepatocellular carcinoma).
It is easy to confuse GI PEComas-NOS with GIST for their sharing epithelioid and/or spindle cell morphologies and their occurring within the abdomen. Morphologically, cells of PEComas and GIST show eosinophilic or clear cytoplasm but PEComa is different form GIST for its distinct perivascular concentric proliferation and representative granular cytoplasm.[15,22] Immunohistochemically, melanocytic markers (HMB-45 and Melan A) are the most helpful tool for their positiveness in PEComas but always negativeness in GIST. For epithelioid GIST, there is a cave that it can be misdiagnosed for their focally positive for melanocytic markers. However, epithelioid GIST can be picked by the absence of perivascular concentric proliferation and the positive staining of CD117. Furthermore, it should be aware that PEComa is prone to misdiagnosed as GIST for their occasionally expressing CD117, so if a pendent case showed CD117-positive cells less than 50%, the detection of melanocytic markers would be needful.[13]
MM involving GI is another important differential diagnosis of GI PEComas-NOS for its ability to imitate almost any morphology except prominent nucleoli and clear cytoplasm.[22] However, the cells of MM are various in shape such as epithelioid, spindle, small round, signet ring or plasmacytoid and it always composed of multiple shape cells mentioned above. For metastatic MM, furthermore, it is quite important to rely on more clinical history than microscopic findings. The clinical history of primary tumor of skin is regarded as the most basic element for differential diagnosis. S-100 is important to distinguish MM form PEComas because it is more frequently positive in MM. Even a fraction of PEComas could be S-100 positive, their expression of S-100 is usually weak and focal.[14]
Smooth muscle tumor should also be regarded as differential diagnosis of GI PEComas-NOS. Given similar appearance of epithelium and spindle, smooth muscle tumor shows disseminate cytoplasmic eosinophilia, vacuoles around the nucleus, and “cigar-shaped” nuclei while PEComa manifests clear to lightly eosinophilic cytoplasm, and round or ovoid nuclei. The negative for melanocytic markers like HMB-45 and Melan-A in smooth muscle tumor may also be useful clues. Multinucleated giant cells and “spider cell”-like cells that promote diagnosis of PEComas might be found under careful detection.[27,42]
RCC, especially the chromophobe cell type, is likely to confuse pathologist for its epithelioid appearance and rich angiogenesis. History of primary renal tumor is straightforward. The positive for CK, CD117, CD10, RCC antigen and the specificity of CD10 in RCC are also helpful.[22,40]
Paraganglioma can mimic PEComas for their semblable morphology though it rarely involves GI. But it is toilless to distinguish it from PEComas for its organoid growth pattern and diffuse synaptophysin and positivity of chromogranin marker but negativeness of myoid and melanocytic markers.[55]
GI clear cell sarcoma always has nests of round or epithelioid cells and mixed osteoclast-like giant cells, it expresses S100 strongly and carries t(2:22)(q34;q12) (EWS-CREB11) or t(12; 22)(q13; q13)(EWS-ATF1) gene fusion in most cases. These characteristics are main differences between GI PEComas-NOS and GI clear cell sarcoma.[56]
ASPS and PEComas show marked morphologic resemblance. Immunohistochemical staining for melanocytic markers and SMA can be applicable for ruling out APSP because cells of APSP are negative for these markers.[24]
For the other carcinomas (adrenocortical or hepatocellular carcinoma), the expression of melanocytic markers (HMB-45 and Melan-A) and the lack of immunoreactivity for cytokeratins help to exclude the diagnosis of carcinoma.[3]
3.7. Management strategy
The consistently effective treatment of GI PEComas-NOS is yet to be established. Complete resection is served as mainstay of treatment. Chemotherapy and immunotherapy are also applied in some cases. Given the potential malignant behavior of GI PEComas-NOS, close surveillance with imaging and endoscope is necessary.
Surgical resection is the most important and common method for primary tumor or local recurrence of GI PEComas-NOS. As a whole, surgery plays a resultful role in benign patients according to the general view of follow-up data. Most of the patients received laparotomy and only one patient underwent laparoscopic surgical procedures.[14] This might attribute to the difficulty of preoperative diagnosis and emergency situations. In consideration of the successful application in other GI tumors, laparoscopic surgery might also act effectively upon GI PEComas-NOS.
Different from PEComas arising in other system, endoscopy could be applied as auxiliary examination and even treatment in GI PEComas-NOS. Endoscopic resections were tried in 4 cases while only 2 succeeded, the others failed for the reason of bleeding or perforation.[11,17,19,26] The former patients had no recurrence of a 6 months or 15 months follow-up. We are, therefore, of opinion that endoscopic resection could be performed in polypoid lesions with superficial origin or infiltration, if with adequate preparation of the conversion to laparotomy.
Chemotherapy had been implemented in some potential or distinct malignant cases but its role remained out of clearity. For the variety of dose of different drugs taken and the heterogeneity of tumor, it would be quite a long way to conclude whether it works. However, it is almost unanimous that chemotherapy should be considered to apply in malignant cases. Immunotherapy such as IFN-α 2b had also been tried in some patients with unproven effect[25] while radiotherapy had not been applied in GI PEComas yet.
3.8. Follow-up
The longest period of follow-up in the articles was 180 months. The patient who had radical excision with a favorable outcome, was considered as a successful example of management.[23] Pisharody et al[19] presented to surveille the patient for physical examination and CT scans every 6 months, simultaneously, a yearly endoscope was also recommended. Since GI PEComas-NOS might represent at an aggressive course and lead to death, a closed and long-term follow-up accompanied by endoscope and imaging for the purpose of ruling out local recurrence or distant metastasis of the tumor, would be necessary.
4. Discussion
As a rare entity, although, increasing numbers of cases of PEComas have been reported and some underlying molecular mechanisms of this neoplasm have been explored, a much more clear picture about GI PEComas-NOS rises. GI PEComa-NOS is a less common type of mesenchymal tumor which expresses myomelanocytic markers, composed of epithelioid and/or spindle cell with perivascular distribution. It usually occurs in the middle age and more prevalent in women, most follow a benign course but some could show malignant progression.
However, there are still many mysteries of GI PEComas-NOS that should be further studied, including the origin of PEC, the nature counterpart of it, the underlying molecular mechanism of this lesion, the firm criteria to categorize, how to reach a correct diagnosis before surgery, the optional management strategy, and its prognosis. What we focus on mostly is the appropriate treatment of GI PEComas-NOS. Different strategies had been adopted in many cases with varying effects. Freeman et al[23] reported a case that they adopted surgery only and had a disease-free survival of 180 months after a long-period follow-up. Then as well, by combining operation and adjuvant chemotherapy, Shi et al[32] announced their patient with no evidence of recurrence or metastasis in the following 60 months surveillance. However, there were also some cases with poor prognosis being described such as one patient who had surgery only and died of tumor in the third month.[36] Researchers also reported that integrated therapy of completed resection and chemotherapy was adopted in a case but the patient had local recurrence and liver metastasis in 22 months and subsequently, died of tumor in 27 months.[27] And we see that, even the most appropriate management strategy is yet to be defined, surgical resection now almost serves as the predominant treatment and chemotherapy or immunotherapy is considered in cases with trait of malignancy. But we can also see that this pattern of treatment is of insufficient evidence that could make survival benefit. This ascribes to the rarity of incidence, uneven reports of case, different surgical methods and chemotherapy regimens.
As more pathologist and physicians realize this disease and the growing numbers of cases being diagnosed, we are of the opinion that there should be more normalized cases reports, more exploration of molecular mechanisms. Thus, since yet no RCTs are available at present, many points of debates still remain open.
5. Conclusions
The biologic behavior of GI PEComas-NOS still remains unclear. After plenty of studies concerning GI PEComas-NOS are brought out, there are still quite a lot unverified clinicopathological issues of this kind of scarce disease need to be clarified. The most appropriate treatment is yet to be defined. There may not be enough cases to be researched and analyzed to further our knowledge of it due to the rareness of this entity. Above all, we propose, the randomized clinical research should be conducted in despite of significant difficulties that we may face.
Acknowledgments
We thank Mr. Chenglong Zheng and Miss Fengjuan Chen for helping polish and improve the language of this paper.
Supplementary Material
Footnotes
Abbreviations: AML = angiomyolipoma, ASPS = alveolar soft part sarcoma, CCST = clear-cell “sugar” tumor, CT = computed tomography, GI = gastrointestinal tract, GI PEComas-NOS = perivascular epithelioid cell tumors of gastrointestinal tract, GIST = gastrointestinal stromal tumor, HPF = high power field, LAM = lymphangioleiomyomatosis, MM = metastatic melanoma, MRI = magnetic resonance imaging, PEC = perivascular epithelioid cell, PEComas = perivascular epithelioid cell tumors, PEComas-NOS = perivascular epithelioid cell tumors–not otherwise specified, PRISMA = Preferred Reporting Items for Systematic Reviews and Meta-Analyses, RCC = metastatic renal cell carcinoma, RCTs = randomized controlled trials, SMA = smooth muscle actin.
ZC, SH, JW, MX and YH contributed equally to this work.
Authors’ contribution: ZC devised the study. ZC, SH, JW, MX, YH, JC, YY and JP extracted, analyzed, and interpreted the data, reviewed the result and wrote the manuscript. WS authorized and tutored the research.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- 1.Bonetti F, Pea M, Martignoni G, et al. PEC and sugar. Am J Surg Pathol 1992; 16:307–308. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. [Google Scholar]
- 3.Armah HB, Parwani AV. Perivascular epithelioid cell tumor. Arch Pathol Lab Med 2009; 133:648–654. [DOI] [PubMed] [Google Scholar]
- 4.Lu B, Wang C, Zhang J, et al. Perivascular epithelioid cell tumor of gastrointestinal tract: case report and review of the literature. Medicine 2015; 94:e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornick JL, Fletcher CDM. PEComa: what do we know so far? Histopathology 2006; 48:75–82. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Flores A. Evidence on the neural crest origin of PEComas. Rom J Morphol Embryol 2011; 52:7–13. [PubMed] [Google Scholar]
- 7.Stone CH, Lee MW, Amin MB, et al. Renal angiomyolipoma: further immunophenotypic characterization of an expanding morphologic spectrum. Arch Pathol Lab Med 2001; 125:751–758. [DOI] [PubMed] [Google Scholar]
- 8.Ardeleanu C, Bussolati G. Telocytes are the common cell of origin of both PEComas and GISTs: an evidence-supported hypothesis. J Cell Mol Med 2011; 15:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Issat T, Maciejewski T, Beta J, et al. Rare case of uterine PEC-oma (perivascular epithelioid cell tumor) recurrence. Case report and literature review. Ginekol Pol 2012; 83:552–554. [PubMed] [Google Scholar]
- 10.Musella A, De Felice F, Kyriacou AK, et al. Perivascular epithelioid cell neoplasm (PEComa) of the uterus: a systematic review. Int J Surg 2015; 19:1–5. [DOI] [PubMed] [Google Scholar]
- 11.Tazelaar HD, Batts KP, Srigley JR. Primary extrapulmonary sugar tumor (PEST): a report of four cases. Mod Pathol 2001; 14:615–622. [DOI] [PubMed] [Google Scholar]
- 12.Genevay M, Mc Kee T, Zimmer G, et al. Digestive PEComas: a solution when the diagnosis fails to “fit”. Ann Diagn Pathol 2004; 8:367–372. [DOI] [PubMed] [Google Scholar]
- 13.Evert M, Wardelmann E, Nestler G, et al. Abdominopelvic perivascular epithelioid cell sarcoma (malignant PEComa) mimicking gastrointestinal stromal tumour of the rectum. Histopathology 2005; 46:115–117. [DOI] [PubMed] [Google Scholar]
- 14.Ryan P, Nguyen VH, Gholoum S, et al. Polypoid PEComa in the rectum of a 15-year-old girl: case report and review of PEComa in the gastrointestinal tract. Am J Surg Pathol 2009; 33:475–482. [DOI] [PubMed] [Google Scholar]
- 15.Im S, Yoo C, Jung JH, et al. Primary perivascular epithelioid cell tumor in the rectum: a case report and review of the literature. Pathol Res Pract 2013; 209:244–248. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto H, Oda Y, Yao T, et al. Malignant perivascular epithelioid cell tumor of the colon: report of a case with molecular analysis. Pathol Int 2006; 56:46–50. [DOI] [PubMed] [Google Scholar]
- 17.Baek JH, Chung MG, Jung DH, et al. Perivascular epithelioid cell tumor (PEComa) in the transverse colon of an adolescent: a case report. Tumori 2007; 93:106–108. [DOI] [PubMed] [Google Scholar]
- 18.Righi A, Dimosthenous K, Rosai J. PEComa: another member of the MiT tumor family? Int J Surg Pathol 2008; 16:16–20. [DOI] [PubMed] [Google Scholar]
- 19.Pisharody U, Craver RD, Brown RF, et al. Metastatic perivascular epithelioid cell tumor of the colon in a child. J Pediatr Gastroenterol Nutr 2008; 46:598–601. [DOI] [PubMed] [Google Scholar]
- 20.Cho HY, Chung DH, Khurana H, et al. The role of TFE3 in PEComa. Histopathology 2008; 53:236–239. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka M, Kato K, Gomi K, et al. Perivascular epithelioid cell tumor with SFPQ/PSF-TFE3 gene fusion in a patient with advanced neuroblastoma. Am J Surg Pathol 2009; 33:1416–1420. [DOI] [PubMed] [Google Scholar]
- 22.Shi HY, Wei LX, Sun L, et al. Clinicopathologic analysis of 4 perivascular epithelioid cell tumors (PEComas) of the gastrointestinal tract. Int J Surg Pathol 2010; 18:243–247. [DOI] [PubMed] [Google Scholar]
- 23.Freeman HJ, Webber DL. Perivascular epithelioid cell neoplasm of the colon. World J Gastrointest Oncol 2010; 2:205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M, Cho KJ, Yu C, et al. Perivascular epithelioid cell tumor of the sigmoid colon with transcription factor E3 expression. Ann Diagn Pathol 2012; 16:306–311. [DOI] [PubMed] [Google Scholar]
- 25.Park SJ, Han DK, Baek HJ, et al. Perivascular epithelioid cell tumor (PEComa) of the ascending colon: the implication of IFN-alpha2b treatment. Korean J Pediatr 2010; 53:975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanazawa A, Fujii S, Godai TI, et al. Perivascular epithelioid cell tumor of the rectum: report of a case and review of the literature. World J Surg Oncol 2014; 12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29:1558–1575. [DOI] [PubMed] [Google Scholar]
- 28.Gross E, Vernea F, Weintraub M, et al. Perivascular epithelioid cell tumor of the ascending colon mesentery in a child: case report and review of the literature. J Pediatr Surg 2010; 45:830–833. [DOI] [PubMed] [Google Scholar]
- 29.Lai CL, Hsu KF, Yu JC, et al. Malignant perivascular epithelioid cell tumor of the mesentery: a case report and literature review. Onkologie 2012; 35:114–117. [DOI] [PubMed] [Google Scholar]
- 30.Fu X, Jiang JH, Gu X, et al. Malignant perivascular epithelioid cell tumor of mesentery with lymph node involvement: a case report and review of literature. Diagn Pathol 2013; 8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wejman J, Nowak K, Gielniewska L, et al. PEComa of the mesentery coexisting with colon cancer: a case report. Diagn Pathol 2015; 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Geng J, Xie H, et al. Malignant perivascular epithelioid cell tumor arising in the mesentery: a case report. Oncol Lett 2015; 9:2189–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapur S, Patel NK, Levin MB, et al. Malignant mesenteric perivascular epithelioid cell neoplasm presenting as an intra-abdominal fistula in a 49-year-old female. Case Rep Oncol Med 2014; 2014:534175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitteldorf CA, Birolini D, da Camara-Lopes LH. A perivascular epithelioid cell tumor of the stomach: an unsuspected diagnosis. World J Gastroenterol 2010; 16:522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada S, Nabeshima A, Noguchi H, et al. Coincidence between malignant perivascular epithelioid cell tumor arising in the gastric serosa and lung adenocarcinoma. World J Gastroenterol 2015; 21:1349–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waters PS, Mitchell DP, Murphy R, et al. Primary malignant gastric PEComa - diagnostic and technical dilemmas. Int J Surg Case Rep 2012; 3:89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson J, Hartke J, Akram S. Metastatic perivascular epithelioid cell tumor of the stomach: an unusual diagnosis. Am J Gastroenterol 2013; 108:S238–S1238. [Google Scholar]
- 38.Mhanna T, Ranchere-Vince D, Hervieu V, et al. Clear cell myomelanocytic tumor (PEComa) of the duodenum in a child with a history of neuroblastoma. Arch Pathol Lab Med 2005; 129:1484–1486. [DOI] [PubMed] [Google Scholar]
- 39.Banerjee S, Premkumar J, Manipadam MT, et al. Perivascular epithelioid cell tumour of the duodenum. Trop Gastroenterol 2013; 34:182–184. [PubMed] [Google Scholar]
- 40.Chen Z, Shi H, Peng J, et al. Perivascular epithelioid cell tumor in the duodenum: challenge in differential diagnosis. Int J Clin Exp Pathol 2015; 8:8555–8562. [PMC free article] [PubMed] [Google Scholar]
- 41.Narayanaswamy S, Venkatanarasimha N, Buckley D, et al. Duodenal PEComa: a review of literature. Eur J Radiol Extra 2008; 67:e121–e123. [Google Scholar]
- 42.Unluoglu S, Bayol U, Korkmaz N, et al. Perivascular epithelioid cell tumor of the ileum presenting as diverticulitis. Case Rep Pathol 2012; 2012:476941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agaimy A, Wunsch PH. Perivascular epithelioid cell sarcoma (malignant PEComa) of the ileum. Pathol Res Pract 2006; 202:37–41. [DOI] [PubMed] [Google Scholar]
- 44.Birkhaeuser F, Ackermann C, Flueckiger T, et al. First description of a PEComa (perivascular epithelioid cell tumor) of the colon: report of a case and review of the literature. Dis Colon Rectum 2004; 47:1734–1737. [DOI] [PubMed] [Google Scholar]
- 45.Scheppach W, Reissmann N, Sprinz T, et al. PEComa of the colon resistant to sirolimus but responsive to doxorubicin/ifosfamide. World J Gastroenterol 2013; 19:1657–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koutlas IG, Pambuccian SE, Jessurun J, et al. Perivascular epithelioid cell tumor of the oral mucosa. Arch Pathol Lab Med 2005; 129:690–693. [DOI] [PubMed] [Google Scholar]
- 47.Fassan M, Cassaro M, Vecchiato M, et al. Malignant perivascular epithelioid cell tumor of the esophagus. Case Rep Pathol 2012; 2012:438505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yanai H, Matsuura H, Sonobe H, et al. Perivascular epithelioid cell tumor of the jejunum. Pathol Res Pract 2003; 199:47–50. [DOI] [PubMed] [Google Scholar]
- 49.Qu GM, Hu JC, Cai L, et al. Perivascular epithelioid cell tumor of the cecum: a case report and review of literatures. Chin Med J 2009; 122:1713–1715. [PubMed] [Google Scholar]
- 50.Pan CC, Jong YJ, Chai CY, et al. Comparative genomic hybridization study of perivascular epithelioid cell tumor: molecular genetic evidence of perivascular epithelioid cell tumor as a distinctive neoplasm. Hum Pathol 2006; 37:606–612. [DOI] [PubMed] [Google Scholar]
- 51.Kenerson H, Folpe AL, Takayama TK, et al. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum Pathol 2007; 38:1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogemann D, Flemming P, Kreipe H, et al. Correlation of MRI and CT findings with histopathology in hepatic angiomyolipoma. Eur Radiol 2001; 11:1389–1395. [DOI] [PubMed] [Google Scholar]
- 53.Tan Y, Zhang H, Xiao EH. Perivascular epithelioid cell tumour: dynamic CT, MRI and clinicopathological characteristics-analysis of 32 cases and review of the literature. Clin Radiol 2013; 68:555–561. [DOI] [PubMed] [Google Scholar]
- 54.Rao Q, Cheng L, Xia QY, et al. Cathepsin K expression in a wide spectrum of perivascular epithelioid cell neoplasms (PEComas): a clinicopathological study emphasizing extrarenal PEComas. Histopathology 2013; 62:642–650. [DOI] [PubMed] [Google Scholar]
- 55.Doyle LA, Hornick JL, Fletcher CD. PEComa of the gastrointestinal tract: clinicopathologic study of 35 cases with evaluation of prognostic parameters. Am J Surg Pathol 2013; 37:1769–1782. [DOI] [PubMed] [Google Scholar]
- 56.Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol 2010; 41:1–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.