

Abstract
Hirayama disease (HD) is characterized by development of asymmetric forearm muscle atrophy during adolescence with or without focal cervical spinal cord atrophy. The purpose of this study is to assess the correlation of clinical symptoms, disease progression, and electrophysiological findings with cervical spine magnetic resonance imaging (MRI) findings.
The medical records, cervical spine MRIs, and electrophysiological findings of 44 HD patients were retrospectively reviewed and analyzed.
Denervation changes in any single C5 to C7 root-innervated muscle (deltoid, biceps, triceps, or extensor digitorum communis) occurred more frequently in the 25 patients with cord atrophy than the 19 patients without cord atrophy (88% vs 53%, P = 0.02). Onset age, duration of disease progression, neurological examinations, nerve conduction study, and electromyographic findings from individual muscles were similar between patient groups.
Compared with HD patients without cord atrophy, HD patients with cord atrophy experience a more severe denervation change in C5 to C7 root-innervated muscles.
Keywords: electromyography, electrophysiology, Hirayama disease, magnetic resonance imaging
1. Introduction
Hirayama disease (HD) is a rare condition characterized by asymmetric weakness and atrophy of cervical 8 (C8)—thoracic (T1) root-innervated muscles during adolescence.[1–3] There are also some HD patients in whom muscular atrophy was noted in the C5 to C6 innervated muscles (e.g., biceps and brachioradialis).[2,4–6] Most HD patients begin to develop progressive upper limb weakness in their teens and early 20s, followed within several years by spontaneous stabilization.[3,7] Although the underlying pathophysiology of HD remains uncertain, the HD-related spinal cord pathology reveals an abundance of small neurons, weak gliosis, central necrosis, and shrinkage of bilateral anterior horns at the C7 and C8 levels, suggesting ischemia.[8] Cervical spinal cord compression by the dural sac during repeated neck flexion with minor trauma, and disparate growth between the vertebral column and spinal cord during puberty may lead to those ischemic changes in the anterior horn cells at the C8 and T1 vertebral levels.[2,3]
Localized cervical spinal cord atrophy is detected in around 49% to 100% of HD patients, and most spinal cord atrophies are located at the C6 to C7 vertebral levels.[2,4,9–15] On the other hand, electromyographic (EMG) studies in HD patients usually show denervation changes in C8 to T1 root-innervated muscles (e.g., abductor pollicis brevis [APB] and first dorsal interosseous [FDI]).[5,14,16,17] However, whether the neuroimaging findings are relevant to the clinical presentation and/or duration of disease progression in HD patients remains unknown. Also uncertain is whether cervical cord atrophy and denervation change in cervical root-innervated muscles are related. To answer these questions, we evaluated the correlation of electrophysiological/neuroimaging findings with clinical information in our HD patients and tried to identify factors that are potentially associated with disease severity or progression.
2. Materials and methods
2.1. Ethics statement and study populations
We retrospectively reviewed the records of all patients with HD who were attended and followed up in our neuromuscular clinics in Chang Gung Memorial Hospital-Linkou Medical Center from 1995 to 2010. Ethics approval was provided by the institutional review boards of the Chang Gung Memorial Hospital (96–1168B).
Diagnostic criteria for HD included onset before the age of 25 years, unilateral or bilateral distal predominant weakness and wasting of the upper limbs without significant sensory function impairment, stationary clinical course after slow insidious progression, no lower limbs involvement, and no history of spinal cord tumor, cervical vertebral abnormality, syringomyelia, multifocal motor neuropathy, congenital muscular dystrophy, trauma, infection, inflammation, or any other cause for the clinical findings.[18–20] Nerve conduction velocity (NCV), EMG, and cervical spine magnetic resonance imaging (MRI) studies were performed in all HD patients. Patients were divided into cord atrophy and noncord atrophy groups by following criteria of MRI studies. Onset age, gender ratios, duration of symptoms onset to first medical examination, duration of symptoms onset to stationary phase, percentage of progression to bilateral limbs involvement, neurological examinations, cervical spine MRIs, and electrophysiological findings of HD patients with and without cord atrophy were retrospectively reviewed and analyzed.
2.2. Magnetic resonance imaging study
The magnetic resonance imagings (MRIs) were carried out on 1.5-T scanners. Localized cord atrophy was defined as a decrease in cord size in comparison with the cord above and below the affected level on sagittal view[9,10] (Fig. 1). The earliest cervical spine MRIs of all patients in the neck neutral position were compared and reviewed by a radiologist and neurologist.
Figure 1.
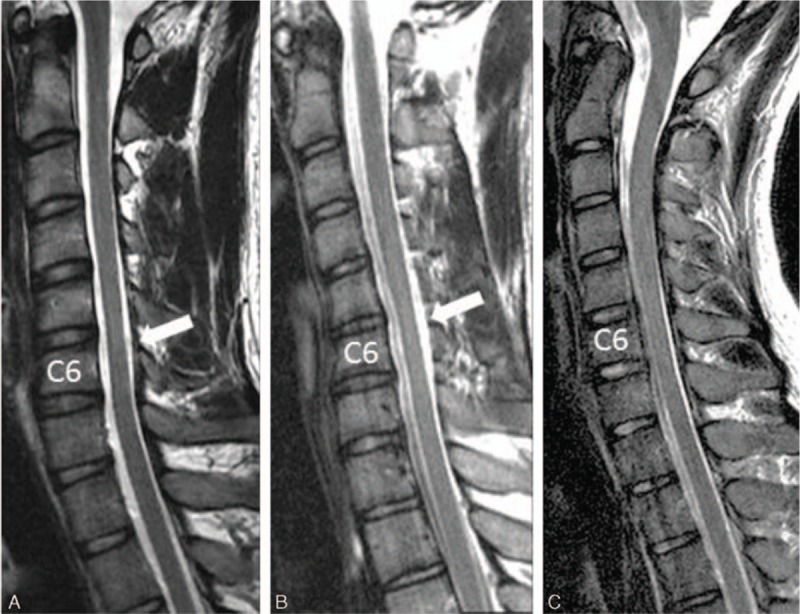
(A) Sagittal T2-weighted cervical magnetic resonance imaging (MRI) of the spinal cord atrophied at the C5 to C6 vertebral body levels (white arrow) in a 22-year-old man with progressive distal atrophy of the right hand for 2 years. (B) Sagittal T2-weighted cervical MRI of the spinal cord atrophied at the C6 vertebral level (white arrow) in a 17-year-old man with progressive distal atrophy of the right hand for 6 months. (C) Sagittal T2-weighted cervical MRI of the spinal cord without cord atrophy in a 19-year-old man with progressive distal atrophy of the right hand for 2 years.
2.3. Nerve conduction study
The NCV and EMG studies in our laboratory were performed using standardized procedures.[16,21] We used Nicolet Viasys Viking Select EMG EP System (MFI Medical Equipment, Inc., San Diego, CA) for electrodiagnostic studies. Original device default filter settings were applied: 2 to 10 K and 2 to 20 K for the motor and sensory conduction studies, respectively, and 20 to 5 KHz with 60-Hz notch filter on for needle EMG study. The sensitivity was 2 or 5 mV for motor conduction study and 10 or 20 μV for sensory conduction study, with sweep duration of 2 or 5 ms per division. The examiner would adjust sensitivity and sweep duration to get the clearest response figure during analysis. For needle EMG study, the sensitivity was 50 μV for spontaneous activity detection and 200 μV for motor unit potential analysis, and sweep duration was 10 ms per division. Motor conduction studies consisted of stimulating the median and the ulnar nerves. The compound muscle action potentials (CMAPs) of the median and the ulnar nerves were recorded from the APB and the abductor digiti minimi (ADM), respectively. The active electrode was placed over the belly of the muscles, and the reference electrode was placed over the tendon of those muscles. We stimulated the median nerve at the wrist and elbow, and the ulnar nerve at the wrist, below, and above the elbow. When stimulating the ulnar nerve, the distance across the elbow segment was fixed at 10 cm, with the elbow in moderate flexion. In sensory conduction studies, we stimulated the median and the ulnar nerves at the wrist and recorded the sensory nerve action potentials on the 2nd and 5th digits, respectively. The ulnar/median (U/M) CMAP amplitude ratio (stimulated at the wrist) was calculated for each patient. The patients diagnosed with carpal tunnel syndrome (median NCV at the wrist <43 m/s[22] with sensory axon loss) and ulnar neuropathy at the elbow (ulnar [forearm − across elbow] NCV > 10 m/s[23] with sensory axon loss) were excluded.
2.4. Electromyography
Concentric needle electrodes were used for needle electromyography (EMG). The following muscles were commonly investigated: deltoid, biceps, triceps, pronator teres, extensor digitorum communis (EDC), flexor carpi ulnaris (FCU), APB, FDI, and ADM. Because not all these muscles were electromyographically studied in all patients, we focused on those muscles studied in over 50% of patients (i.e., the deltoid, biceps, triceps, EDC, FCU, APB, and FDI muscles). Acute denervation change was defined as the presence of abnormal spontaneous activity (fibrillation potentials or positive sharp waves) in at least 2 spots, while chronic reinnervation was defined as the presence of abnormal long-duration (>15 ms) or large-amplitude (>6 mV) motor unit action potentials with reduced recruitment. The frequency of denervation changes in individual muscles was analyzed. Individual root-innervated muscles were designated C5 to C7 (biceps or deltoid, triceps, or EDC) or C8 to T1 (APB, FDI, or FCU).
2.5. Statistical analysis
The Statistical Program for Social Sciences statistical software (version 19.0; IBM, North Castle, NY) was used for statistical analysis. P values <0.05 were considered statistically significant. Noncategorical variables were compared using the Mann–Whitney U test. Categorical variables were compared using the 2-tailed Fisher exact test.
3. Results
Forty-four patients with HD fitting the above diagnostic criteria and having available cervical MRI, NCV, and EMG information were recruited to this study. Cervical spinal cord atrophy was detected in 25 (56.8%) HD patients – mostly at the C5 (47.7%), C6 (45.5%), and a few at the C7 vertebral level (11.4%). In the patients with cord atrophy, 8 (32%) patients had cord atrophy at a single vertebral level, 12 (48%) at both the C5 and C6 vertebral levels, 1 (4%) at both C6 and C7 vertebral levels, and 4 (16%) from C5 to C7 vertebral levels.
In these 44 patients, needle EMG studies were done in the deltoid, biceps, triceps, EDC, APB, FDI, and FCU, respectively, of 22 (50%), 42 (95.5%), 22 (50%), 28 (63.6%), 41 (93.2%), 29 (65.9%), and 26 (59.1%) patients. The percentage of active denervation change (i.e., in deltoid [0.0%], biceps [2.4%], triceps [18.2%], EDC [32.1%], APB [25%], FDI [34.5%], and FCU [50%]) is less than the percentage of chronic denervation change (i.e., in deltoid [10%], biceps [29.3%], triceps [81%], EDC [65%], APB [69.2%], FDI [96.6%], and FCU [96%]; Fig. 2) in patients with HD. Furthermore, chronic denervation changes were more commonplace and most frequent in the FDI and FCU muscles.
Figure 2.
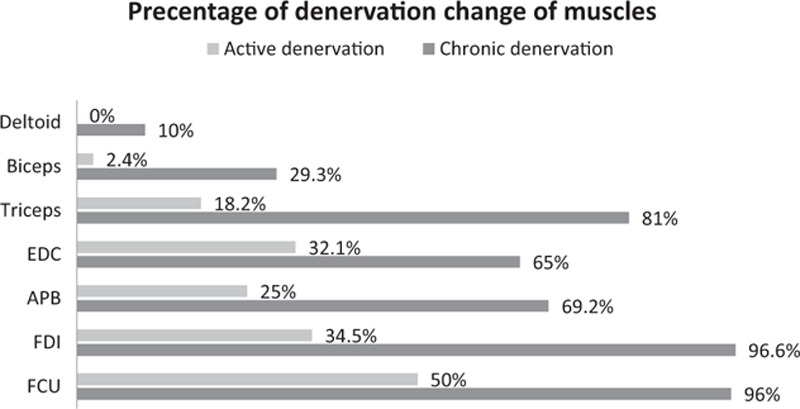
Percentage of denervation change in individual muscles in Hirayama disease patients.
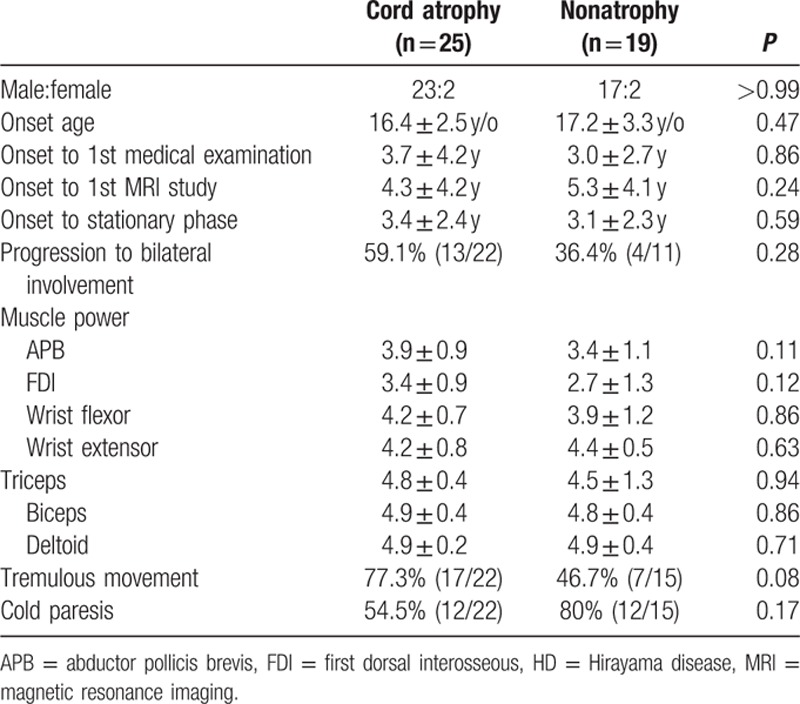
Patients with and without cord atrophy had similar age at symptoms onset, gender ratios, interval between symptoms onset and first presentation at our institution, interval between symptoms onset and the stationary phase of the disease, percentage of patients with tremor and cold paresis, and percentage of patients progressing to bilateral limb involvement. Muscle strengths based on Medical Research Council grade were also similar between HD patients with and without cord atrophy (Table 1).
Table 1.
Clinical data of HD patients with and without spinal cord atrophy.
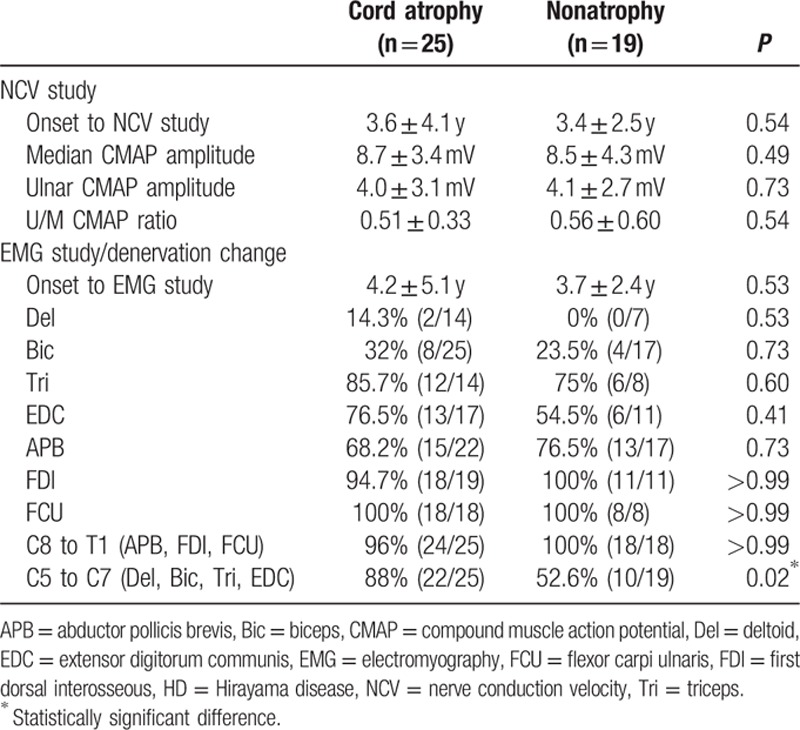
The median and ulnar CMAP amplitudes (stimulated at the wrist), U/M CMAP amplitude ratio, and incidences of denervation change (acute and chronic) in individual muscles (deltoid, biceps, triceps, EDC, APB, FDI, and FCU) and in any single C8 to T1 root-innervated muscle (APB, FDI, or FCU) were similar in HD patients with and without cord atrophy (Table 2). However, the incidence of denervation change in any single C5 to C7 root-innervated muscle (deltoid, biceps, triceps, or EDC) was significantly greater in HD patients with cord atrophy (88% vs 53%, P = 0.02) (Table 2), indicating that denervation change in C5 to C7 root-innervated muscles is more strongly associated with cord atrophy in patients with HD.
Table 2.
Nerve conduction and EMG findings in HD patients with and without spinal cord atrophy.
4. Discussion
Previous studies have demonstrated that around 49% to 100% of HD patients have cervical spinal cord atrophy, mostly located at the C6 to C7 vertebral level.[2,4,9–15] Pathologic studies also show reduction in the anterior–posterior diameter of the spinal cord and shrinkage of the bilateral anterior horns at the C7 and C8 vertebral levels.[8] Similar to previous studies, our study showed that 57% of HD patients had cervical cord atrophy, often located at the C5 (48%) and C6 (45%) vertebral levels. The characteristic features of HD (focal atrophy and regional denervation change of the cervical cord) indicate that HD is a focal spinal cord disorder rather than a diffused motor neuron degenerative disorder.
Our previous study found that U/M CMAP ratio was reduced in HD patients and increased in amyotrophic lateral sclerosis patients, indicating the different underlying pathophysiologies of those 2 diseases.[21] In the present study, HD patients with and without cord atrophy had similar median and ulnar CMAP amplitudes and similar (U/M) ratios, suggesting that the fundamental spinal cord pathophysiological mechanism was the same in HD patients with and without cord atrophy.
In a study of the clinical course and cervical MRIs of 7 HD patients, Biondi et al[10] found 5 patients (71%) with spinal cord atrophy, mostly at the C6 to C7 vertebral levels, and poor correlation of the rate of clinical symptom progression and stabilization periods with MRI findings. Our study with a larger patient number also showed similar results, suggesting that the course and duration of HD symptom progression is similar in patients with and without cord atrophy. Also as previously reported,[7] our study showed that periods of disease stabilization in both groups were the same (i.e., around 3 years). Collectively, these findings again indicate that HD with or without cord atrophy has a similar underlying pathophysiology.
Pradhan[6] described 11 HD patients in India with bilaterally severe symmetric forearm muscles involvement and severe weakness and wasting in the bilateral C7, C8, and T1 myotomes that frequently spilled over to the C6 segment. Nine patients (82%) had typical lower cervical cord atrophy on MRIs, while all 11 patients had severe flattening of the lower cervical spinal cord near the C5 to C6 vertebral levels and a crescent-shaped enhancing epidural space extending from the C4 to T2 vertebral levels when the neck was flexed. Pradhan concluded that spinal cord compression by the dural sac near the C6 vertebral body was more severe in HD patients with bilateral symmetric involvement than in other HD patients. The more severe spinal cord compression may be due to the shorter length of the dural canal, which cannot compensate for the increased length of the vertebral canal during growth in the juvenile period.[9,14] Our study showed that denervation change in C5 to C7 root-innervated muscles occurred more frequently in HD patients with cord atrophy (mostly at the C5–C6 vertebral levels). The focal spinal cord atrophy indicated a more severe underlying pathological change in the spinal cord at the C5 to C6 vertebral levels. Anatomically, the anterior horn cells of C6 to C7 root-innervated muscles are located near the C5 to C6 vertebral levels. Thus, focal anterior horn cells proximal to the C5 to C6 vertebral level may contribute to the denervation changes in the C5 to C7 root-innervated muscles of HD patients with spinal cord atrophy. Focal ischemic change of the spinal cord at the C5 to C6 vertebral levels, leading to prominent denervation change in C5 to C7 root-innervated muscles, may be more severe in HD patients with cord atrophy than in HD patients without cord atrophy. On the other hand (but consistent with other studies),[5,14,16,17] our study found that the frequency of denervation change in C8 to T1 root-innervated muscles is similar between HD patients with and without cord atrophy, suggesting that similar ischemic damage occurs at the T1 vertebral level in HD patients with and without cord atrophy.
Our study showed that HD with and without cord atrophy could be differentiated on the basis of EMG findings in a group of muscles innervated at the C5 to C7 root level. EMG findings of any single muscle or muscles from single roots were similar between these 2 groups, reflecting the multiple cervical segment involvement in HD and frequent extension of cord atrophy to more than one segment. Unfortunately, in this retrospective study, the muscles monitored by EMG were not the same in every HD patient, and hence we were unable to identify the “best” combination of muscles to use for EMG prediction of the presence of cord atrophy.
In conclusion, our study found that over 50% HD patients had cervical spinal cord atrophy at the C5 to C6 vertebral levels. HD patients with and without cervical spinal cord atrophy had similar characteristic clinical features, course, and disease progression. HD patients with cervical cord atrophy had a more severe denervation change in muscles innervated by C5 to C7 segments. These findings suggest that pathological changes are more extensive in patients with cord atrophy than in those without cord atrophy.
Footnotes
Abbreviations: ADM = abductor digiti minimi, APB = abductor pollicis brevis, CAMPs = compound muscle action potentials, EDC = extensor digitorum communis, EMG = electromyography, FCU = flexor carpi ulnaris, FDI = first dorsal interosseous, HD = Hirayama disease, MRI = magnetic resonance imaging, NCV = Nerve conduction velocity, SNAPs = sensory nerve action potentials, Tri = Triceps.
The authors have no conflicts of interest to disclose.
References
- 1.Sobue I, Saito N, Iida M, et al. Juvenile type of distal and segmental muscular atrophy of upper extremities. Ann Neurol 1978; 3:429–432. [DOI] [PubMed] [Google Scholar]
- 2.Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med 2000; 39:283–290. [DOI] [PubMed] [Google Scholar]
- 3.Huang YL, Chen CJ. Hirayama disease. Neuroimaging Clin N Am 2011; 21:939–950.ix–x. [DOI] [PubMed] [Google Scholar]
- 4.Tashiro K, Kikuchi S, Itoyama Y, et al. Nationwide survey of juvenile muscular atrophy of distal upper extremity (Hirayama disease) in Japan. Amyotroph Lateral Scler 2006; 7:38–45. [DOI] [PubMed] [Google Scholar]
- 5.Peiris JB, Seneviratne KN, Wickremasinghe HR, et al. Non familial juvenile distal spinal muscular atrophy of upper extremity. J Neurol Neurosurg Psychiatry 1989; 52:314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pradhan S. Bilaterally symmetric form of Hirayama disease. Neurology 2009; 72:2083–2089. [DOI] [PubMed] [Google Scholar]
- 7.Gourie-Devi M, Nalini A. Long-term follow-up of 44 patients with brachial monomelic amyotrophy. Acta Neurol Scand 2003; 107:215–220. [DOI] [PubMed] [Google Scholar]
- 8.Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease): focal cervical ischemic poliomyelopathy. Neuropathology 2000; 20 (suppl):S91–S94. [DOI] [PubMed] [Google Scholar]
- 9.Chen CJ, Hsu HL, Tseng YC, et al. Hirayama flexion myelopathy: neutral-position MR imaging findings—importance of loss of attachment. Radiology 2004; 231:39–44. [DOI] [PubMed] [Google Scholar]
- 10.Biondi A, Dormont D, Weitzner I, Jr, et al. MR Imaging of the cervical cord in juvenile amyotrophy of distal upper extremity. AJNR Am J Neuroradiol 1989; 10:263–268. [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder R, Keller E, Flacke S, et al. MRI findings in Hirayama's disease: flexion-induced cervical myelopathy or intrinsic motor neuron disease? J Neurol 1999; 246:1069–1074. [DOI] [PubMed] [Google Scholar]
- 12.Misra UK, Kalita J, Mishra VN, et al. A clinical, magnetic resonance imaging, and survival motor neuron gene deletion study of Hirayama disease. Arch Neurol 2005; 62:120–123. [DOI] [PubMed] [Google Scholar]
- 13.Sonwalkar HA, Shah RS, Khan FK, et al. Imaging features in Hirayama disease. Neurol India 2008; 56:22–26. [DOI] [PubMed] [Google Scholar]
- 14.Gandhi D, Goyal M, Bourque PR, et al. Case 68: Hirayama disease. Radiology 2004; 230:692–696. [DOI] [PubMed] [Google Scholar]
- 15.Pradhan S, Gupta RK. Magnetic resonance imaging in juvenile asymmetric segmental spinal muscular atrophy. J Neurol Sci 1997; 146:133–138. [DOI] [PubMed] [Google Scholar]
- 16.Huang YC, Ro LS, Chang HS, et al. A clinical study of Hirayama disease in Taiwan. Muscle Nerve 2008; 37:576–582. [DOI] [PubMed] [Google Scholar]
- 17.Kao KP, Wu ZA, Chern CM. Juvenile lower cervical spinal muscular atrophy in Taiwan: report of 27 Chinese cases. Neuroepidemiology 1993; 12:331–335. [DOI] [PubMed] [Google Scholar]
- 18.Hirayama K, Tsubaki T, Toyokura Y, et al. Juvenile muscular atrophy of unilateral upper extremity. Neurology 1963; 13:373–380. [DOI] [PubMed] [Google Scholar]
- 19.Oryema J, Ashby P, Spiegel S. Monomelic atrophy. Can J Neurol Sci [Le journal canadien des sciences neurologiques] 1990; 17:124–130. [DOI] [PubMed] [Google Scholar]
- 20.Singh N, Sachdev KK, Susheela AK. Juvenile muscular atrophy localized to arms. Arch Neurol 1980; 37:297–299. [DOI] [PubMed] [Google Scholar]
- 21.Lyu RK, Huang YC, Wu YR, et al. Electrophysiological features of Hirayama disease. Muscle Nerve 2011; 44:185–190. [DOI] [PubMed] [Google Scholar]
- 22.Kimura J. The carpal tunnel syndrome: localization of conduction abnormalities within the distal segment of the median nerve. Brain 1979; 102:619–635. [DOI] [PubMed] [Google Scholar]
- 23.Chaudhry V, Clawson LL. Entrapment of motor nerves in motor neuron disease: does double crush occur? J Neurol Neurosurg Psychiatry 1997; 62:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]