

Abstract
To further investigate into the relapses of Henoch–Schönlein purpura (HSP), we analyzed the frequency, clinical features, and predictors of relapses in series of 417 unselected patients from a single center. After a median follow-up of 12 (interquartile range [IQR]: 2–38) years, almost one-third of the 417 patients (n = 133; 32%; 85 men/48 women) had experienced at least 1 relapse. At the time of disease diagnosis, patients who later experienced relapses had less commonly infections than those who never suffered flares (30.8% vs 41.9%; P = 0.03). In contrast, patients who experienced relapses had a longer duration of the first episode of palpable purpura than those without relapses (palpable purpura lasting >7 days; 80.0% vs 68.1%; P = 0.04). Abdominal pain (72.3% vs 62.3%; P = 0.03) and joint manifestations (27.8% vs 15.5%; P = 0.005) were also more common in patients who later developed relapses. In contrast, patients who never suffered relapses had a slightly higher frequency of fever at the time of disease diagnosis (9.3% vs 3.8%; P = 0.06). At the time of disease diagnosis, corticosteroids were more frequently given to patients who later had relapses of the disease (44% vs 32% in nonrelapsing patients; P = 0.03). Relapses generally occurred soon after the first episode of vasculitis. The median time from the diagnosis of HSP to the first relapse was 1 (IQR: 1–2) month. The median number of relapses was 1 (IQR 1–3). The main clinical features at the time of the relapse were cutaneous (88.7%), gastrointestinal (27.1%), renal (24.8%), and joint (16.5%) manifestations. After a mean ± standard deviation follow-up of 18.9 ± 9.8 years, complete recovery was observed in 110 (82.7%) of the 133 patients who had relapses. Renal sequelae (persistent renal involvement) was found in 11 (8.3%) of the patients with relapses. The best predictive factors for relapse were joint and gastrointestinal manifestations at HSP diagnosis (odds ratio [OR]: 2.22; 95% confidence interval [CI]: 1.34–3.69, and OR: 1.60; 95% CI: 1.01–2.53, respectively). In contrast, a history of previous infection was a protective factor for relapses (OR: 0.60; 95% CI: 0.38–0.94). In conclusion, joint and gastrointestinal manifestations at the time of diagnosis of HSP are predictors of relapses.
Keywords: Henoch–Schönlein purpura, IgA vasculitis, predictors, relapses
1. Introduction
Henoch–Schönlein purpura (HSP) is a systemic vasculitis characterized by the presence of palpable purpura, joint manifestations, abdominal pain, and nephritis.[1–6] HSP is typically a childhood disorder that can also occur in adults.[1,7,8] Whereas in children is usually a self-limited benign disease, in adults it has been associated with more severe clinical features and a poorer outcome.[9–11] Nonetheless, the overall prognosis of the disease is usually good.
Relapses are common in HSP. However, information specifically focused on relapses is scarce and results are often discordant. In this regard, the frequency of relapses reported in previous studies ranges from 2.7% to 51.7%.[12,13] Relapses present in most cases as a new episode of cutaneous rash that is often associated with gastrointestinal and renal manifestations.[12,13] Patient selection due to severity, mainly for the presence of renal manifestations at the time of diagnosis, may be one of the main reasons for the discrepancy in the frequency and clinical spectrum of relapses in HSP. In some cases, the numbers of patients included in the studies are small. More importantly, in most series full information on the clinical manifestations of the vasculitis at the time of the relapses, the median time to relapse since the diagnosis, and the average number and duration of relapses is not available. Furthermore, there are series that only include adults,[12,17] whereas others are exclusively focused on children.[13–16]
Taking into account these considerations, we aimed to assess the frequency clinical features and predictors of relapses in a large series of unselected patients with HSP from a single center.
2. Patients and methods
Retrospective study of 417 consecutive patients attended at a single tertiary care center between January 1975 and December 2012. Patients were diagnosed with HSP according to the criteria proposed by Michel et al, based on the American College of Rheumatology (ACR) database and methodology.[19,20] We used these criteria because we have recently reported that the European League Against Rheumatism criteria showed low concordance with previous sets of HSP classification criteria.[21] HSP was confirmed by a skin biopsy in 110 cases (most of them adults), showing the typical histological findings of leukocytoclastic vasculitis, including neutrophilic infiltration, leukocytoclasia, and fibrinoid necrosis into the vessel wall of arterioles, capillaries, and postcapillary venules, and red cell extravasation.[22,23] The remaining 307 patients, in whom a skin biopsy was not performed, had typical nonthrombocytopenic symmetric palpable purpura. In addition, all of them fulfilled the criteria proposed by Michel et al.[19]
This is an observational study on patients with HSP with relapsing disease. In cases like this, according to Spanish National Regulation, an Ethical Committee approval is not mandatory.
2.1. Clinical definitions
Classification by age groups: As previously reported,[9,19,20,24,25] for the purpose of the present study patients older than 20 were considered adults, and those 20 years of age or younger were considered children. As previously discussed in former studies of our group, the cut-off age of 20 was chosen because this age was proposed as a criterion for HSP by the ACR (age ≤ 20 years at disease onset),[20] and because this age best discriminated HSP from hypersensitivity vasculitis in the study by Michel et al.[19]
Precipitating factors: As previously reported when we assessed the clinical spectrum of HSP in northern Spain,[2] we considered a drug or a mild infection (in most cases an upper respiratory tract infection) as the probable precipitating factor(s) for a cutaneous vasculitis if the drug intake or the onset of the infectious process started within the week before the development of the vasculitis cutaneous lesions. Nevertheless, if the cutaneous vasculitis occurred after antibiotic therapy for a mild infection, both the infection and the drug were considered as the possible precipitating factors. Fever was considered to be present if the temperature was >37.7°C, regardless of the site where the temperature was assessed and the age of the patient.[2] Joint manifestations were defined if the patients had arthralgia with or without joint swelling (arthritis). As described in a former study of our group, nephropathy was divided in 2 categories: mild and severe.[25] Mild nephropathy was defined if the patients had microhematuria (≥5 red cells/high power field) and/or proteinuria that did not reach the nephrotic range. Severe nephropathy was considered to be present when the patient had 1 of the following findings: nephrotic syndrome: defined as plasma albumin level ≤ 25 g/L and either 1 g of proteinuria/d per m2 of body surface area in children or >3.5 g of proteinuria/d in adults, with or without the presence of edema; or acute nephritic syndrome that was defined as hematuria with at least 2 of the following features hypertension, elevated plasma urea or creatinine serum levels, and oliguria. Renal insufficiency was defined as creatinine level >125% of the upper limit of normal. The spectrum of gastrointestinal manifestations encompassed the presence of at least 1 of the following manifestations: bowel angina (characterized by the presence of diffuse abdominal pain that worsened after the meals), gastrointestinal bleeding (if the patient suffered melena or hematochezia or he/she had a positive stool Guaiac test), and nausea and vomiting in the context of the vasculitis. Constitutional symptoms were considered to be present if the patient had asthenia and/or anorexia and weight loss of at least 4 kg. A relapse was defined when a patient previously diagnosed with HSP and asymptomatic for at least 4 weeks, presented again a new flare of cutaneous lesions or other systemic manifestations of the vasculitis.[2,9]
2.2. Laboratory studies
As previously described when we assessed the clinical spectrum of HSP in northern Spain,[2] we performed full blood cell count, Westergren erythrocyte sedimentation rate (ESR)/h, and urinalysis in most patients. Also, as previously described,[2] most adults with HSP were tested for rheumatoid factor (RF) (which was initially performed by quantitative latex agglutination test, and later on by nephelometry), antinuclear antibodies (ANAs) (initially by indirect immunofluorescence initially using rodent tissues as substrate and more recently by Hep-2 cells), and serum C3 and C4 complement levels (initially by radial immunodiffusion and more recently by using nephelometry). Other testing such as antineutrophil cytoplasmic antibodies was determined in patients seen after 1992. Antineutrophil cytoplasmic antibodies were initially performed by indirect immunofluorescence on alcohol fixed neutrophils and later on using ELISA with purified proteinase-3 and myeloperoxidase.[2] As previously reported, cryoglobulins as well as the composition of the cryoprecipitate were assessed by double immunodiffusion with specific antibodies, and immunoglobulins were studied by nephelometry.[2] Other laboratory tests such as anti-DNA antibodies (by immunofluorescence using Chrithidia luciliae as a substrate), blood cultures, Guaic test to disclose occult blood, hepatitis B, hepatitis C, or HIV infection serology, were only performed when considered by the physician who was in charge of the patient.[2] Anemia was defined if the hemoglobin level was ≤11 g/dL and leukocytosis if the white blood cell count was ≥11,000/mm3. An ESR was defined as elevated if it was higher than 20 or 25 mm/h for men or women, respectively.[9,26] IgA levels were considered to be increased if the total serum IgA level was >400 mg/dL.
To reduce the risk of selection bias, in the present study we included all the patients from our area that fulfilled definitions for HSP, regardless of whether a biopsy was performed or not. In this regard, a skin biopsy was carried out in most adults with skin lesions whereas it was only done in a small number of children.[2] Biopsies were not generally performed to children with typical HSP vasculitis who did not suffer severe renal involvement.[2] We feel that the inclusion of children with typical HSP without tissue biopsy would reduce the risk of selection bias providing more accurate information on the actual clinical spectrum of this vasculitis.[2] A renal biopsy was usually done whenever that a feature suggestive of severe renal disease, such as protein excretion above 1 g/d or elevated plasma creatinine concentration, was present. Light microscopy often disclosed mesangial hypercellularity as well as increased deposition of extracellular matrix proteins in patients with nephropathy. The typical finding in these patients was the presence of prominent granular IgA deposits in the mesangium seen by immunoflourescence.
The duration of the follow-up, time from the diagnosis of HSP to the first relapse, number of relapses, clinical features at the time of relapses, treatment, and outcome were recorded in all the patients.
2.3. Data collection and statistical analysis
Data were first reviewed in an attempt to retrieve the following information: precipitating factors; clinical, laboratory, and histopathological features; treatment; and prognosis. These data were extracted from the clinical records according to a specific protocol, reviewed for confirmation of the diagnosis, and stored in a computerized database. To minimize entry error all the data were double checked.
Besides chart review, most patients were interviewed by phone to know if relapses had occurred after the last visit at the hospital. The statistical analysis was performed with the STATISTICA software package (Statsoft Inc., Tulsa, OK). All continuous variables were tested for normality. Results were expressed as mean ± standard deviation (SD) or as median and range or interquartile range (IQR) as appropriate. Student t test or Mann–Whitney U test was used to compare continuous variables, and χ2 test for categorical variables. Univariate and then multivariate stepwise logistic regression analyses were conducted to identify the independent predictors of HSP relapse. A P value < 0.05 was considered statistically significant in all the calculations.
3. Results
We included 417 patients diagnosed with HSP. After a median follow-up of 12 (IQR: 2–38) years almost one-third of them (n = 133; 32%; 85 men/48 women) had experienced at least 1 relapse. One hundred three of 133 patients with relapses were children or young adults (age ≤20 years).
The main demographic, clinical, and laboratory findings of the patients with relapsing and nonrelapsing HSP are summarized in Tables 1 and 2.
Table 1.
Main demographic, etiological, and clinical features’ differences between patients with HSP who experienced further relapses and those who did not suffer this complication.
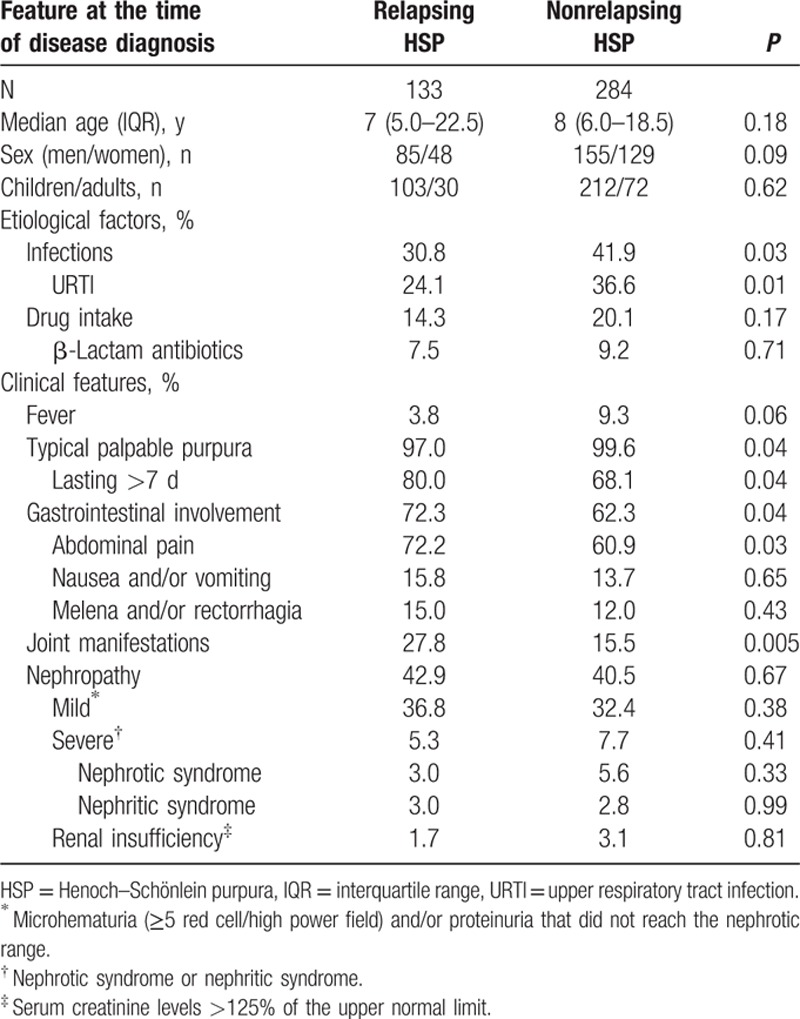
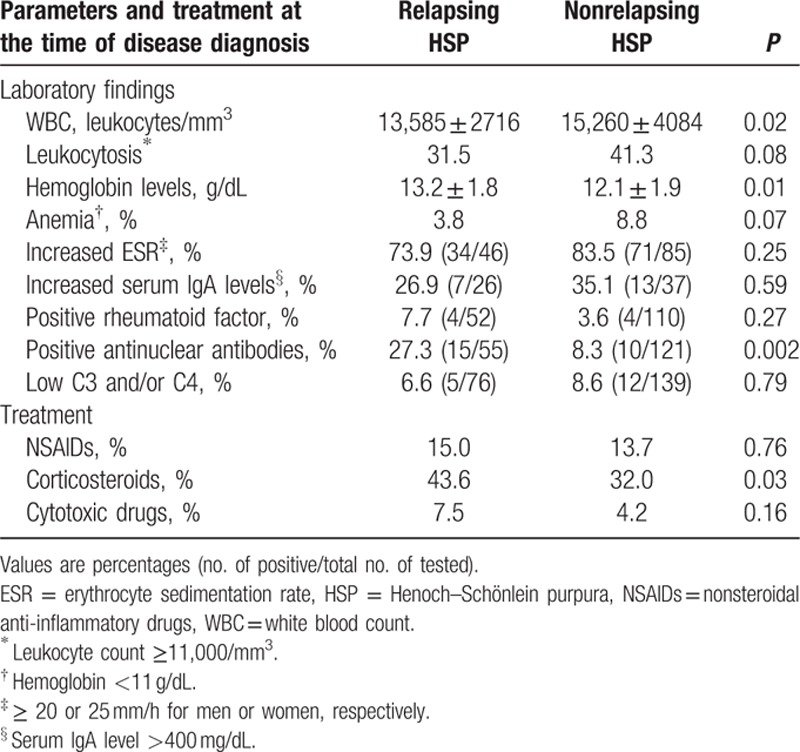
Table 2.
Laboratory findings and treatment differences between patients with HSP who experienced further relapses and those who did not suffer this complication.
3.1. Demographic, etiologic, and clinical differences at the time of disease diagnosis between patients with and without relapses
No differences according to gender or age at the time of disease diagnosis between patients with HSP who had relapses and those who did not experience this complication were found. Nevertheless, several clinical differences between these 2 groups were observed. With regard to potential precipitating factors for the development of the disease, infections, in particular upper respiratory tract infections, were less commonly observed in the subgroup of patients who later suffered relapses (30.8% vs 41.9% in those without relapses; P = 0.03). However, acute otitis media and mumps were more commonly observed in the subgroup of patients who experienced relapses (2.26% vs 0.35% and 0.75% vs 0.35%, respectively) but the difference did not achieve statistical significance. Other infections were uncommon in both groups. On the other hand, the frequency of drug intake shortly before the onset of the disease was similar in both groups (Table 1).
With respect to differences in the clinical spectrum of the disease at the time of disease diagnosis between those who over the extended follow-up developed relapses and those who did not have this complication, patients who experienced relapses had a longer duration of the first episode of palpable purpura than those without relapses (palpable purpura lasting >7 days; 80.0% vs 68.1%; P = 0.04). It was also the case for abdominal pain (72.3% vs 62.3%; P = 0.03) and joint manifestations (27.8% vs 15.5%; P = 0.005). In contrast, patients who never suffered relapses had a slightly higher frequency of fever at the time of disease diagnosis (9.3%) than those with relapses (3.8%); P = 0.06. No significant differences in the frequency of nephritis and renal insufficiency were seen between both groups of patients (Table 1).
3.2. Differences in the laboratory data found at the time of diagnosis of the disease between HSP patients with and without relapses
A raised ESR was frequently seen at the time of disease diagnosis. Nevertheless, no significant differences were seen between the group of patients who experienced relapses and those who never had flares of the disease (Table 2).
Relapsing patients had significantly lower white blood count (mean value: 13,585 leukocytes/mm3) and higher hemoglobin (mean value: 13.2 g/dL) than those who did not suffer flares (mean values 15,260 leukocytes/mm3 and 12.1 g/dL, respectively) (Table 2). ANA positivity was more commonly disclosed in the relapsed group (27.3% vs 8.3%; P = 0.002). Nevertheless, none of the patients with positive ANA met classification criteria for systemic lupus erythematosus. There were no significant differences in the frequency of RF. It was also the case for IgA elevation or lower levels of serum complement (Table 2).
3.3. Treatment and outcome
Corticosteroids were the drugs more commonly used in both the patients with and those without relapses of the disease. Nevertheless, they were more frequently given to patients who later had relapses (44% vs 32 % in nonrelapsing patients; P = 0.03). At the time of the disease diagnosis, the median initial corticosteroid dose in the subgroup who later experienced relapses was 30 (IQR: 20–45) mg whereas in those who never suffered relapses it was 30 (IQR 30–45) mg (P = 0.6). The duration of corticosteroids at the time of disease diagnosis in the subgroup of patients with HSP who later had relapses was 1 (0.5–3) month versus 1.5 (0.7–4) months in those who never suffered relapses (P = 0.9). At that time, corticosteroids were prescribed because of persistence of skin lesions or visceral involvement, including severe abdominal pain, gastrointestinal bleeding, or nephropathy. Cytotoxic immunosuppressive drugs were given usually either as corticosteroid-sparing agents or as add-on therapy in patients with severe renal involvement. However, no differences in the use of immunosuppressive drugs between both groups were seen (Table 2). In this regard, 10 of the 133 who experienced relapses were treated with immunosuppressive drugs at the time of disease diagnosis (azathioprine 5, oral cyclophosphamide 3, and mycophenolate 2). Twelve of the 284 who did not have relapses were also treated with immunosuppressive drugs (azathioprine 5, oral cyclophosphamide 6, and oral methotrexate 1; P = 0.16).
After a mean ± SD follow-up of 18.9 ± 9.8 years, complete recovery was observed in 110 (82.7%) of the 133 patients who had relapses. Renal sequelae (persistent renal involvement) was found in 11 (8.3%) of the patients who suffered relapses. In most cases, renal damage was of mild severity, not associated with impairment of renal function and manifested by hematuria or hematuria plus proteinuria. At last follow-up chronic renal insufficiency was only observed in 2 (1.5%) patients from the group with relapses. During the first episode of the vasculitis (at time of diagnosis) and then throughout the course of the disease, including the extended follow-up, dialysis was only required in 2 patients from the group with relapses.
Out of 5 patients with relapses, 1 received medication at the time of a flare of the disease. As occurred at the time of disease diagnosis, corticosteroids were the drugs more commonly given in the episodes of relapses (17%). Other drugs used at that time were immunosuppressive drugs (3%) and nonsteroidal anti-inflammatory drugs (3%).
3.4. Frequency, clinical features, and predictive factors of relapses
Relapses generally occurred soon after the first episode of vasculitis. In this regard, the median time from the diagnosis of HSP to the first relapse was 1 (IQR: 1–2) month. The median number of relapses was 1 (IQR: 1–3). In general, the duration of the vasculitis at the time of the relapses was shorter than at the time of the disease diagnosis (median duration of the vasculitic episodes of relapses 3.5 [IQR: 3–5] days).
As shown in Fig. 1, cutaneous involvement in almost 90% of patients (in the majority of the cases presenting as typical palpable purpura), either isolated or in combination with other clinical manifestations, was the most common feature observed at the time of the relapses of the disease. Other clinical manifestations observed at the time of the relapses in this subgroup of 133 patients with HSP were gastrointestinal (27.1%), renal (24.8%), and joint (16.5%) manifestations. Regarding gastrointestinal manifestations at the time of the relapses, abdominal pain was the manifestation observed in most patients followed by nausea and/or vomiting. Melena and/or rectorrhagia only occurred in a few patients at the time of the relapses.
Figure 1.
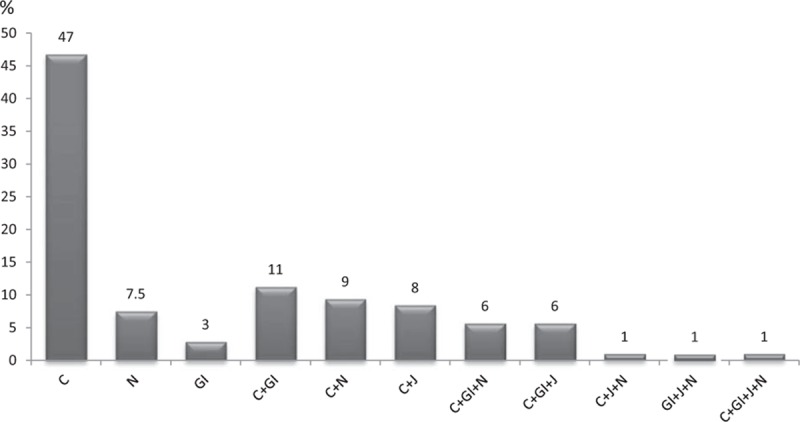
Clinical spectrum at the time of the relapses in 133 patients with Henoch–Schönlein purpura who experienced flares after the diagnosis of disease. Data are expressed as percentages. C = cutaneous lesions, GI = gastrointestinal involvement, J = joint involvement, N = nephropathy.
A regression model for the risk of relapses in HSP is shown in Table 3. The multivariate analysis disclosed that the best predictors of relapses were joint (odds ratio [OR]: 2.22; 95% confidence interval [CI]: 1.34–3.69; P = 0.002) and gastrointestinal manifestations (OR: 1.60; 95% CI: 1.01–2.53; P = 0.04) at the time of diagnosis of HSP. In contrast, a history of previous infection at the time of disease diagnosis was found to be a protective factor against relapses of HSP (OR: 0.60; 95% CI: 0.38–0.94; P = 0.02).
Table 3.
Regression models for the risk of relapses in patients with HSP from northern Spain.
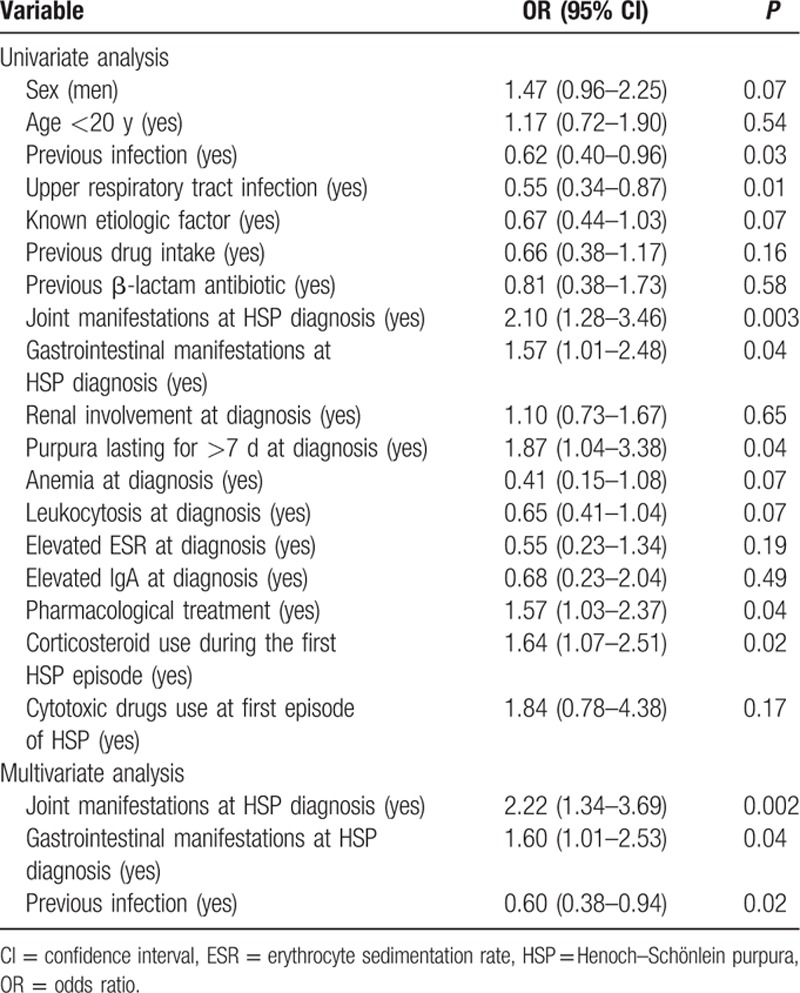
In a further step, to determine if there were potential differences in what predicts relapses between children and adults, we performed a multivariable logistic regression analysis in each age subgroup of patients with HSP (children and adults). In children (age ≤ 20 years at disease onset), a history of infection shortly before the onset of HSP manifestations was associated with a lower risk of relapses (OR: 0.57; 95% CI: 0.35–0.93; P = 0.025). In contrast, the presence of joint manifestations at the time of disease diagnosis was the best predictor of relapses of HSP in children (OR: 2.51; 95% CI: 1.42–4.44; P = 0.002). In adults, however, the presence of gastrointestinal manifestations at the time of disease diagnosis was the best predictor of relapses of HSP (OR: 4.00; 95% CI: 1.38–11.62; P = 0.011).
Finally, as shown in Table 3, the use of corticosteroids during the first HSP episode was associated with an increased risk of relapses of HSP (OR: 1.64; 95% CI: 1.07–2.51; P = 0.02). However, the association of corticosteroid use with relapses of the disease was only statistically significant in the subgroup of children with HSP (OR: 2.25; 95% CI: 1.34–3.77; P = 0.002).
4. Discussion
HSP is considered a benign and self-limited disorder, being renal involvement the most severe complication.[1–6] Many reports have assessed the frequency of nephropathy in HSP,[1,2,6,9,27] and some of them have tried to identify predictive factors for renal involvement.[17,28–30] However, relapses, a well-known complication of HSP,[2] have been scarcely assessed.[12–16] Moreover, studies dealing with relapses were focused mainly on patients with renal involvement, included a relatively small number of cases or were performed in pediatric or adult populations separately (Table 4).[12–18] Therefore, the frequency, type, and predictive factors for HSP relapses are not well established.
Table 4.
Relapses in Henoch–Schönlein purpura in previous studies: literature review.
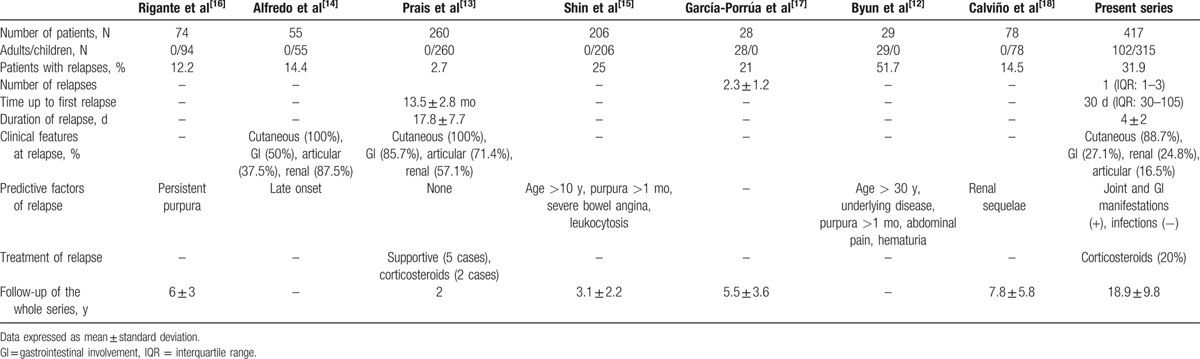
Our study encompassed a large and unselected series of 417 patients with HSP seen in a single tertiary care center during a 37-year period. The main features of our patients are similar to those from other series published in the literature.[10,31–34]
The frequency of patients with HSP who experience relapses varies greatly from a series to another. It ranges from 2.7% to 51.7% (Table 4).[12–18] This is probably due to the wide differences in the selection of patients in the different studies. In addition, in some of them, the number of relapses is not specified.
Clinical manifestations during relapses have shown great variability in the different studies. Nevertheless, the most common feature is the presence of cutaneous lesions, either isolated or in combination with gastrointestinal or renal involvement (Table 4). As shown in our series, the relapsing episode usually tends to mimic the initial one.[13,14]
In our series, we have identified 3 independent predictive factors for HSP relapses; joint and gastrointestinal manifestations at and a history of previous infection at the time of the diagnosis of HSP. Interestingly when patients were stratified according to the age of disease onset in children (age 20 years or younger) and adults, we observed that relapses in children occurred more commonly in those presenting with joint manifestations whereas in adults the relapses of the disease were more commonly seen in those patients who experienced gastrointestinal manifestations at the time of disease diagnosis. In addition, the protective role of infection against the risk of relapses in HSP was mainly related to children with HSP.
In keeping with Trapani et al,[35] we also observed an association between the use of corticosteroids at the time of disease diagnosis and the risk of relapses. However, this association was only observed in children but not in adults with HSP. Table 4 shows the factors associated with HSP relapses in other published series. Thus, Shin et al[15] reported severe bowel angina, age >10 years, persistent purpura (>1 month), and leukocytosis as the best set of predictors for relapse. Byun et al[12] described age > 30 years, the presence of an underlying disease, persistent purpura (>1 month), abdominal pain, and the presence of hematuria as the predictors of relapsing HSP. Moreover, there are other studies in which only a predictive factor, such as persistent purpura[16] or late onset of HSP,[14] was found. In this regard, Calviño et al[18] observed that children who experienced renal sequelae were at greater risk relapses than those patients without renal impairment.
The good renal prognosis observed in our cohort might be at odds with other reports of long-term risk of renal insufficiency among patients diagnosed with HSP. This fact was previously highlighted when we described the clinical spectrum of HSP in northern Spain.[2] In this regard, there is a great variability in clinical features and outcome in different series. It may be related to several factors including selection bias because some studies are mainly based on patients with nephritis.[36] On the other hand, the use of variable criteria for the classification of HSP among different series may be another factor to explain differences in the renal outcome. Another factor that may explain differences in the renal outcome is the absence of a standardized therapeutic management, especially in cases of nephropathy. In our study, we assessed a large series of unselected patients with HSP. In this regard, to avoid selection bias, we included all patients seen in the different departments attending patients with HSP in our hospital (Rheumatology, Pediatrics, Nephrology, Dermatology and Internal Medicine). Therefore, we included not only cases presenting with severe disease or with atypical presentation but also cases with mild disease. Moreover, in our study, we included patients of all ages, adults and children. However, it is known that the frequency of nephritis and the risk of renal insufficiency are more common in adults.[1,9,10,36]
The pathogenesis of relapses in HSP still remains unknown. Therefore, it would be desirable to search for novel biomarkers that could predict relapses more accurately. In this sense, several studies have revealed the relevant role of some genetic variants (including those located in the human leukocyte antigen [HLA] system) in both, HSP susceptibility and clinical heterogeneity.[37–40] However, a minority of studies are focused on the predisposition to relapse. Thus, HLA B35 haplotype has been related to relapsing HSP.[41] Noteworthy, we have found that an infection shortly before the onset of the vasculitis, which is a predisposing factor for the first episode of HSP is, by contrast, a protective factor for a relapsing disease. The reason for this finding is unknown. It is possible that factors associated with infection in HSP could be of transient nature and, thus, lead to a more limited, nonrelapsing disease. On the other hand, it is possible that patients fulfilling definitions for HSP in whom an infection, in particular an upper respiratory tract infection, was closely related to or associated with the onset of the disease may represent a different subset of this vasculitis, different from that unrelated to infection. In this regard, abnormal autoimmune tests (RF and ANAs) were more frequently observed in patients with HSP who experienced relapses than in those who never had flares of the disease.
Finally, given the nature of this retrospective cohort, changing techniques used for assays is an understandable consequence. However, we acknowledge this as a potential limitation of the study.
In conclusion, we have assessed the frequency, type, and risk factors of relapses in a large series of unselected patients with HSP from a single center. They occur in almost one-third of the patients and the clinical features at the time of the relapses are similar to those found at disease diagnosis. We should pay special attention to those patients presenting with joint or gastrointestinal manifestations at the time of diagnosis of HSP, because the relapsing rate seems to be higher in these cases. In contrast, an infection shortly before the onset of HSP appears to be a protective factor for relapsing disease. Further population-based studies including large series of patients are needed to confirm our data and identify other possible factors that allow us to predict more accurately relapses in patients with HSP.
Acknowledgment
The authors thank to the members of the Rheumatology, Dermatology, Pediatrics Internal Medicine and Pathology Services of Hospital Universitario Marqués de Valdecilla, Santander, Spain.
Footnotes
Abbreviations: CI = confidence interval, ESR = erythrocyte sedimentation rate, HSP = Henoch–Schönlein purpura, IQR = interquartile range, OR = odds ratio, RF = rheumatoid factor, SD = standard deviation.
VC-R and JLH had equal contribution.
MAG-G and RB shared senior authorship.
This study was supported by grants from “Fondo de Investigaciones Sanitarias” PI12/00193 (Spain). This work was also partially supported by RETICS Program, RD08/0075 (RIER) from “Instituto de Salud Carlos III” (ISCIII) (Spain).
The authors have no conflicts of interest to disclose.
References
- 1.Saulsbury FT. Clinical update: Henoch–Schönlein purpura. Lancet 2007; 369:976–978. [DOI] [PubMed] [Google Scholar]
- 2.Calvo-Río V, Loricera J, Mata C, et al. Henoch–Schönlein purpura in northern Spain: clinical spectrum of the disease in 417 patients from a single center. Medicine (Baltimore) 2014; 93:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González-Gay MA, García-Porrúa C. Systemic vasculitis in adults in northwestern Spain, 1988–1997. Clinical and epidemiologic aspects. Medicine (Baltimore) 1999; 78:292–308. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Gay MA, Blanco R, Pina T. IgA vasculitis (Henoch–Schönlein purpura). In: Ball GV, Fessler BJ, Bridges SL Jr. eds. Vasculitis. 3rd ed. New York: Oxford University Press; 2014. Chapter 39. [Google Scholar]
- 5.Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol 1999; 10:2637–2644. [DOI] [PubMed] [Google Scholar]
- 6.Saulsbury FT. Henoch–Schönlein purpura. Curr Opin Rheumatol 2001; 13:35–40. [DOI] [PubMed] [Google Scholar]
- 7.Cassidy JT, Petty RE. Cassidy JT, Petty RE. Henoch-Schonlein purpura. Textbook of Pediatric Rheumatology 3rd EdPhiladelphia: WB Saunders; 1995; 384–388. [Google Scholar]
- 8.Brogan P, Bagga A. Cassidy J, Laxer RM, Petty RE. Leukocytoclastic vasculitis. Textbook of Pediatric Rheumatology 6th EdPhiladelphia: Elsevier; 2011; 483–497. [Google Scholar]
- 9.Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch–Schönlein purpura in adulthood and in childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864. [DOI] [PubMed] [Google Scholar]
- 10.García-Porrúa C, Calvo MC, Llorca J, et al. Henoch–Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum 2002; 32:149–156. [DOI] [PubMed] [Google Scholar]
- 11.García-Porrúa C, González-Gay MA. Comparative clinical and epidemiologic study of hypersensitivity vasculitis versus Henoch–Schönlein purpura in adults. Semin Arthritis Rheum 1999; 28:404–412. [DOI] [PubMed] [Google Scholar]
- 12.Byun JW, Song HJ, Kim L, et al. Predictive factors of relapse in adult with Henoch–Schönlein purpura. Am J Dermatopathol 2012; 34:139–144. [DOI] [PubMed] [Google Scholar]
- 13.Prais D, Amir J, Nussinovitch M. Recurrent Henoch–Schönlein purpura in children. J Clin Rheumatol 2007; 13:25–28. [DOI] [PubMed] [Google Scholar]
- 14.Alfredo CS, Nunes NA, Len CA, et al. Henoch–Schönlein purpura: recurrence and chronicity. J Pediatr (Rio J) 2007; 83:177–180. [DOI] [PubMed] [Google Scholar]
- 15.Shin JI, Park JM, Shin YH, et al. Predictive factors for nephritis, relapse, and significant proteinuria in childhood Henoch–Schönlein purpura. Scand J Rheumatol 2006; 35:56–60. [DOI] [PubMed] [Google Scholar]
- 16.Rigante D, Candelli M, Federico G, et al. Predictive factors of renal involvement or relapsing disease in children with Henoch–Schönlein purpura. Rheumatol Int 2005; 25:45–48. [DOI] [PubMed] [Google Scholar]
- 17.García-Porrúa C, González-Louzao C, Llorca J, et al. Predictive factors for renal sequelae in adults with Henoch–Schönlein purpura. J Rheumatol 2001; 28:1019–1024. [PubMed] [Google Scholar]
- 18.Calviño MC, Llorca J, García-Porrúa C, et al. Henoch–Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore) 2001; 80:279–290. [DOI] [PubMed] [Google Scholar]
- 19.Michel BA, Hunder GG, Bloch DA, et al. Hypersensitivity vasculitis and Henoch–Schonlein purpura: a comparison between the 2 disorders. J Rheumatol 1992; 19:721–728. [PubMed] [Google Scholar]
- 20.Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch–Schonlein purpura. Arthritis Rheum 1990; 33:1114–1121. [DOI] [PubMed] [Google Scholar]
- 21.Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification of Henoch–Schönlein purpura. An analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol 2015; 33 suppl 89:S44–S47. [PubMed] [Google Scholar]
- 22.Gibson LE. Cutaneos vasculitis. Approach to diagnosis and systemic associations. Mayo Clin Proc 1990; 65:221–229. [DOI] [PubMed] [Google Scholar]
- 23.Lie JT. Illustrated histopathologic classification criteria for selected vasculitis syndromes. American College of Rheumatology Subcommittee on Classification of Vasculitis. Arthritis Rheum 1990; 33:1074–1087. [DOI] [PubMed] [Google Scholar]
- 24.Calvo-Río V, Loricera J, Ortiz-Sanjuán F, et al. Revisiting clinical differences between hypersensitivity vasculitis and Henoch–Schönlein purpura in adults from a defined population. Clin Exp Rheumatol 2014; 32 suppl 82:S34–S40. [PubMed] [Google Scholar]
- 25.Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Cutaneous vasculitis in children and adults: associated diseases and etiologic factors in 303 patients. Medicine (Baltimore) 1998; 77:403–418. [DOI] [PubMed] [Google Scholar]
- 26.Pillebout E, Thervet E, Hill G, et al. Henoch–Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278. [DOI] [PubMed] [Google Scholar]
- 27.Calvo-Río V, Loricera J, Martín L, et al. Henoch–Schönlein purpura nephritis and IgA nephropathy: a comparative clinical study. Clin Exp Rheumatol 2013; 31 suppl 75:S45–S51. [PubMed] [Google Scholar]
- 28.Shrestha S, Sumingan N, Tan J, et al. Henoch–Schönlein purpura with nephritis in adults: adverse prognostic indicators in a UK population. QJM 2006; 99:253–265. [DOI] [PubMed] [Google Scholar]
- 29.Coppo R, Andrulli S, Amore A, et al. Predictors of outcome in Henoch–Schönlein nephritis in children and adults. Am J Kidney Dis 2006; 47:993–1003. [DOI] [PubMed] [Google Scholar]
- 30.Tancrede-Bohin E, Ochonisky S, Vignon-Pennamen MD, et al. Schönlein–Henoch purpura in adult patients. Predictive factors for IgA glomerulonephritis in a retrospective study of 57 cases. Arch Dermatol 1997; 133:438–442. [DOI] [PubMed] [Google Scholar]
- 31.Hung SP, Yao-Hsu Y, Yu-Tsan L, et al. Clinical manifestations and outcomes of Henoch–Schönlein purpura: comparison between adults and children. Pediatr Neonatol 2009; 50:162–216. [DOI] [PubMed] [Google Scholar]
- 32.Lin SJ, Huang JL. Henoch–Schonlein purpura in Chinese children and adults. Asian Pac J Allergy Immunol 1998; 16:21–25. [PubMed] [Google Scholar]
- 33.Ilan Y, Naparstek Y. Schönlein–Henoch syndrome in adults and children. Semin Arthritis Rheum 1991; 21:103–109. [DOI] [PubMed] [Google Scholar]
- 34.Uppal SS, Hussain MA, Al-Raqum HA, et al. Henoch–Schönlein's purpura in adults versus children/adolescents: a comparative study. Clin Exp Rheumatol 2006; 24:26–30. [PubMed] [Google Scholar]
- 35.Trapani S, Micheli A, Grisolia F, et al. Henoch–Schönlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum 2005; 35:143–153. [DOI] [PubMed] [Google Scholar]
- 36.Faull RJ, Aarons I, Woodroffe AJ, et al. Adult Henoch–Schönlein nephritis. N Z J Med 1987; 17:396–401. [DOI] [PubMed] [Google Scholar]
- 37.Amoli MM, Thomson W, Hajeer AH, et al. HLA-DRB1∗01 association with Henoch–Schönlein purpura in patients from northwest Spain. J Rheumatol 2001; 28:1266–1270. [PubMed] [Google Scholar]
- 38.López-Mejías R, Genre F, Pérez BS, et al. HLA-DRB1 association with Henoch–Schonlein purpura. Arthritis Rheumatol 2015; 67:823–827. [DOI] [PubMed] [Google Scholar]
- 39.López-Mejías R, Genre F, Sevilla-Pérez B, et al. Association of HLA-B∗41:02 with Henoch–Schönlein purpura (IgA vasculitis) in Spanish individuals irrespective of the HLA-DRB1 status. Arthritis Res Ther 2015; 17:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amoli MM, Thomson W, Hajeer AH, et al. Interleukin 1 receptor antagonist gene polymorphism is associated with severe renal involvement and renal sequelae in Henoch–Schönlein purpura. J Rheumatol 2002; 29:1404–1407. [PubMed] [Google Scholar]
- 41.Nathwani D, Laing RB, Smith CC, et al. Recurrent post-infective Henoch–Schönlein syndrome: a genetic influence related to HLA B35? J Infect 1992; 25:205–210. [DOI] [PubMed] [Google Scholar]