

Abstract
We summarized the clinical characteristics of patients presenting with seizures and limbic encephalitis (LE) associated with leucine-rich glioma inactivated-1 protein antibody (LGI1) in order help recognize and treat this condition at its onset.
We analyzed clinical, video electroencephalogram (VEEG), magnetic resonance imaging (MRI), and laboratory data of 10 patients who presented with LGI1-LE and followed up their outcomes from 2 to 16 (9.4 ± 4.2) months.
All patients presented with seizures onset, including faciobrachial dystonic seizure (FBDS), partial seizure (PS), and generalized tonic-clonic seizure (GTCS). Four patients (Cases 3, 5, 7, and 8) had mild cognitive deficits. Interictal VEEG showed normal patterns, focal slowing, or sharp waves in the temporal or frontotemporal lobes. Ictal VEEG of Cases 4, 5, and 7 showed diffuse voltage depression preceding FBDS, a left frontal/temporal origin, and a bilateral temporal origin, respectively. Ictal foci could not be localized in other cases. MRI scan revealed T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity and evidence of edema in the right medial temporal lobe in Case 3, left hippocampal atrophy in Case 5, hyperintensities in the bilateral medial temporal lobes in Case 7, and hyperintensities in the basal ganglia and frontal cortex in Case 10. All 10 serum samples were positive for LGI1 antibody, but it was only detected in the cerebrospinal fluid (CSF) of 7 patients. Five patients (Cases 2, 4, 6, 7, and 8) presented with hyponatremia. One patient (Case 2) was diagnosed with small cell lung cancer. While responses to antiepileptic drugs (AEDs) were poor, most patients (except Case 2) responded favorably to immunotherapy.
LGI1-LE may initially manifest with various types of seizures, particularly FBDS and complex partial seizures (CPS) of mesial temporal origin, and slowly progressive cognitive involvement. Clinical follow-up, VEEG monitoring, and MRI scan are helpful in early diagnosis. Immunotherapy is effective for the treatment of both seizure and LE associated with LGI1 antibody. Although mostly nonparaneoplastic, tumor screening is recommended in some cases.
Keywords: cognitive deficits, faciobrachial dystonic seizure, hyponatremia, leucine-rich gliomainactivated-1 protein, limbic encephalitis
1. Introduction
Leucine-rich glioma-inactivated-1 (LGI1) protein antibody-associated limbic encephalitis (LE) is the most common type of voltage-gated potassium channel (VGKC) complex antibody-associated LE (VGKC-LE); it was first reported in 2001. LGI1-LE is characterized by faciobrachial dystonic seizure (FBDS), cognitive decline, hyponatremia, and T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity in the mesial temporal lobe on magnetic resonance imaging (MRI).[1] It is a relatively rare and complex disease, and little is known about the nature or course of this condition. Furthermore, LE-associated antibody analyses are not routinely performed. Early detection is thus difficult in some cases, but early diagnosis and immunotherapy can improve the clinical outcome.[2] We recruited 10 cases who presented with seizures onset from March 2013 to June 2015; confirmed LGI1-LE at our epilepsy center; and analyzed clinical features, treatment response, and prognosis to investigate the diagnosis and treatment of this condition at its onset.
2. Methods and materials
2.1. Clinical data
This study was reviewed and approved by the Ethics Committee of Beijing Tian tan Hospital, Capital Medical University in the People's Republic of China in accordance with the Declaration of Helsinki. Informed consent was obtained from each subject enrolled in this study.
Diagnostic criteria for LGI1-LE[3] were as follows: at least 1 of amnesia, FBDS, or seizures with temporal lobe semiology; serum and/or cerebrospinal fluid (CSF) LGI1 antibody positivity; and with or without T2 or FLAIR hyperintensity in the mesial temporal lobe on MRI. Ten patients with an age range of 34–78 (58.2 ± 15.6) years and a male:female ratio of 3:2 were included. No patient showed a history of epilepsy, encephalitis, head trauma, or other central nervous diseases.
The Mini-Mental State Examination (MMSE) was administered to all patients. Systemic and neurological examinations, interictal and ictal video electroencephalogram (VEEG), cranial MRI, CSF (cell counting; protein, glucose, and chloride levels; oligoclonal bands; anti-Hu, Yo, Ri, and Ma; anti-NMDA-R, AMPA-R, GABAB-R, CASPR2, and LGI1) and serum (sodium and anti-NMDA-R, AMPA-R, GABAB-R, CASPR2, and LGI1) analyses, and tumor screenings were conducted for all patients. Tumor screenings consisted of chest computed tomography (CT) scan; abdominal, thyroid, and pelvic ultrasounds; and tumor marker test.
2.2. Treatment
In all cases, antiepileptic drugs (AEDs) were the initial treatment and were administered solely or in combination (range 1–3 AEDs) for 8–360 (114.2 ± 121.8) days. Nine patients were treated with intravenous immunoglobulin (IVIg; 0.4 g/kg daily for 5 days). The patient in Case 2 was discharged after IVIg treatment and discontinued further treatment due to a diagnosis of lung cancer; the other 8 patients continued treatment with prednisone (60 mg daily for 1 month, then tapering by 5 mg/week). One patient (Case 3) was treated with prednisone alone (60 mg daily for 1 month, then tapering by 5 mg/week).
3. Results
We studied 10 patients with LGI1-LE. The initial presenting symptom was epileptic seizures. The time from onset to diagnosis was 0.3–12 (3.3 ± 4.1) months, with a follow-up period of 2–16 (9.4 ± 4.2) months. Table 1 lists the patients’ clinical details.
Table 1.
Clinical features and treatment of patients.
3.1. Seizures
Most patients (7/10) presented with FBDS; some (4/10) presented with complex partial seizures (CPS), the subjective symptoms of which suggested a mesial temporal origin. Four patients (Cases 1, 2, 6, and 10) presented with FBDS as the onset seizure, manifested as sudden, short, and predominantly tonic contractions of the upper limbs. The end of the moment was accompanied by a dystonic posture and ipsilateral facial grimacing, lasting 1–2 seconds. These seizures typically occurred hundreds of times per day. There were no prodromal symptoms and no loss of consciousness. Case 2 manifested with a fall due to simultaneous lower limb involvement. Four patients (Cases 4, 7, 8, and 9) manifested with generalized tonic-clonic seizures (GTCS) at onset. In this subgroup, 1 patient (Case 4) experienced CPS and FBDS, 1 patient (Case 7) experienced CPS, and 2 patients (Cases 8 and 9) experienced FBDS in the course of disease progression. The time from GTCS to FBDS was 0.3–6 months and from GTCS to CPS was 0.5–1 month. Case 4 presented with GTCS at onset and 4 times in total. Two weeks later, this patient experienced CPS with paroxysmal blank staring, which lasted 1–2 minutes, followed 2 months later by serious FBDS and an autonomic aura with palpitations that lasted about 5 seconds preceding FBDS. One patient (Case 5) presented with CPS at onset, manifested as disturbance of awareness, with arrested speech and motion automatisms that lasted for 20–50 seconds and occurred 10–20 times per day. Three months later, this patient experienced 1 GTCS. One patient (Case 3) presented with simple partial seizure (SPS) at onset, manifested as a sudden numbness of the left side of the body and face, lasting 2–5 seconds and occurring 10–20 times daily. Two months later, this patient experienced complex partial seizure (CPS) with paroxysmal confusion, forgetting her name or home address, which lasted for 30–60 seconds and occurred 6 times in total. Five months later, this patient experienced a GTCS during sleep and was admitted to our center for treatment.
3.2. Cognitive assessment
Four patients (Cases 3, 5, 7, and 8) complained of memory impairment. The time from seizure onset to memory impairment was from 0.5 to 6 months. MMSE results indicated mild cognitive deficits in 4 patients (Case 3: 22/30, Case 5: 21/30, Case 7: 23/30, and Case 8:22/30).
3.3. Systemic and neurological examinations
Systemic and neurological examinations were normal in 6 patients. Four patients had mild mental deficits.
3.4. VEEG
Interictal VEEG showed normal findings (Cases 1, 6, and 10), unilateral or bilateral frontotemporal slowing (Cases 2, 3, 5, and 8), or temporal sharp waves (Cases 4, 7, and 9). Ictal VEEG of FBDS (Cases 1, 2, 6, 8, 9, and 10) and SPS (Case 3) showed multiple events associated with movement and muscle artifacts, but no rhythmic changes or sharp waves. Ictal VEEG of Case 4 showed a diffuse voltage depression with palmodic aura preceding FBDS (Fig. 1). Ictal VEEG of Case 5 revealed a left frontal origin of CPS 6 months after onset and a left mesial temporal origin of CPS 12 months after onset. Ictal VEEG of Case 7 revealed a bilateral mesial temporal lobe origin of CPS.
Figure 1.
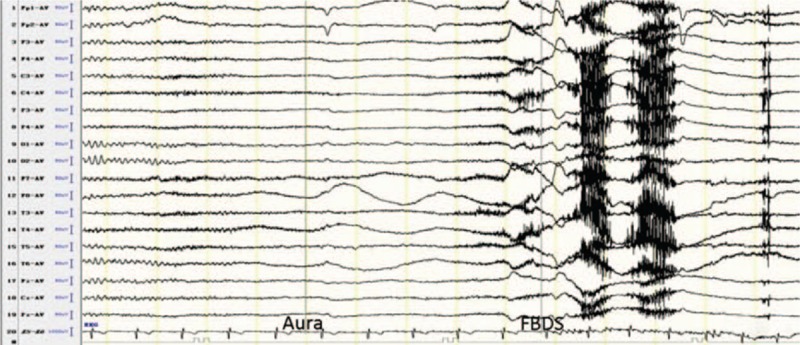
Ictal VEEG of Case 4 showed a diffuse voltage depression with a palmodic aura preceding FBDS.
3.5. MRI
Case 3 showed T2/FLAIR hyperintensity and evidence of edema in the right mesial temporal lobe 5 months after onset (Fig. 2). Case 5 showed left hippocampal atrophy 12 months after onset (Fig. 3). Case 7 showed T2/FLAIR hyperintensities in the bilateral mesial temporal lobes 1.2 months after onset. These 3 patients all had normal MRI findings at seizure onset. Case 10 showed T2/FLAIR hyperintensities in the basal ganglia and frontal cortex 0.5 months after onset (Figs. 4 and 5). MRI in the other 6 patients showed normal findings (Case 9), hydatoncus (Case 1), mild generalized atrophy (Case 2), and small vessel disease (Cases 4, 6, and 8), and no changes in limbic structures at 0.3–9 months after onset, respectively.
Figure 2.
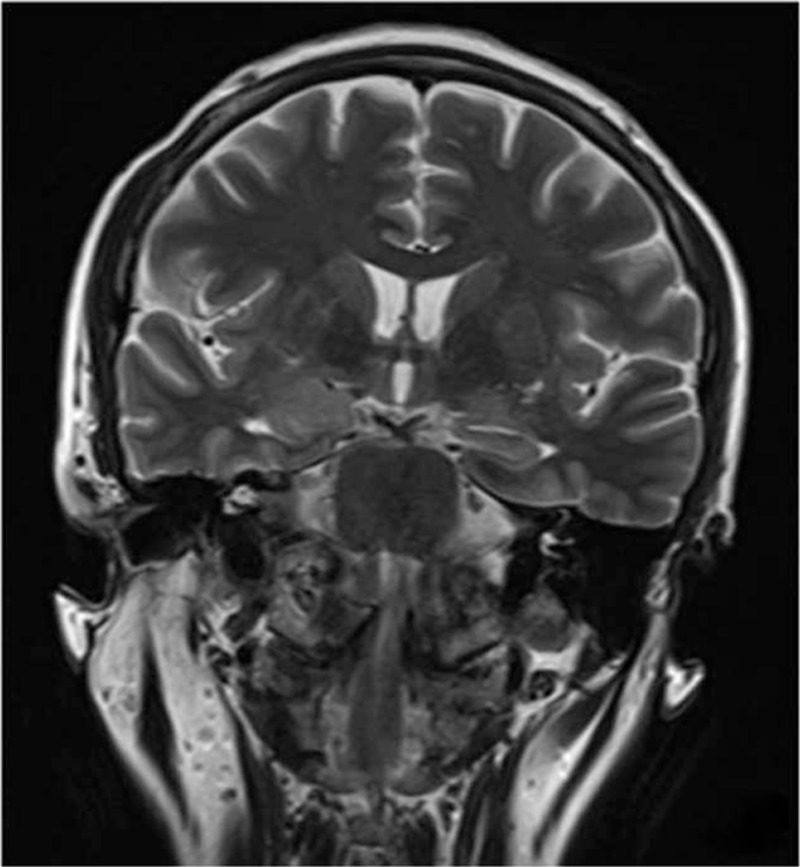
Case 3 showed T2 hyperintensity and evidence of edema in the right mesial temporal lobe 5 months after onset.
Figure 3.
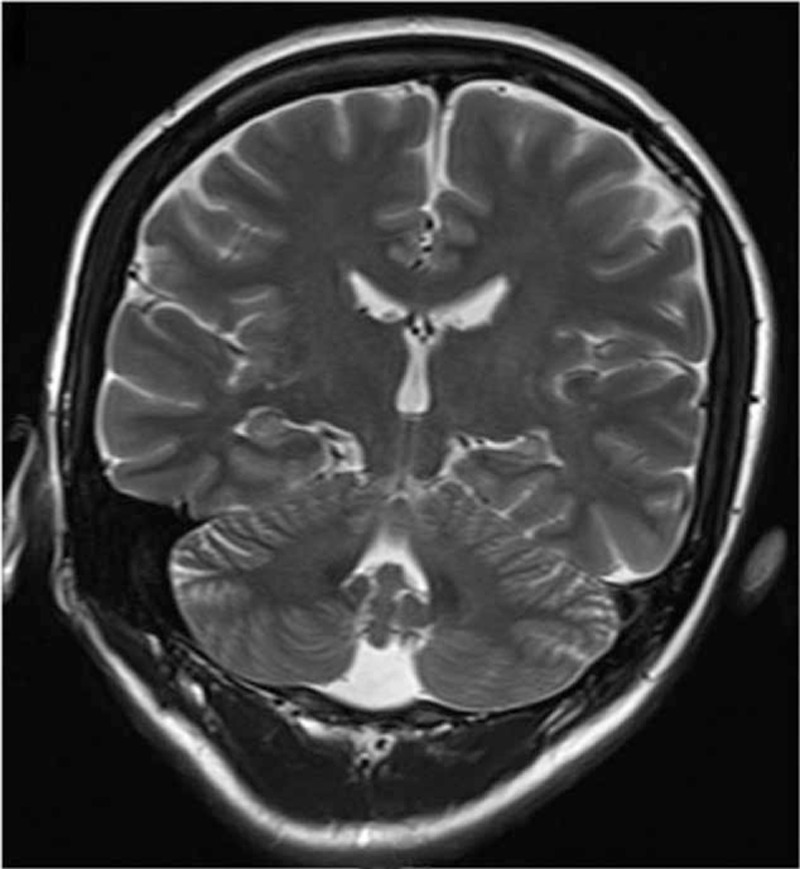
Case 5 showed left hippocampal atrophy 12 months after onset.
Figure 4.
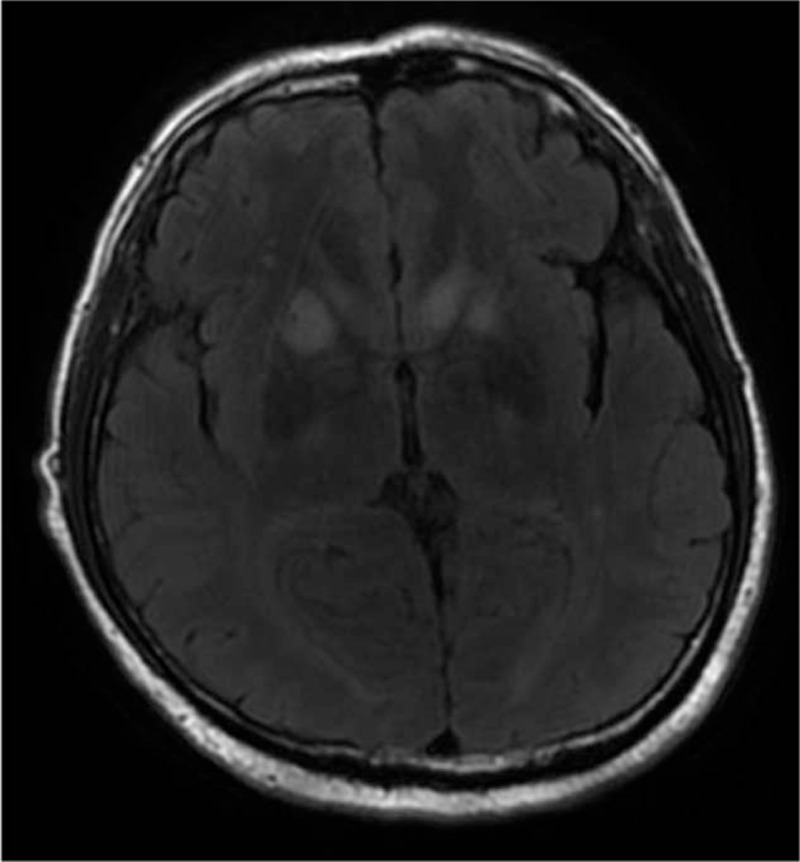
Case 10 showed FLAIR hyperintensities in the basal ganglia and frontal cortex after 0.5 month after onset.
Figure 5.
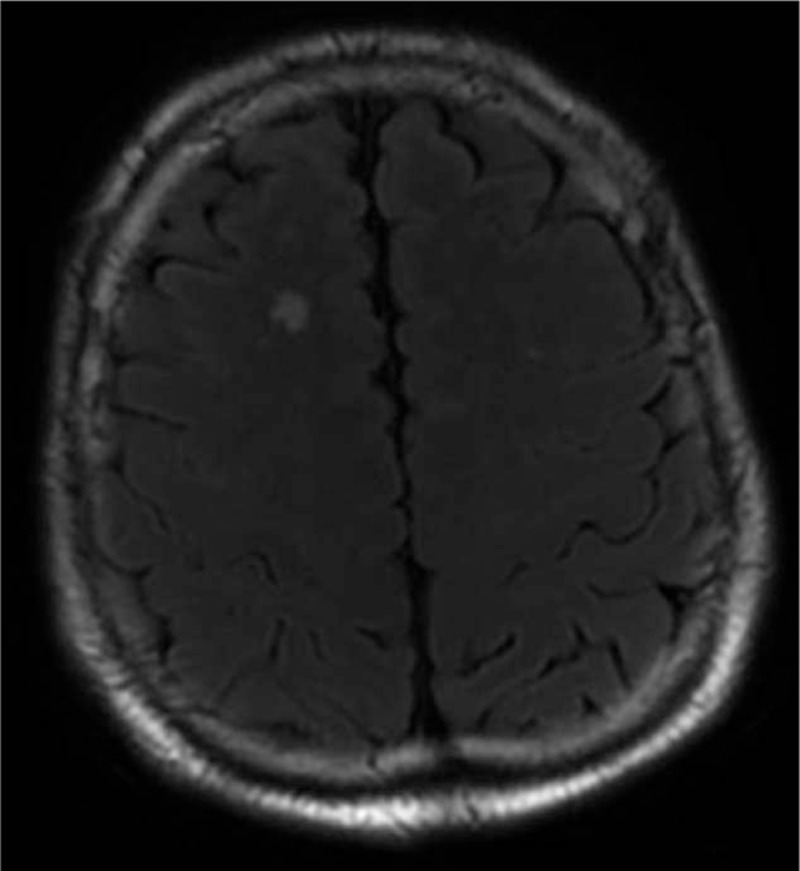
Case 10 showed FLAIR hyperintensities in the basal ganglia and frontal cortex 0.5 month after onset.
3.6. Positron emission tomography
One patient (Case 10) underwent brain positron emission tomography (PET) with 18F-fluorodeoxyglucose (18F-FDG), which revealed basal ganglia hypermetabolism and extensive cortical hypometabolism 0.5 month after onset (Fig. 6).
Figure 6.
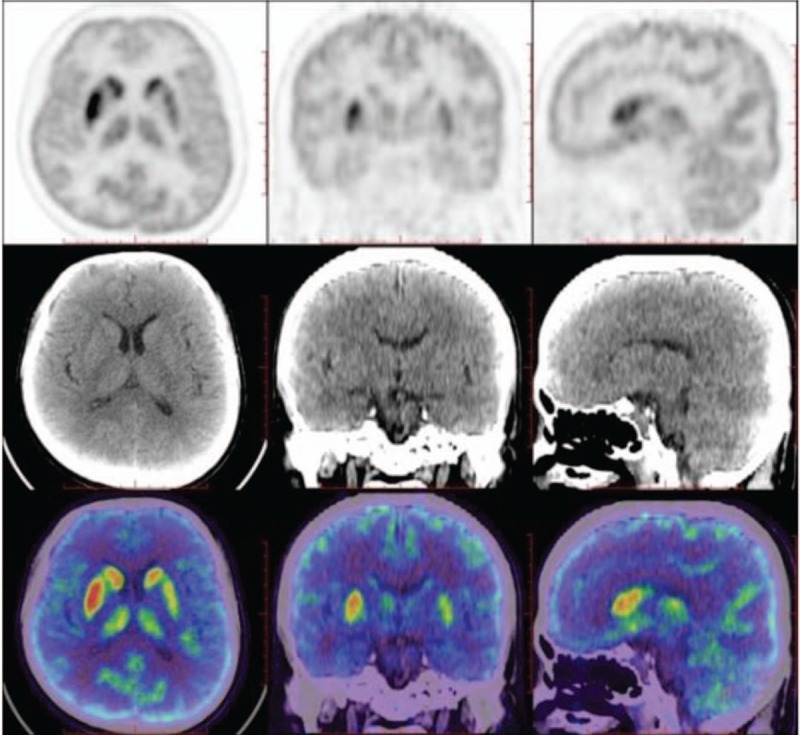
Case 10 showed 18F-FDG-PET/CT hypermetabolism in the basal ganglia and extensive cortical hypometabolism 0.5 month after onset.
3.7. CSF analysis
CSF analysis results were positive for LGI1 antibodies in 7 patients (Cases 2, 3, 4, 5, 7, 8, and 9). Cell counts, glucose, protein, and chloride levels, and oligoclonal band measurements were normal or negative.
3.8. Serum analysis
Serum analysis results were positive for LGI1 antibodies in all patients. Four patients (Cases 2, 4, 6, and 8) showed mild serum hyponatremia (130–134 mmol/L), and 1 patient (Case 7) showed modest serum hyponatremia (115 mmol/L) without clinical manifestations. All recovered normally with oral rehydration salts.
3.9. Tumor screening
Chest CT scan; abdominal, thyroid, and pelvic ultrasounds; and tumor marker test were performed for all patients. A high-density shadow on the right lung and multiple enlarged mediastinal lymph nodes were found in the chest CT scan of Case 2. Small cell lung cancer was confirmed through lymph node biopsy.
3.10. Seizure outcomes
In 7 patients (Cases 1, 2, 4, 6, 8, 9, and 10), FBDS did not respond to AEDs. In Case 4, GTCS and CPS ceased after treatment with a combination of 3 AEDs (levetiracetam, oxcarbazepine [OTX], and valproic acid), but FBDS emerged. The frequencies of SPS in Case 3 and CPS in Case 7 also did not decrease with AEDs. In Case 5, CPS frequency was reduced by 50% after treatment with a combination of 2 AEDs (OTX, topiramate [TPM]). Eight patients (Cases 1, 4, 5, 6, 7, 8, 9, and 10) were seizure free after treatment with IVIg and prednisone. One patient (Case 3) was seizure free after prednisone treatment alone. The period from initial immunotherapy to seizure remission was from 1 to 8 (3.4 ± 2.6) weeks. These 9 patients showed no relapse after withdrawal of immunotherapy, with a 6–16 (10.2 ± 3.2) months follow-up. One patient (Case 2) showed no response to IVIg; this patient was diagnosed with small cell lung cancer and refused further treatment. Seizure frequency did not decrease in the following 2 months, after which point contact was lost.
3.11. Cognition outcomes
No further cognitive deficits were found during follow-up. Six months after the initial immunotherapy, MMSE scores were as follows: Case 3, 27/30; Case 5, 22/30; Case 7, 30/30; and Case 8, 28/30.
4. Discussion
VGKC-LE is the most common nonparaneoplastic LE in adults,[4] and LGI1-LE is the most common subtype. It typically occurs in middle-aged males, with a mean onset age of 60 years. The onset age of our cases varied from 34 to 78 years, with a mean of 58.2 ± 15.6 years.
Seizure is the most common manifestation of LGI1-LE, in more than 80% of cases. FBDS was recently described as a characteristic and often presenting feature of LGI1-LE.[5] Our cases all presented with seizures at onset, but had varying individual characteristics. Seven (70%) patients presented with FBDS, 4 of whom (40%) manifested FBDS as an initial and unique syndrome. In this subgroup, the time from FBDS onset to immunotherapy varied from 0.5 to 1.5 months, and all patients experienced a therapeutic effect. Recognition of this specific seizure type thus offers an opportunity for early diagnosis, and immunotherapy may prevent the development of other manifestations of LE including temporal origin seizures and cognitive impairment. The other 3 (30%) patients presented with GTCS at onset, and FBDS emerged during the disease course, indicating that FBDS might not be the initial manifestation and might coexist with other seizure types. Three (30%) patients manifested with SPS, CPS, or GTCS and without FBDS. Four (40%) patients presented with CPS, the subjective symptoms of which suggested a mesial temporal origin. Navarro et al[6] reported that LGI1-LE could be linked to involvement of the motor cortex and hippocampus. Similar to their study of seizure type, our results showed that FBDS and mesial temporal origin CPS were the 2 most typical seizure subtypes in patients with LGI1-LE, although we did not observe the characteristic EEG frontal focal slow wave preceding FBDS. The possible relationship between these seizures and LGI1-LE should be investigated in more detail. Our findings also suggested that LGI1-LE involvement might be considered in patients presenting with GTCS and SPS at onset. Case 3 presented with frequent brief episodes of left hemianesthesia early in the disease course, with normal physical examination, neuroimaging, and routine test results. These findings confused the diagnosis and delayed immunotherapy to some extent. Some patients with poorly AEDs-responsive epilepsy may also have an immune-mediated etiology.
Cognitive impairment is the main neurological disorder in LGI1-LE. In our study, 4 (40%) patients had mild memory deficits 0.5–6 months after seizure onset, and 6 (60%) showed no cognitive impairment in the acute phase, indicating that memory deficits are the most common manifestation of cognitive impairment. It maybe insidious at onset and associated with the course of LGI1-LE. Malter et al[7] reported that 89% of patients manifested memory deficits at an 8-month follow-up, and about 65% of patients still showed deficits in at least 1 memory domain at a 26-month follow-up. Majoie et al[8] also proposed that memory and verbal fluency were most affected during the course of LGI1-LE. Three patients (Cases 3, 5, and 7) with memory deficits also showed abnormal MRI signals in the mesial temporal lobe. This indicated that abnormal structures in the mesial temporal lobe might be associated with memory dysfunction. The absence of cognitive deficits at onset may increase the difficulty of early diagnosis for LGI1-LE; considering the clinical features and test results may therefore be helpful. VGKC/LGI1-associated antibodies should be measured in adults with sudden unexplained seizures, even without cognitive deficits, which might help early diagnosis and treatment.[9,10]
Hyponatremia due to the syndrome of inappropriate antidiuretic hormone secretion is another characteristic feature of LGI1-LE,[11,12] with a reported occurrence of 60%-88%.[5,13] Coexpression of LGI1 antibodies in the hypothalamus and kidney may be the mechanism. Generally, hyponatremia is mild to modest and not difficult to rectify.[14] Five (50%) patients in our study showed mild to modest hyponatremia and recovered with sodium supplements.
CSF positivity for the LGI1 antibody is the specific indicator of LGI1-LE. Consistent with other studies, our patients’ CSF samples showed no pleocytosis, protein elevation, or positive oligoclonal bands.[8,15] Unremarkable CSF examination results should thus not dissuade consideration of a diagnosis of LGI1-LE. Three (30%) patients in our study manifested LGI1 antibody positivity only in the serum, not the CSF. Vincent et al[16] reported that VGKC antibody values in the CSF varied between 1% and 10% of serum values. Furthermore, LGI1 antibodies have been observed in the serum of patients without the typical LE presentation. Evaluating LGI1 antibodies in CSF instead of serum may increase diagnostic specificity.[13] In the meantime, we recommend that LGI1 antibody tests should be performed on both serum and CSF if available.
Interictal and ictal VEEG were recorded for all patients in our study. Interictal VEEG showed normal patterns, focal slowing, or sharp waves without unique changes. The ictal localization of FBDS remained uncertain in our patients. FBDS is primarily an epileptic phenomenon; the seizure semiologies are suggestive of frontal lobe involvement but differ from common frontal or temporal lobe semiologies, and basal ganglia imaging changes are observed. To date, the origin of FBDS remains unclear; some authors suspect a subcortical origin,[17] others a cortical one,[5,12] and still others contributions from both sites.[18] Navarro et al[6] recently reported a characteristic EEG frontal focal slow wave preceding FBDS. Due to the limitations of scalp VEEG, more investigations of larger samples should be carried out to determine the origins of FBDS. A video recorder, however, is more helpful in the diagnosis of FBDS and differential diagnosis with movement disorders.[5,17] The ictal VEEG of 2 patients (Cases 5 and 7) revealed mesial temporal lobe CPS origins. Interestingly, we observed a change in epileptic origin in Case 5, from the frontal to temporal lobe, although the clinical manifestations of CPS were similar. The VEEG changes indicated temporal lobe involvement and possible immune etiology before LGI1-LE confirmation. We propose that it is necessary to follow-up VEEG results in the setting of uncontrolled seizures or uncertain etiology.
Abnormal MRI signals in the mesial temporal lobe are important evidence in the diagnosis of LGI1-LE. In 7 of our cases, there were no limbic structure abnormalities on MRI. We therefore agree with the argument by Irani et al[5] that the presence of abnormal mesial temporal lobe changes on MRI is not a prerequisite for a diagnosis of LE. We observed dynamic changes in the mesial temporal lobe signal in the MRI from 3 cases with memory deficits. This indicates that abnormal signals in the temporal lobe might be present in the normal course of the disease, rather than in the acute phase. The abnormal hippocampal structure relates to memory deficits, so follow-up MRI scan and careful evaluation of the temporal area are recommended. Furthermore, 1 patient (Case 10) showed T2/FLAIR hyperintensities in the basal ganglia and frontal cortex. This patient presented with FBDS, and imaging evidence might indicate involvement of the basal ganglia and frontal cortex in FBDS.
LGI1-LE may show different abnormalities on 18F-FDG PET/CT imaging. One patient (Case 10) underwent brain PET/CT that showed basal ganglia hypermetabolism and extensive cortical hypometabolism. This patient had T2/FLAIR hyperintensities signal in the basal ganglia and frontal cortex on MRI. Irani et al[5] reported abnormal glucose metabolism in the temporal region or basal ganglia, even when the MRI was normal. Brain MRI and 18F-FDG PET/CT can complement each other. Given the limited number of brain PET/CT studies in LGI-LE, more investigations are needed.
LGI1-LE is mostly nonparaneoplastic.[16,19,20] Case 2 was diagnosed with small cell lung cancer through mediastinal lymph node biopsies. He presented with FBDS and had no treatment response to immunotherapy. In consideration of this individual case, we are unsure of the relationship between LGI1 antibody positivity and small cell lung cancer. In fact, some patients with VGKC-LE have been reported to have malignancies including thymomas or lung cancer; the paraneoplastic proportion ranges from 9% to 20%.[21,22] Searching for possible tumors in patients with LGI1-LE is suggested, especially in elderly patients or those who do not respond to immunotherapy. There is no consensus regarding the extent and degree of sophistication of the diagnostic procedures for these tumor searches. The most sensitive procedure for finding even small tumors is whole-body FDG-PET, ideally in conjunction with CT scans. If PET examination is not available, conventional tumor investigation is necessary, including chest CT scan and abdominal and pelvic ultrasounds. For paraneoplastic LE, the best result is achieved by successful tumor therapy. This can lead to improvement, or at least cessation, of neurological deficit progression.[23] In this study, however, the patient with lung cancer refused further therapy, and we could not follow-up his prognosis.
In this study, after IVIg and low dose oral steroids or oral steroids alone, the 9 nonparaneoplastic patients became seizure-free. The period from treatment to seizure remission was 1–8 weeks. In the 4 cases with memory deficits, 3 showed cognitive improvement after immunotherapy. Case 5, however, did not significantly improve even with immunotherapy; the condition may already have progressed too far.
FBDS and other seizures in patients with LGI1-LE usually respond poorly to AEDs. In our study, only 1 patient showed a 50% decreased seizure frequency with a combination of 3 AEDs. With immunotherapy, most patients became seizure free and showed improved memory performance, consistent with other studies.[9,20] Delays in diagnosis and starting immunotherapy often mean that patients are not restored to their baseline and may have ongoing functional limitations due to memory deficits.[8] There is no fixed therapeutic schedule for immunotherapy; first-line immunotherapy includes IVIg, steroids, and plasma exchange.[10] We administered IVIg followed by low-dose prednisone to reduce relapses. There were no relapses after immunotherapy withdrawal over 6–16 months follow-up. We argue for immediate initiation of immunotherapy once LGI1-LE is diagnosed.
LGI1-LE is a relatively rare disease, and our study had a limited number of cases. It is necessary to investigate and follow-up more cases to fully understand this complex disorder. Recognition of the characteristic FBDS is an important step in making a definite diagnosis of LGI1-LE, but some patients present with different seizures and without distinct cognitive deficits at onset. Accordingly, LGI1-LE should be considered in adults with sudden mesial temporal origin, unexplained, and drug-resistant seizures, even without observed cognitive deficits. MRI scan and VEEG monitoring are helpful for dynamic observation of the manifestations. Although most cases of LGI1-LE are nonparaneoplastic, tumor screening should be considered, especially in elderly patients and those who do not respond to immunotherapy.
Footnotes
Abbreviations: AED = antiepileptic drug, CPS = complex partial seizure, CSF = cerebrospinal fluid, CT = computed tomography, FBDS = faciobrachial dystonic seizure, FDG = fluorodeoxyglucose, FLAIR = fluid-attenuated inversion recovery, GTCS = generalized tonic-clonic seizure, LE = limbic encephalitis, LGI1 = leucine-rich glioma inactivated-1 protein, MMSE = Mini-Mental State Examination, MRI = magnetic resonance imaging, OTX = oxcarbazepine, PET = positron emission tomography, PS = partial seizure, SPS = simple partial seizure, TPM = topiramate, VEEG = video electroencephalogram, VGKC = voltage-gated potassium channel.
The authors have no conflicts of interest to declare.
References
- 1.Krastinova E, Vigneron M, Le Bras P, et al. Treatment of limbic encephalitis with anti-glioma-inactivated 1 (LGI1) antibodies. J Clin Neurosci 2012; 19:1580–1582. [DOI] [PubMed] [Google Scholar]
- 2.Anderson NE, Barber PA. Limbic encephalitis—a review. J Clin Neurosci 2008; 15:961–971. [DOI] [PubMed] [Google Scholar]
- 3.Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav 2008; 10:529–538. [DOI] [PubMed] [Google Scholar]
- 4.Haberlandt E, Bast T, Ebner A, et al. Limbic encephalitis in children and adolescents. Arch Dis Child 2011; 96:186–191. [DOI] [PubMed] [Google Scholar]
- 5.Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede LGI1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900. [DOI] [PubMed] [Google Scholar]
- 6.Navarro V, Kas A, Apartis E, et al. Motorcortex and hippocampus are the two main cortical targets inLGI1-antibody encephalitis. Brain 2016; 139:1079–1093. [DOI] [PubMed] [Google Scholar]
- 7.Malter MP, Frisch C, Schoene-Bake JC, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol 2014; 261:1695–1705. [DOI] [PubMed] [Google Scholar]
- 8.Majoie HJ, de Baets M, Renier W, et al. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141. [DOI] [PubMed] [Google Scholar]
- 9.Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013; 136:3151–3162. [DOI] [PubMed] [Google Scholar]
- 10.Szots M, Marton A, Kover F, et al. Natural course of LGI1 encephalitis: 3-5 years of follow up. Neurol Sci 2014; 343:198–202. [DOI] [PubMed] [Google Scholar]
- 11.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010; 9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrade DM, Tai P, Dalmau J, et al. Tonic seizures: a diagnostic clue of anti-LGI1 encephalitis? Neurology 2011; 76:1355–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lancaster E, Dalmau J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012; 8:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincent A. Developments in autoimmune channelopathies. Autoimmunity Rev 2013; 12:678–681. [DOI] [PubMed] [Google Scholar]
- 15.Jarius S, Hoffmann L, Clover L, et al. CSF findings in patients with voltage gated potassium channel antibody associated limbic encephalitis. J Neurol Sci 2008; 15:74–77. [DOI] [PubMed] [Google Scholar]
- 16.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712. [DOI] [PubMed] [Google Scholar]
- 17.Striano P. Faciobrachial dystonic attacks: seizures or movement disorder? Ann Neurol 2011; 70:179–180. [DOI] [PubMed] [Google Scholar]
- 18.Boesebeck F, Schwarz O, Dohmen B, et al. Faciobrachial dystonic seizures arise from cortico-subcortical abnormal brain areas. J Neurol 2013; 260:1684–1686. [DOI] [PubMed] [Google Scholar]
- 19.Thieben MJ, Lennon VA, Boeve BF, et al. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 2004; 62:1177–1182. [DOI] [PubMed] [Google Scholar]
- 20.Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78. [DOI] [PubMed] [Google Scholar]
- 21.Pozo-Rosich P, Clover L, Saiz A, et al. Voltage-gated potassium channel antibodies in limbic encephalitis. Ann Neurol 2003; 54:530–533. [DOI] [PubMed] [Google Scholar]
- 22.Zuliani L, Saiz A, Tavolato B, et al. Paraneoplastic limbic encephalitis associated with potassium channel antibodies: value of anti-glial nuclear antibodies in identifying the tumour. J Neurol Neurosurg Psychiatry 2007; 78:204–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keime-Guibert F, Graus F, Broët P, et al. Clinical outcome of patients with anti-Hu-associated encephalomyelitis after treatment of the tumor. Neurology 1999; 53:1719–1723. [DOI] [PubMed] [Google Scholar]