

Abstract
Rac1 is a small GTPase and plays key roles in multiple cellular processes including the production of reactive oxygen species (ROS). However, whether Rac1 activation during myocardial ischaemia and reperfusion (I/R) contributes to arrhythmogenesis is not fully understood. We aimed to study the effects of Rac1 inhibition on store overload‐induced Ca2+ release (SOICR) and ventricular arrhythmia during myocardial I/R. Adult Rac1f/f and cardiac‐specific Rac1 knockdown (Rac1ckd) mice were subjected to myocardial I/R and their electrocardiograms (ECGs) were monitored for ventricular arrhythmia. Myocardial Rac1 activity was increased and ventricular arrhythmia was induced during I/R in Rac1f/f mice. Remarkably, I/R‐induced ventricular arrhythmia was significantly decreased in Rac1ckd compared to Rac1f/f mice. Furthermore, treatment with Rac1 inhibitor NSC23766 decreased I/R‐induced ventricular arrhythmia. Ca2+ imaging analysis showed that in response to a 6 mM external Ca2+ concentration challenge, SOICR was induced with characteristic spontaneous intracellular Ca2+ waves in Rac1f/f cardiomyocytes. Notably, SOICR was diminished by pharmacological and genetic inhibition of Rac1 in adult cardiomyocytes. Moreover, I/R‐induced ROS production and ryanodine receptor 2 (RyR2) oxidation were significantly inhibited in the myocardium of Rac1ckd mice. We conclude that Rac1 activation induces ventricular arrhythmia during myocardial I/R. Inhibition of Rac1 suppresses SOICR and protects against ventricular arrhythmia. Blockade of Rac1 activation may represent a new paradigm for the treatment of cardiac arrhythmia in ischaemic heart disease.
Keywords: Rac1, arrhythmia, store overload‐induced calcium release, ischaemia and reperfusion, reactive oxygen species
Introduction
Cardiac arrhythmia is a leading cause of death after myocardial infarction (MI) 1. Intracellular Ca2+ homoeostasis is critical to the heart in the maintenance of normal cardiac rhythms and contractility 2, 3, 4. Prolonged myocardial ischaemia during MI increases intracellular Ca2+, which is further exacerbated with reperfusion, leading to cardiomyocyte Ca2+ overload and cardiac arrhythmias 5, 6. Despite significant knowledge gained on cardiac Ca2+ cycling mechanisms, factors regulating cardiomyocyte Ca2+ and their roles in cardiac arrhythmia are still not fully understood.
Rac1 is a small GTPase protein and plays a key role in multiple cellular processes including cell migration, proliferation, survival and trafficking 7. Rac1 is also an essential regulatory subunit of NADPH oxidase for the generation of reactive oxygen species (ROS) 8, 9. In the heart, activation of Rac1 induces cardiac hypertrophy 10, 11, and genetic overexpression of Rac1 results in hypertension 12 and atrial fibrillation 13, 14. Rac1 activation also contributes to myocardial I/R injury in the diabetic hearts 15. However, the role of Rac1 in arrhythmogenesis is not fully understood.
Ryanodine receptor 2 (RyR2), an intracellular Ca2+ release channel in the sarcoplasmic reticulum (SR), is a key component in cardiac excitation‐contraction coupling. Under normal physiological conditions, a small Ca2+ influx through the L‐type Ca2+ channel upon membrane depolarization activates the RyR2 channel, resulting in a large Ca2+ release from the SR and subsequent muscle contraction 16. In the ischemic heart, phosphorylation and redox modifications of RyR2 increase RyR2 open probability causing SR Ca2+ leak 17, 18, 19. In addition, SR overload during myocardial I/R induces spontaneous Ca2+ release and leak mediated by RyR2, a term previously defined as store overload‐induced Ca2+ release (SOICR), leading to spontaneous Ca2+ waves in cardiomyocytes, which induce cardiac arrhythmias 20, 21, 22. Our recent studies show that activation of Rac1 increases intracellular Ca2+ in cardiomyocytes 23. However, how Rac1 disrupts Ca2+ homeostasis and promotes ventricular arrhythmia in the ischaemic heart remains elusive.
In this study, we hypothesized that inhibition of Rac1 protects against ventricular arrhythmia during myocardial I/R through decreases in ROS, RyR2 oxidation and SOICR. To test this hypothesis, cardiac‐specific Rac1 knockdown (Rac1ckd) mice were subjected to myocardial I/R. Rac1 activation, ROS production and oxidation of RyR2 were determined, SOICR in cardiomyocytes were analysed. Our data show for the first time that inhibition of Rac1 suppresses SOICR and protects against ventricular arrhythmia during myocardial I/R. Thus, Rac1 may represent a potential therapeutic target for the treatment of cardiac arrhythmia in ischaemic heart disease.
Materials and methods
Animals
Rac1f/f mice with C57BL/6 background were obtained from Jackson Laboratory (Bar Habor, Maine, USA). In these mice, exon 1 of Rac1 gene is flanked by loxP sites. The generation of cardiac‐specific Rac1 knockdown mice (Rac1ckd, that is, Cre‐Tg;Rac1f/f) was carried out by breeding Rac1f/f with cardiac‐specific α‐myosin heavy chain (α‐MHC) promoter controlled Cre recombinase overexpression transgenic mice (α‐MHC‐Cre), resulting in deletion of Rac1 gene in adult cardiomyocytes as previously described 23. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (8th Edition, 2011) and all experimental protocols were approved by Animal Use Subcommittee at the University of Western Ontario.
Induction of myocardial I/R
Myocardial I/R were performed as previously described 24, 25. Briefly, Rac1f/f and Rac1ckd male mice (3–4 months old) were anaesthetized with ketamine (50 mg/kg, i.p.) and xylazine (12.5 mg/kg, i.p.). The adequacy of anaesthesia was monitored by the absence of withdrawal reflex to tail pinch. Subsequently mice were intubated and artificially ventilated with a respirator (SAK‐830, CWE, Ardmore, PA, USA). The left coronary artery was occluded by positioning a 8‐0 suture around it with a PE‐50 tubing. After 45 min., the PE tubing was removed to allow reperfusion for either 60 min. (Rac1 activation assay, ECG recording, infarct size and ROS generation measurement) or 15 min. (for free thiol content measurement). To study the effect of Rac1 inhibition, some Rac1f/f and Rac1ckd mice were treated with saline or a Rac1 inhibitor NSC23766 (2.5 mg/kg, IP) for 30 min. before I/R 26, 27.
ECG monitoring
The ECG was monitored 5 min. before and throughout the entire I/R by limb lead I, with needle electrodes inserted subcutaneously in mice and recorded with PowerLab Chart 7.0 (AD Instrument, Colorado, Springs, CO, USA) as previously described 25. Premature ventricular contractions (PVCs) were defined as singlet or doublet premature QRS complexes in relation to the P wave. Ventricular tachycardia (VT) was defined as a run of three or more premature QRS complexes. The number of PVCs and incidence of VT were quantified. Additionally, PR interval, RR interval, QRS and QT durations were recorded.
Infarct size measurement
After I/R, infarct size was determined as described previously 24, 28. In brief, after 45 min. ischaemia and 1 h reperfusion the left coronary artery was religated using the same suture. One per cent Evans blue dye (0.7 ml) was injected into the heart through the cannulated aorta to determine the non‐ischaemic (perfused, blue) and ischaemic (not perfused, not blue) areas of the heart. The heart was serially sectioned into four pieces and incubated with 1% triphenyltetrazolium chloride (TTC) for 10 min at room temperature. The viable, ischaemic tissue was stained into a red colour while infarct area was unstained as a white colour. Each heart section was pictured and weighed. The weight of non‐risk area (blue), infarct area (white) and area at risk (not blue) was calculated. Infarct size was measured as percentage of the weight of infarct area to the area at risk.
Rac1 activity assay
Rac1 activity was measured by GTP‐bound Rac1 GTPase through specific protein interaction with the Pak1 protein‐binding domain (Active Rac1 Pull‐Down and Detection Kit; Thermo Scientific Pierce, Rockford, IL, USA) according to the manufacturer's instructions 29. Total homogenates of the heart tissue were centrifuged at 10,000 g for 10 min., and the supernatant was incubated with agarose beads coated with a GST‐Pak1 protein‐binding domain for 2 hrs at 4°C. The pulled down beads were resuspended in SDS sample buffer and boiled for 5 min. to elute Rac1‐GTP. Rac1‐GTP and total Rac1 protein levels were detected by western blot analysis.
Western blot analysis
Protein lysates were subjected to separation on a 10% SDS‐PAGE gel, followed by electrotransfer to nitrocellulose membranes. Blots were probed with specific antibodies against Rac1 (Active Rac1 Pull‐Down and Detection Kit), RyR2 (MA3‐916, Affinity Bioreagents, Golden CO, USA), α‐actinin (A7811, Sigma, St. Louis, MO, USA) respectively. Signals were detected by the enhanced chemiluminescence detection method and quantified by densitometry using FluorChem 8000 software (Alpha Innotech, San Leandro, CA, USA).
Isolation of adult cardiomyocytes
Adult cardiomyocytes were enzymatically isolated from the heart as described before 23. Briefly, mice were heparinized (5000 U/kg, i.p.) and killed by cervical dislocation under ketamine (50 mg/kg, i.p.) and xylazine (12.5 mg/kg, i.p.) anaesthesia. Hearts from Rac1f/f and Rac1ckd mice were isolated and perfused with digestion buffer containing 50 μg/ml of liberase TH (Roche) through a Langendorff system. After enzymatic digestion, healthy and rod‐shaped cardiomyocytes were incubated with buffer containing increasing concentrations of Ca2+ (12.5 μM–1 mM).
Single cell Ca2+ imaging of SOICR
Ventricular cardiomyocytes isolated from Rac1f/f and Rac1ckd mice were loaded with 1 μM Fura‐2AM (Invitrogen, Waltham, MA, USA) for 30 min. at room temperature. To inhibit Rac1 activity, Rac1f/f myocytes were perfused with a Rac1 inhibitor, NSC23766 (10 μM) 26, 27. SOICR was measured as previously described 21, 22. Briefly, cardiomyocytes were incubated in Krebs‐Ringer‐Hepes (KRH) buffer containing 1 mM CaCl2, 125 mM NaCl, 5 mM KCl, 6 mM glucose, 1.2 mM MgCl2 and 25 mM Hepes (pH7.4) and stimulated at 0.5 Hz for 20 sec. CaCl2 was added to the bath to final extracellular Ca2+ concentrations (1, 3 and 6 mM) followed by caffeine (10 mM) treatment to assess SR Ca2+ content.
ROS production and RyR2 free thiol content
Changes in superoxide production from myocardium after I/R were detected with a fluorescent indicator dihydroethidium (DHE; Invitrogen) 30. In brief, fresh transverse croysections (8 μm) of the heart were placed on glass slides and incubated with DHE (2 μmol/l) for 30 min. at 37°C followed by washing with PBS for three times. Images were taken at a fixed exposure time under a fluorescence microscope (Observer D1, Zeiss, Germany) using an AxioCam high resolution camera. The fluorescence signal was measured with the AxioVision 4 program. RyR2 free thiols were determined with fluorescent probe for sulfhydryl (SH) groups of cysteines by monobromobimane (mBB) as described 18. Briefly, total homogenates of the risk area of the heart were prepared and heavy SR vesicles were isolated under non‐reducing conditions 18. Equal amount of protein samples were incubated with 500 μM mBB (Invitrogen) for 30 min. in the dark at room temperature. Samples were electrophoresed in 5% SDS‐PAGE gels and transilluminated with UV light. Monobromobimane fluorescence signal was normalized to total RyR2 measured by Coomassie blue staining of the gels run in parallel. The RyR2 band was confirmed by Western blotting using an anti‐RyR2 antibody.
Reagents
Ketamine and xylazine were purchased from Bioniche Animal Health Canada Inc. (Belleville, ON, USA) and Bachem AG, Switzerland respectively. NSC23766 was from Calbiochem (San Diego, CA, USA). All other reagents if unspecified were obtained from Sigma, St. Louis, MO, USA.
Statistical analysis
Data are presented as mean ± SEM. Results were analysed using one or two‐way anova followed by Newman–Keuls or Bonferroni post‐hoc test for multigroup comparisons. P < 0.05 was considered statistically significant.
Results
Rac1 activation during myocardial I/R
Rac1ckd mice were generated through breeding of alpha‐myosin heavy chain (MHC)‐Cre mice with Rac1f/f mice and baseline cardiac function was not significantly different between adult Rac1ckd and Rac1f/f mice as we previously reported 23, 29. To assess myocardial Rac1 activity, Rac1ckd and Rac1f/f mice were subjected to 45 min. of myocardial ischaemia followed by 1 hr of reperfusion (I/R). Hearts were isolated, and Rac1‐GTP, total Rac1 and α‐actinin protein levels were determined using Western blot analysis. Myocardial Rac1 activity was induced after I/R in Rac1f/f mice. The response was diminished in Rac1ckd mice (Fig. 1A and B). Myocardial total Rac1 to α‐actinin protein ratios were decreased by 56% in Rac1ckd compared to Rac1f/f mice (Fig. 1C). In isolated cardiomyocytes, total Rac1 protein levels were reduced by 53% in Rac1ckd compared to Rac1f/f cells (Fig. 1D and E). These results show that a significant down‐regulation of total Rac1 protein and I/R‐induced Rac1 activity in Rac1ckd mice.
Figure 1.
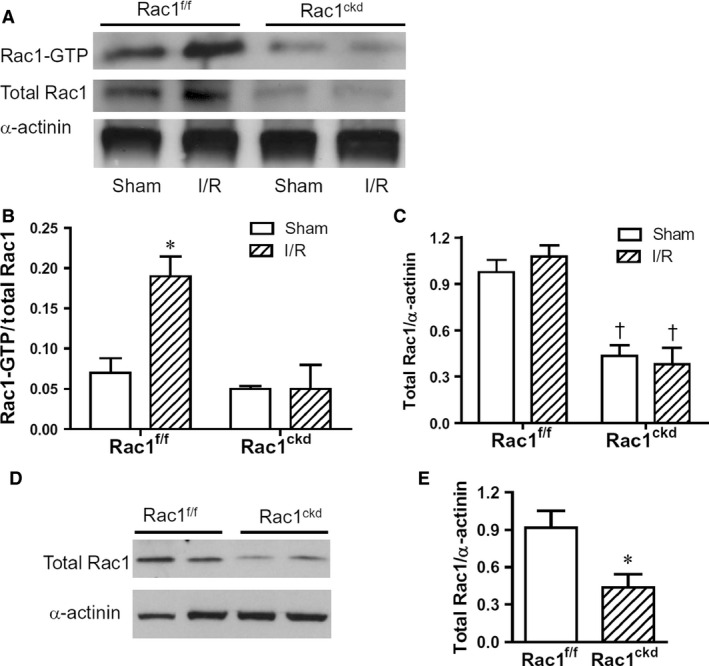
Myocardial Rac1 activity after I/R. (A) Representative Western blots of Rac1‐GTP with α‐actinin as loading control in Rac1f/f and Rac1ckd hearts after myocardial I/R. (B) Quantification of Rac1 activity using Rac1‐GTP to total Rac1 protein ratios. (C) Quantification of total Rac1 to α‐actinin protein ratios in the heart. (D) Representative Western blots of total Rac1 and α‐actinin proteins in isolated cardiomyocytes from adult Rac1f/f and Rac1ckd mice. (E) Quantification of total Rac1 to α‐actinin protein ratios in the isolated cardiomyocytes. Data are mean ± SEM from three to four mice per group. * P < 0.05 versus Rac1f/f sham in B or Rac1f/f in E, †P < 0.05 versus respective Rac1f/f.
Knockdown of Rac1 inhibits ventricular arrhythmia during myocardial I/R
To study the role of Rac1 in ventricular arrhythmia during I/R, Rac1f/f and Rac1ckd mice were subjected to 45 min. of myocardial ischaemia followed by 1 hr of reperfusion, and lead I ECG was monitored. Heart rate, PR interval, QRS duration and QTc interval were similar at baseline and during I/R between Rac1f/f, Rac1f/f treated with NSC23766 and Rac1ckd mice (Table 1). Notably, total number of ventricular premature beats (PVC), the incidence of VT and duration of VT were all significantly decreased in Rac1f/f treated with NSC23766 and Rac1ckd compared with Rac1f/f mice (Fig. 2A and B, Table 2). These data show knockdown of Rac1 inhibits ventricular arrhythmias during myocardial I/R. However, infarct size determined at 1 hr after reperfusion was not significantly different among three groups (Fig. 2C and D), suggesting a similar level of ischaemic injury among the groups.
Table 1.
Changes of heart rate, PR interval, QRS duration and QT interval following myocardial ischaemia and reperfusion
| Rac1f/f | Rac1f/f+NSC23766 | Rac1ckd | P value | |
|---|---|---|---|---|
| Mouse numbers | 10 | 7 | 13 | |
| Heart rate, bpm | ||||
| Basal | 346 ± 20 | 332 ± 14 | 343 ± 24 | 0.917 |
| Ischaemia | 345 ± 20 | 352 ± 11 | 343 ± 18 | 0.946 |
| Reperfusion | 340 ± 18 | 359 ± 16 | 313 ± 15 | 0.173 |
| PR interval, ms | ||||
| Basal | 40.9 ± 0.8 | 41.4 ± 1.9 | 44.4 ± 1.0 | 0.798 |
| Ischaemia | 43.1 ± 3.6 | 45.1 ± 3.2 | 43.8 ± 4.2 | 0.239 |
| Reperfusion | 42.3 ± 1.5 | 51.2 ± 5.7 | 44.9 ± 1.7 | 0.958 |
| QRS duration, ms | ||||
| Basal | 21.3 ± 1.3 | 18.6 ± 2.1 | 21.6 ± 1.6 | 0.661 |
| Ischaemia | 22.6 ± 0.9 | 19.8 ± 1.4 | 21.3 ± 1.1 | 0.656 |
| Reperfusion | 21.4 ± 0.9 | 20.7 ± 1.1 | 22.1 ± 2.4 | 0.958 |
| QTc interval, ms | ||||
| Basal | 72.0 ± 1.9 | 64.9 ± 4.4 | 63.7 ± 3.3 | 0.264 |
| Ischaemia | 75.6 ± 1.6 | 68.5 ± 4.3 | 69.2 ± 3.3 | 0.250 |
| Reperfusion | 76.1 ± 2.0 | 74.2 ± 2.3 | 71.0 ± 2.2 | 0.230 |
Data are mean ± SEM. Mice were anaesthetized with ketamine and xylazine, and artificially ventilated with a respirator. Heart rate‐corrected QT interval (QTc) was calculated according to the formula QTc = QT/(RR/100)1/2. P values were analysed by one way anova. No significant difference was found in any of the parameters among three groups of mice.
Figure 2.
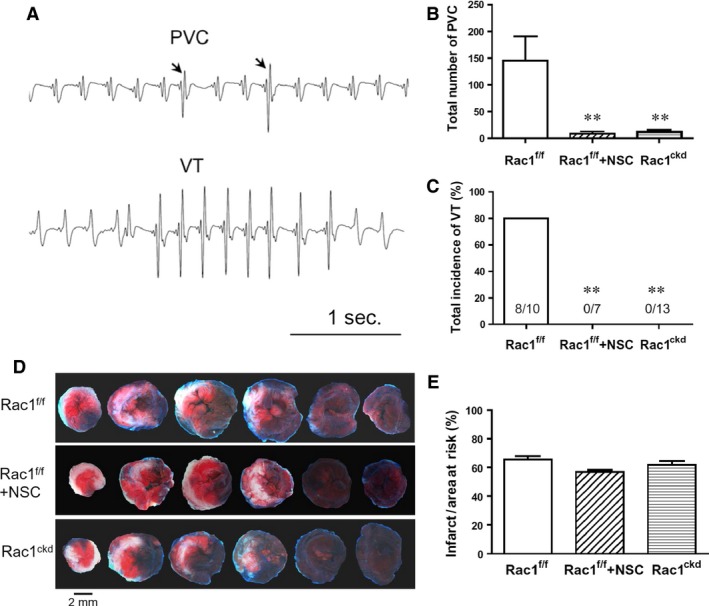
Inhibition of Rac1 decreases cardiac arrhythmia during 45 min. of ischaemia followed by 1 hr of reperfusion (I/R). (A) Representative ECG tracings showing premature ventricular complex (PVC) and ventricular tachycardia (VT). (B) Total number of PVC. (C) Total incidence of VT. (D) Representative sections of Evans Blue and triphenyltetrazolium chloride stained hearts showing area at risk (blue) and infarct area (white) respectively. (E) Quantification of infarct area to area at risk. Data are mean ± SEM. n = 7–13 mice per group in B and C, n = 6 mice per group in E. **P < 0.01 versus Rac1f/f.
Table 2.
Inhibition of Rac1 reduces ventricular arrhythmia following myocardial ischaemia and reperfusion in mice
| Rac1f/f | Rac1f/f+NSC23766 | Rac1ckd | P value | |
|---|---|---|---|---|
| Mouse numbers | 10 | 7 | 13 | |
| Number of PVC | ||||
| Ischaemia | 44.6 ± 14.3 | 2.0 ± 1.1** | 6.3 ± 2.0** | 0.0025 |
| Reperfusion | 100.2 ± 48.3 | 6.3 ± 3.3* | 5.4 ± 2.9* | 0.0351 |
| Incidence of VT | ||||
| Ischaemia | 3/10 | 0/7 | 0/13 | 0.0678 |
| Reperfusion | 5/10 | 0/7** | 0/13** | 0.0025 |
| VT Duration, sec. | ||||
| Ischaemia | 4.3 ± 0.3 | 0** | 0** | 0.0001 |
| Reperfusion | 3.9 ± 2.1 | 0** | 0** | 0.0001 |
Data are mean ± SEM. Mice were anaesthetized with ketamine and xylazine, and artificially ventilated with a respirator. P values on number of premature ventricular contraction (PVC) and ventricular tachycardia (VT) duration were analysed by one way anova followed by Newman–Keuls test. Incidence of VT was analysed by Chi‐square test. *P < 0.05, **P < 0.01 versus Rac1f/f control mice.
Inhibition of Rac1 decreases SOICR in cardiomyocytes
Intracellular Ca2+ is increased in prolonged ischaemia. During reperfusion, SR Ca2+ ATPase activity is restored and increases Ca2+ uptake into the SR, leading to SR overload 4, 5. SOICR is a spontaneous SR Ca2+ release via RyR2 under conditions of SR Ca2+ overload, which is thought to be arrhythmogenic for interference of cardiac electrical activity 20, 21, 22. To study the role of Rac1 in Ca2+ release under SR overload, isolated Rac1f/f and Rac1ckd cardiomyocytes were loaded with fura‐2AM and Ca2+ concentration was monitored as the fluorescence ratio. Ca2+ store overload was induced in quiescent myocytes by elevating extracellular Ca2+ concentrations as previously described 31, and the occurrence of SOICR, that is, spontaneous intracellular Ca2+ waves, was monitored in single cells (Fig. 3A,B and C). The threshold extracellular Ca2+ concentration was ~3 mM for Rac1f/f cardiomyocytes, whereas it was doubled at 6 mM for Rac1ckd cardiomyocytes and for Rac1f/f cardiomyocytes treated with a Rac1 inhibitor NSC23766. In response to 6 mM external Ca2+ concentration, the frequency and amplitude of SOICR were significantly decreased in Rac1ckd and NSC23766‐treated Rac1f/f cardiomyocytes (Fig. 3D and E). Ca2+ imaging confirmed that these events were calcium waves (see video S1 for normal uniform contractions in response to 0.5 Hz pacing in a Rac1f/f myocyte and video S2 for spontaneous wave contractions of a Rac1f/f myocyte in response to 6 mM extracellular Ca2+ concentration). Furthermore, at 6 mM external Ca2+ concentration, over 50% of Rac1f/f cardiomyocytes exhibited SOICR while the occurrence of SOICR was seen in only 20–30% of Rac1ckd and NSC23766‐treated Rac1f/f cells (P < 0.01, Fig. 3F). SR Ca2+ content as assessed by caffeine (10 mM) stimulation at the end of 6 mM external Ca2+ treatment was significantly lower in Rac1ckd and NSC23766‐treated Rac1f/f cells (P < 0.01, Fig. 3G). These data show that Rac1 promotes SR Ca2+ release and induction of SOICR in response to Ca2+ overload.
Figure 3.
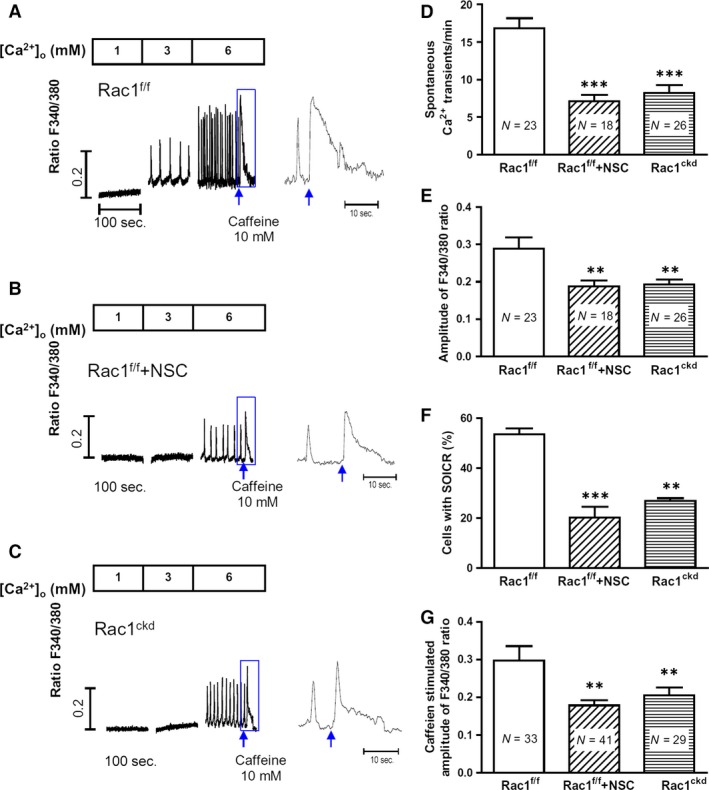
Effects of Rac1 inhibition decreases store overload‐induced Ca2+ release (SOICR) of adult ventricular myocytes. (A–C) Representative Fura‐2 ratios of Rac1f/f, NSC23766‐treated Rac1f/f and Rac1ckd myocytes in response to increasing extracellular Ca2+ concentrations (1, 3 and 6 mM) followed by caffeine (10 mM) stimulation (arrows). The boxed areas are enlarged. The threshold of SOICR was increased after Rac1 inhibition with NSC23766 and in Rac1ckd myocytes. (D–F) Quantification of frequency, amplitude and occurrence of Ca2+ transients in Rac1f/f, NSC23766‐treated Rac1f/f (Rac1f/f +NSC) and Rac1ckd cardiomyocytes in response to 6 mM extracellular Ca2+. (G) sarcoplasmic reticulum (SR) Ca2+ content assessed by caffeine stimulation. Data are mean ± SEM. Numbers in bars indicate myocyte numbers from three mice per group, **P < 0.01, ***P < 0.001 versus Rac1f/f.
To further assess changes in Ca2+ transients and SR Ca2+ content, myocytes were paced at 0.5 Hz followed by caffeine (10 mM) stimulation at 1 and 6 mM extracellular Ca2+ concentrations (Fig. 4A–D). Our data show that Ca2+ transients induced by pacing and SR Ca2+ content assessed by caffeine were significantly smaller in Rac1ckd compared to Rac1f/f myocytes (Fig. 4E and F).
Figure 4.
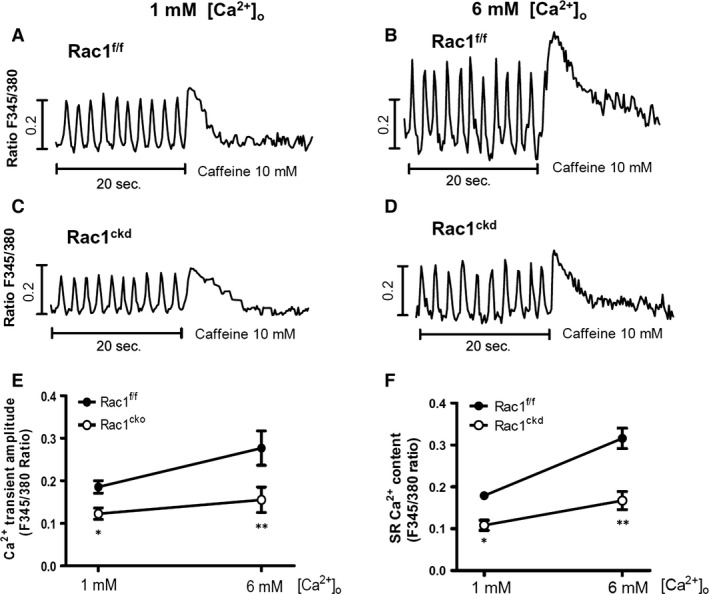
Rac1 inhibition reduces cardiomyocyte Ca2+ transients and sarcoplasmic reticulum (SR) Ca2+ load. (A–D) Representative Fura‐2 ratios in Rac1f/f and Rac1ckd myocytes, which were perfused at 1 and 6 mM extracellular Ca2+ concentrations and paced at 0.5 Hz for 20 sec. followed by treatment with caffeine. (E) Ca2+ transient amplitude in Rac1f/f and Rac1ckd myocytes during pacing. (F) SR Ca2+ load assessed by caffeine (10 mM) in Rac1f/f and Rac1ckd myocytes. Data are mean ± SEM of 10–16 myocytes from 3 mice per group, *P < 0.05, **P < 0.01 versus corresponding Rac1f/f.
Inhibition of Rac1 reduces ROS generation and RyR2 oxidation during I/R
Reactive oxygen species are generated in the heart during ischaemia and a significant burst of ROS formation occurs upon reperfusion 3, 4, 32. NADPH oxidases (Nox2 and Nox4) play a key role in ROS generation in the ischemic heart, and Rac1 is an important regulatory subunit of the Nox2 enzyme 33, 34. In the present study, superoxide generation during myocardial I/R was assessed using DHE staining on cryosections of the heart. The results show that myocardial I/R significantly increased ROS levels in Rac1f/f hearts, which was reduced in Rac1ckd hearts (Fig. 5A and B).
Figure 5.
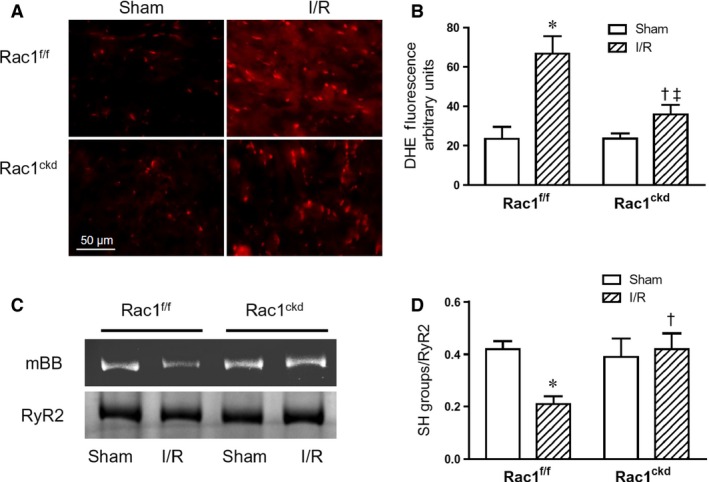
Rac1 inhibition decreases myocardial superoxide generation and oxidation of RyR2 after I/R. (A) Representative dihydroethidium (DHE) fluorescence staining (red) of heart sections after 45 min. of ischaemia followed by 1 hr of reperfusion (I/R). (B) Quantification of the red fluorescence intensity after I/R in Rac1ckd and Rac1f/f hearts. (C) Representative blots of RyR2 oxidation after 45 min. of ischaemia followed by 15 min. of reperfusion (I/R) using monobromobimane (mBB). (D) Quantification of free thiols of RyR2 in Rac1f/f and Rac1ckd hearts after I/R. Data are mean ± SEM. N = 3–5 mice per group, *P < 0.05 versus Rac1f/f sham, †P < 0.05 versus Rac1f/f I/R, ‡P < 0.05 versus Rac1ckd sham.
Since elevated oxidative stress induces RyR2 oxidation, which stimulates Ca2+ release, we propose that this could account for Ca2+ overload in cardiomyocytes 18, 35. Next we assessed RyR2 oxidation using monobromobimane (mBB), which labels free thiols of RyR2 18. Following myocardial I/R, mBB labelling was significantly decreased in Rac1f/f hearts, and notably, the response was reversed in Rac1ckd hearts (Fig. 5C and D). There were no significant changes in RyR2 band density (data not shown), suggesting equal RyR2 protein levels among all groups. The preservation of RyR2 free thiols is consistent with decreased ROS generation in Rac1ckd hearts during I/R.
Discussion
The major finding of this study is that inhibition of Rac1 protects against ventricular arrhythmia during myocardial I/R. We further demonstrated that genetic knockdown of Rac1 reduces SOICR in cardiomyocytes. Moreover, down‐regulation of Rac1 decreases ROS production and RyR2 oxidation during myocardial I/R. Our study suggests that Rac1 activation contributes to RyR2 oxidation, SOICR and ventricular arrhythmia during myocardial I/R. Thus, inhibition of Rac1 may have a therapeutic potential for clinical treatment of cardiac arrhythmia in ischaemic heart disease.
Intracellular Ca2+ homeostasis in cardiomyocytes is critical in maintaining normal electrical activities of the heart and Ca2+ overload is known to induce ventricular arrhythmia, cardiac hypertrophy and heart failure 5, 6, 32. Rac1 is a small GTPase involved in multiple physiological and cellular processes 7. Our recent studies show that activation of Rac1 increases intracellular Ca2+ in cardiomyocytes in response to cytokine stimulation 23. Myocardial I/R result in Ca2+ overload and cardiac arrhythmia 4, 5. However, the role of Rac1 in cardiomyocyte Ca2+ handling and ventricular arrhythmia during myocardial I/R remains elusive. We suggested that Rac1 is activated during myocardial I/R and inhibition of Rac1 protects against ventricular arrhythmia during I/R. To test this hypothesis, cardiac‐specific Rac1 knockdown mice were employed. We demonstrated that Rac1 is activated during myocardial I/R in Rac1f/f mice and I/R‐induced Rac1 activity is decreased in Rac1ckd mice. To monitor cardiac arrhythmia, ECG recordings were made during myocardial I/R. Our data show that the number of singlet and doublet ventricular premature beats and the incidence of ventricular tachycardia were significantly decreased by both pharmacological and genetic inhibition of Rac1. Importantly, infarct size following 45 min. of ischaemia and 1 hr of reperfusion was not significantly different among three groups of mice, indicating a similar level of cardiac injury. We did not assess infarct size beyond this period as most cardiac arrhythmia occurred within 40 min. after reperfusion. Thus, a beneficial effect of Rac1 inhibition on infarct size cannot be ruled out at a later time period. Collectively, these results suggest that activation of Rac1 in the heart contributes to ventricular arrhythmia during myocardial I/R. Previous studies showed that cardiac‐specific overexpression of constitutively active Rac1 induces atrial fibrillation in mice and higher Rac1 expression levels are associated with permanent atrial fibrillation in patients with atrial fibrosis 13, 14. Our study is the first demonstration of a definitive role of endogenous Rac1 activation in ventricular arrhythmogenesis during myocardial I/R. Studies have shown that NSC23766 inhibits M2 muscarinic receptors 36, and auto‐antibodies against M2 receptors, which enhance M2 receptor signalling, are associated with atrial fibrillation 37, 38. However, selective inhibition of M2 receptors has no significant effects on ischaemia‐induced ventricular arrhythmia 39. Therefore, the effects of NSC23766 on M2 receptor function are unlikely to contribute to the anti‐arrhythmic effects of NSC23766 observed during myocardial I/R in our study. Additionally, NSC23766 at 100 μM has been shown to have off‐target effects including inhibition of p21‐activated kinase (PAK) activity 40. In the present study, 2.5 mg/kg NSC23766 was used in mice, which is about 5 μM tissue concentration if it is evenly distributed in all tissues. This is 20 times lower than the concentration to achieve an off‐target effect. Thus, the anti‐arrhythmic effects of NSC23766 we observed are unlikely because of its off‐target effects.
To understand the mechanism by which Rac1 activation induces ventricular arrhythmia, SOICR, that is, spontaneous SR Ca2+ release via RyR2 under the conditions of SR Ca2+ overload, was investigated in this study. In response to increasing extracellular Ca2+ concentrations (3 and 6 mM), spontaneous intracellular Ca2+ oscillations were observed in both Rac1f/f and Rac1ckd cardiomyocytes, indicating induction of SOICR. However, the threshold extracellular Ca2+ to induce SOICR in Rac1ckd cardiomyocytes was doubled. Furthermore, the frequency and amplitude of SOICR were significantly decreased in Rac1ckd cardiomyocytes. Similar results were observed in Rac1f/f cardiomyocytes treated with a pharmacological Rac1 inhibitor NSC23766. Our results demonstrated that Rac1 promotes the occurrence of SOICR in cardiomyocytes, which induces ventricular arrhythmia.
RyR2 is sensitive to redox modifications and oxidation of RyR2 by ROS generation induces SR Ca2+ leak in cardiomyocytes 18, 35. RyR2 is a tetrameric complex contains up to 89 cysteine residues per monomer, from which approximately 21 are redox sensitive 41. Rac1 is an important component of NADPH oxidase, which is a major ROS generating enzyme in the heart. In the present study, ROS production was significantly increased in Rac1f/f hearts after I/R but was inhibited in Rac1ckd mice. To assess thiol oxidation of RyR2, monobromobimane labelling assay was employed. Our data show that the amount of free thiols in RyR2 labelled by monobromobimane was significantly decreased after myocardial I/R in Rac1f/f mice. Knockdown of Rac1 restored the amount of free thiols of RyR2. Our results suggest that Rac1 activation promotes ROS generation and RyR2 oxidation, leading to SOICR in cardiomyocytes during myocardial I/R. This study is limited to address the role of Rac1 in RyR2‐mediated cardiac Ca2+ handling. Further studies are required to examine whether Rac1 affects other key players including Na+/Ca2+ exchanger, SERCA and L‐type Ca2+ channels in cardiomyocytes during myocardial I/R.
Oberhofer et al. generated a mouse model that overexpresses a constitutively active mutant of Rac1, V12Rac1 in cardiomyocytes (RacET) 42. The RacET mice displayed cardiac dysfunction and dilated cardiomyopathy. Unexpectedly, a lower ROS production was found in cardiomyocytes. They further demonstrated that overexpression of V12Rac1 uncouples Rac1 activity from ROS production as a result of decreased p47phox plasma membrane to cytosol ratio, which is critical to NADPH oxidase activity. The study showed that Rac1 overexpression results in a ROS‐independent Ca2+ dyshomeostasis and cardiac dysfunction 42. In our study, mice with cardiac‐specific knockdown of Rac1 show normal cardiac function 29. As a result of lower intracellular Ca2+ and SR Ca2+ content, the Rac1 knockdown cardiomyocytes are more resistant to Ca2+ overload during I/R injury. Data from our study are consistent with an important role of endogenous Rac1 in ROS‐dependent modulation of Ca2+ handling in cardiomyocytes.
In summary, this study showed that myocardial I/R activates Rac1 and induces ventricular arrhythmia. Inhibition of Rac1 decreases ROS production, RyR2 oxidation, and cardiac arrhythmia during I/R. Furthermore, SOICR, an important mechanism of arrhythmogenesis can be effectively inhibited by Rac1 inhibitor NSC23766 in cardiomyocytes. Collectively, our study demonstrated that inhibition of Rac1 activation protects against arrhythmogenesis during myocardial I/R, and may represent a new paradigm for the treatment of cardiac arrhythmia in ischemic heart disease.
Funding
This study was supported by operating grants to Q.F. from the Canadian Institutes of Health Research (CIHR, MOP‐119600), to S.M.S. from CIHR (MOP‐64453) and to G.W. from National Natural Science Foundation of China (#81270176 and #81570254). L.Z. was a visiting PhD student from Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. Q.F. was a Career Investigator of the Heart Stroke Foundation of Ontario.
Conflict of Interest
The authors declare that there are no conflicts of interest.
Author contributions
S.M.S., G.W. and Q.F. contributed to conception and design of research; L.Z., X.L., L.G., and Y.W. performed experiments and analysed data. L.Z., L.G., S.M.S. and Q.F. interpreted results; L.Z. and L.G. prepared figures; L.Z. drafted manuscript; S.M.S., G.W. and Q.F. approved final version of manuscript; S.M.S. and Q.F. edited and revised manuscript.
Supporting information
Video S1. Normal uniform contractions elicited by pacing isolated adult Rac1f/f cardiomyocytes.
Video S2. Spontaneous waves in isolated adult Rac1f/f cardiomyocytes in response to Ca2+ overload.
References
- 1. Go AS, Mozaffarian D, Roger VL, et al Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013; 127: e6–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verma S, Fedak PW, Weisel RD, et al Fundamentals of reperfusion injury for the clinical cardiologist. Circulation. 2002; 105: 2332–6. [DOI] [PubMed] [Google Scholar]
- 3. Mozaffari MS, Liu JY, Abebe W, et al Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am J Cardiovasc Dis. 2013; 3: 180–96. [PMC free article] [PubMed] [Google Scholar]
- 4. Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011; 301: H1723–41. [DOI] [PubMed] [Google Scholar]
- 5. Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007; 87: 457–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Venetucci LA, Trafford AW, O'Neill SC, et al The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008; 77: 285–92. [DOI] [PubMed] [Google Scholar]
- 7. Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009; 66: 370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002; 100: 2692–6. [DOI] [PubMed] [Google Scholar]
- 9. Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 2012; 110: 1364–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satoh M, Ogita H, Takeshita K, et al Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006; 103: 7432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Higuchi Y, Otsu K, Nishida K, et al The small GTP‐binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal‐regulating kinase 1 and nuclear factor‐kappa B. J Biol Chem. 2003; 278: 20770–7. [DOI] [PubMed] [Google Scholar]
- 12. Hassanain HH, Gregg D, Marcelo ML, et al Hypertension caused by transgenic overexpression of Rac1. Antioxid Redox Signal. 2007; 9: 91–100. [DOI] [PubMed] [Google Scholar]
- 13. Adam O, Frost G, Custodis F, et al Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol. 2007; 50: 359–67. [DOI] [PubMed] [Google Scholar]
- 14. Reil JC, Hohl M, Oberhofer M, et al Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodelling. Cardiovasc Res. 2010; 87: 485–93. [DOI] [PubMed] [Google Scholar]
- 15. Shan L, Li J, Wei M, et al Disruption of Rac1 signaling reduces ischemia‐reperfusion injury in the diabetic heart by inhibiting calpain. Free Radic Biol Med. 2010; 49: 1804–14. [DOI] [PubMed] [Google Scholar]
- 16. Fabiato A. Time and calcium dependence of activation and inactivation of calcium‐induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985; 85: 247–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fauconnier J, Meli AC, Thireau J, et al Ryanodine receptor leak mediated by caspase‐8 activation leads to left ventricular injury after myocardial ischemia‐reperfusion. Proc Natl Acad Sci U S A. 2011; 108: 13258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Terentyev D, Gyorke I, Belevych AE, et al Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008; 103: 1466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belevych AE, Terentyev D, Viatchenko‐Karpinski S, et al Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009; 84: 387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang D, Zhu WZ, Xiao B, et al Ca2+/calmodulin kinase II‐dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007; 100: 399–407. [DOI] [PubMed] [Google Scholar]
- 21. Jiang D, Xiao B, Yang D, et al RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store‐overload‐induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004; 101: 13062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Q, Xiao J, Jiang D, et al Carvedilol and its new analogs suppress arrhythmogenic store overload‐induced Ca2+ release. Nat Med. 2011; 17: 1003–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang T, Lu X, Li J, et al Inhibition of Na/K‐ATPase promotes myocardial tumor necrosis factor‐alpha protein expression and cardiac dysfunction via calcium/mTOR signaling in endotoxemia. Basic Res Cardiol. 2012; 107: 254. [DOI] [PubMed] [Google Scholar]
- 24. Burger D, Lei M, Geoghegan‐Morphet N, et al Erythropoietin protects cardiomyocytes from apoptosis via up‐regulation of endothelial nitric oxide synthase. Cardiovasc Res. 2006; 72: 51–9. [DOI] [PubMed] [Google Scholar]
- 25. Burger DE, Lu X, Lei M, et al Neuronal nitric oxide synthase protects against myocardial infarction‐induced ventricular arrhythmia and mortality in mice. Circulation. 2009; 120: 1345–54. [DOI] [PubMed] [Google Scholar]
- 26. Gao Y, Dickerson JB, Guo F, et al Rational design and characterization of a Rac GTPase‐specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004; 101: 7618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen E, Li Y, Li Y, et al Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009; 58: 2386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiang FL, Lu X, Liu Y, et al Cardiomyocyte‐specific overexpression of human stem cell factor protects against myocardial ischemia and reperfusion injury. Int J Cardiol. 2013; 168: 3486–94. [DOI] [PubMed] [Google Scholar]
- 29. Zhang T, Lu X, Beier F, et al Rac1 activation induces tumour necrosis factor‐alpha expression and cardiac dysfunction in endotoxemia. J Cell Mol Med. 2011; 15: 1109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moazzen H, Lu X, Ma NL, et al N‐Acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014; 13: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diaz ME, Trafford AW, O'Neill SC, et al Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997; 501: 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997; 58: 95–117. [DOI] [PubMed] [Google Scholar]
- 33. Altenhofer S, Kleikers PW, Radermacher KA, et al The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012; 69: 2327–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsushima S, Tsutsui H, Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia‐reperfusion. Trends Cardiovasc Med. 2014; 24: 202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta‐adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012; 590: 3291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levay M, Krobert KA, Wittig K, et al NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J Pharmacol Exp Ther. 2013; 347: 69–79. [DOI] [PubMed] [Google Scholar]
- 37. Baba A, Yoshikawa T, Fukuda Y, et al Autoantibodies against M2‐muscarinic acetylcholine receptors: new upstream targets in atrial fibrillation in patients with dilated cardiomyopathy. Eur Heart J. 2004; 25: 1108–15. [DOI] [PubMed] [Google Scholar]
- 38. Stavrakis S, Yu X, Patterson E, et al Activating autoantibodies to the beta‐1 adrenergic and m2 muscarinic receptors facilitate atrial fibrillation in patients with Graves’ hyperthyroidism. J Am Coll Cardiol. 2009; 54: 1309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Naito H, Furukawa Y, Hashimoto K. Effects of a cardioselective M2 receptor antagonist, AF‐DX 116, on ventricular arrhythmias in dogs. Heart Vessels. 1997; 12: 229–33. [DOI] [PubMed] [Google Scholar]
- 40. Dutting S, Heidenreich J, Cherpokova D, et al Critical off‐target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J Thromb Haemost. 2015; 13: 827–38. [DOI] [PubMed] [Google Scholar]
- 41. Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006; 71: 310–21. [DOI] [PubMed] [Google Scholar]
- 42. Oberhofer M, Tian Q, Ruppenthal S, et al Calcium dysregulation in ventricular myocytes from mice expressing constitutively active Rac1. Cell Calcium. 2013; 54: 26–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Normal uniform contractions elicited by pacing isolated adult Rac1f/f cardiomyocytes.
Video S2. Spontaneous waves in isolated adult Rac1f/f cardiomyocytes in response to Ca2+ overload.