

Abstract
Peroxisome proliferation occurs by at least two routes, division of existing peroxisomes and de novo biogenesis from the endoplasmic reticulum (ER). The proteins and molecular mechanisms governing peroxisome emergence from the ER are poorly characterized. In this study, we report that two integral membrane peroxins (proteins required for peroxisome biogenesis) in Saccharomyces cerevisiae, Pex29 and Pex30, reside in distinct regions of the ER and associate with Rtn1 and Yop1, reticulon family members that contribute to ER morphology, to govern peroxisome emergence from the ER. In vivo and in vitro analyses reveal that peroxisome proliferation is therefore not restricted to the peroxisome but begins at the level of the ER.
Keywords: endoplasmic reticulum (ER), microscopy, peroxisome, subcellular fractionation, subcellular organelle, yeast, peroxin, peroxisomes, reticulon
Introduction
Peroxisomes are organelles found across the diversity of eukaryotes. Bound by a single lipid bilayer, peroxisomes contain enzymes involved in lipid metabolism that are coupled to the production of hydrogen peroxide and the scavenging of reactive oxygen species. Peroxisome number, size, and volume are dynamically linked to cell type, its developmental state, and environmental stimuli. Control of these processes is critical to human health. Defects in peroxisome membrane formation, protein import, and organelle proliferation cause severe pathologies in humans, and peroxisome function is linked to numerous human health issues (1–4). Studies in many different model systems, including notably different species of yeast, have led to the identification of peroxins, which are defined as proteins involved in peroxisome biogenesis (5). The mechanisms of action of the more than 30 peroxins in peroxisomal biogenesis and proliferation are the subject of intense investigation.
There has been much debate as to whether peroxisomes are semi-autonomous organelles like mitochondria or are derived from internal membrane systems such as the secretory pathway. Evidence supports that peroxisome biogenesis occurs through two separate pathways, de novo biogenesis in which new peroxisomes bud from the ER3 and the division of existing peroxisomes (6–12). These biogenesis pathways are tightly regulated spatially and temporally and involve a host of molecular interactions that mediate the assembly of proteins and lipids at the peroxisome membrane and the ER (13–16). In the budding yeast Saccharomyces cerevisiae, growth and division of peroxisomes are the dominant forms of peroxisome proliferation. However, contribution from the ER is an essential albeit poorly understood process. When peroxisomes are absent, cells form peroxisomes de novo from the ER. Presumably, interactions between proteins and lipids at the ER result in de novo biogenesis of nascent preperoxisomes that undergo a series of steps to ultimately form mature peroxisomes. Several peroxisomal membrane proteins (PMPs) insert into the ER dependent on components of the Sec61 complex, and the subsequent formation of preperoxisomes is dependent on the peroxins Pex3 and Pex19. Pex3 is an integral membrane protein that accumulates initially at an ER subdomain and is then released in a Pex19-dependent fashion (11, 17–19). The cytosolic protein Pex19 appears to interact with Pex3 at the ER, leading to the release of the protein-membrane complex to form nascent peroxisomes, which then fuse and mature into functional peroxisomes capable of importing matrix proteins (8, 12, 20). Thus, although it is apparent that the ER plays a primary role in trafficking key peroxins essential for de novo peroxisome biogenesis, the function of ER-resident proteins and the ER structure itself in peroxin trafficking and the formation of peroxisomes remain to be determined.
In S. cerevisiae, the ER is an extensive tubular structure with distinct domains, including the perinuclear ER surrounding the nucleus and a dynamic meshwork of membranes at the cell periphery called the cortical ER (21, 22). Proteins involved in establishing and maintaining the cortical ER membrane in yeast include the reticulon-like A subfamily of integral membrane proteins, Rtn1, Rtn2, and Yop1 (23, 24). These proteins are proposed to form a wedge-like structure within the ER membrane, inducing membrane curvature and facilitating the formation of tubular structures (25). Furthermore, the meshwork of the cortical ER is maintained by physical interaction between Rtn1 and Sey1 (26), the yeast orthologue of the dynamin-like GTPase atlastin (27). Numerous activities, including de novo peroxisome biogenesis, occur at the cortical ER (28), which raises questions regarding the role of this structure and the reticulon family of proteins in ER function and peroxisome dynamics and biogenesis.
Based on phenotypes of mutants, one can classify two groups of peroxins that influence peroxisome proliferation as follows: one that promotes it, and one that restricts it. In S. cerevisiae, the first class includes Pex11/25/27 proteins that function to elongate and constrict the peroxisome before its fission (29–35). This class of peroxins also functions in poorly understood processes required for de novo peroxisome biogenesis from the ER (36, 37). The homologue of Pex11 in Yarrowia lipolytica was recently shown to be required for the de novo formation of peroxisomes (38). The second class includes Pex28/29/30/31/32 proteins (39, 40), but how these peroxins regulate peroxisome numbers remains unknown. Cells harboring single gene deletions of PEX28, PEX31, or PEX32 have fewer and slightly enlarged peroxisomes, whereas a single deletion of PEX29 or PEX30 results in increased numbers of smaller peroxisomes (39, 40). Epistasis experiments between deletion mutants of all five genes demonstrated that the phenotype of fewer and enlarged peroxisomes is hypostatic to the phenotype of increased numbers of smaller peroxisomes (40). This suggests that Pex29 and Pex30 function in the same pathway upstream of Pex28, -31, and -32. Two homologues of the Pex30/31/32 and Pex28/29 protein families in Y. lipolytica, Pex23 and Pex24, have been shown to be essential for growth in the presence of oleic acid, which requires peroxisomes for its metabolism, and to accumulate numerous small preperoxisomal vesicles or a few enlarged peroxisomes, respectively (41, 42). In the yeast Pichia pastoris, Pex30 is localized to both the ER and peroxisomes, and its absence leads to reduced numbers of enlarged peroxisomes in cells grown in oleic acid-containing medium (43).
Pex29 and Pex30 were recently shown to interact with reticulon proteins to form focal points at the ER from which new peroxisomes arise (44). Here, we confirm that Pex29 and Pex30 are ER-resident proteins that physically interact with a subset of ER-resident proteins, including the reticulon proteins Rtn1 and Yop1. We show that subdomains of the ER containing Pex29 and Pex30 dynamically associate with peroxisomes when cells are grown in the presence of glucose, but they more stably associate with peroxisomes when cells are grown in the presence of oleic acid. An in vivo egression assay demonstrates that Rtn1p and Yop1 restrict peroxisomal vesicle egression, whereas in vitro analysis shows that absence of the reticulon proteins, or of Pex29 and Pex30, leads to peroxisomal proliferation. Our studies reveal that peroxisome proliferation is not restricted to the level of the peroxisome but begins at the ER and results in biochemically distinct and dynamic associations between peroxisomes and the ER in a carbon source-dependent manner.
Experimental Procedures
Yeast Strains and Plasmids
The yeast strains used in this study are listed in Table 1 and were derived from the parental strains BY4741 and BY4742, the corresponding gene deletion strain library (Invitrogen), or the GFP clone library (Invitrogen) (45). Yeast insertions and deletions were made by targeted PCR disruption using chemical transformation or electroporation of respective PCR-amplified fragments. The following plasmids were used for PCR amplification with the appropriate primers and have been previously described: pRSETB-mRFP (46); pGFP/HIS5 (47); pProtA/HIS5 (48); pCM159/G418 (TetO7) (49); pBS34/HPH (mCherry) (50); and pFA6a-natNT2 and pFA6a-hphNT1 (51).
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Ref. |
|---|---|---|
| BY4741 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | 45 |
| BY4742 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0 | 45 |
| PEX30-GFP+ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex30::PEX30-GFP (HIS5) | This study |
| MDH2-GFP+ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, mdh2::MDH2-GFP (HIS5) | This study |
| pex3Δ/MDH2-GFP+ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex3::KanMX4, mdh2::MDH2-GFP (HIS5) | This study |
| pex5Δ/MDH2-GFP+ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex5::KanMX4, mdh2::MDH2-GFP (HIS5) | This study |
| pex7Δ/MDH2-GFP+ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex7::KanMX4, mdh2::MDH2-GFP (HIS5) | This study |
| PEX3-GFP+/MDH2-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex30::PEX30-GFP (HIS5)/PEX30, mdh2::MDH2-mRFP (URA3)/MDH2 | This study |
| PEX30-GFP+/CHC1-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), chc1::CHC1-mRFP (KanMX4)/CHC1 | This study |
| PEX30-GFP+/COP1-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5),cop1::COP1-mRFP (KanMX4)/COP1 | This study |
| PEX30-GFP+/ERG6-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5),erg6::ERG6-mRFP (KanMX4)/ERG6 | This study |
| PEX30-GFP+/NIC96-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), nic96::NIC96-mRFP (KanMX4)/NIC96 | This study |
| PEX30-GFP+/PEX3-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), pex3::PEX3-mRFP (KanMX4)/PEX3 | This study |
| PEX30-GFP+/SAC6-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), sac6::SAC6-mRFP (KanMX4)/SAC6 | This study |
| PEX30-GFP+/SEC13-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), sec13::SEC13-mRFP (KanMX4)/SEC13 | This study |
| PEX30-GFP+/SNF7-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, PEX30/pex30::PEX30-GFP (HIS5), snf7::SNF7-mRFP (KanMX4)/SNF7 | This study |
| PEX30-GFP+/PEX3-mRFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex30::PEX30-GFP (HIS5), pex3::PEX3-mRFP (URA3) | This study |
| PEX30-GFP+/KAR2-mRFP-HDEL | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex30::PEX30-GFP (HIS5), kar2::KAR2-mRFP-HDEL (URA3) | This study |
| pex3Δ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex3::KanMX4 | 45 |
| pex19Δ | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex19::KanMX4 | 45 |
| PEX30-PrA | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex30::PEX30-PrA (HIS5) | This study |
| PEX29-PrA | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex29::PEX29-PrA (HIS5) | This study |
| RTN1-PrA | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, rtn1::RTN1-PrA (HIS5) | This study |
| YOP1-PrA | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, yop1::YOP1-PrA (HIS5) | This study |
| PEX30-GFP+/MDH2-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex30::PEX30-GFP (HIS5)/PEX30, mdh2::MDH2-mRFP (URA3)/MDH2 | This study |
| PEX30-GFP+/POT1-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex30::PEX30-GFP (HIS5)/PEX30, pot::POT1-mRFP (URA3)/POT1 | This study |
| PEX29-GFP+/MDH2-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex29::PEX29-GFP (HIS5)/PEX29, mdh2::MDH2-mRFP (URA3)/MDH2 | This study |
| PEX29-GFP+/POT1-mRFP | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex29::PEX29-GFP (HIS5)/PEX29, pot::POT1-mRFP (URA3)/POT1 | This study |
| PEX30-GFP+/pmRFP-SKL | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex30::PEX30-GFP (HIS5), pmRFP-SKL (URA3) | This study |
| PEX30-GFP/RTN1-mCherry | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex30::PEX30-GFP (kanMX4)/PEX30, rtn1::RTN1-mCherry (hphNT1)/RTN1 | This study |
| PEX30-GFP/YOP1-mCherry | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex30::PEX30-GFP (kanMX4)/PEX30, yop1::YOP1-mCherry (hphNT1)/YOP1 | This study |
| PEX29-GFP/PEX30-mCherry | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex29::PEX29-GFP (kanMX4)/PEX29, pex30::PEX30-mCherry (hphNT1)/PEX30 | This study |
| PEX29-GFP/RTN1-mCherry | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex29::PEX29-GFP (kanMX4)/PEX29, rtn1::RTN1-mCherry (hphNT1)/RTN1 | This study |
| PEX29-GFP/YOP1-mCherry | MATα/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, pex29::PEX29-GFP (kanMX4)/PEX29, yop1::YOP1-mCherry (hphNT1)/YOP1 | This study |
| ptetO7-TATA-Pex19/GFP1-GFP+ | MATa, his3Δ1, leu2Δ0, met15Δ0, URA3::CMV-tTa, pPEX19::KanMX4-tet07-TATA, gpd1::GPD1-GFP (HIS5) | This study |
| ptetO7-TATA-Pex19/GFP1-GFP+/rtn1Δ/rtn2Δ/yop1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, URA3::CMV-tTa, pPEX19::KanMX4-tet07-TATA, gpd1::GPD1-GFP (HIS5), rtn1::natNT2, rtn2::hphNT1, yop1::leu2 | This study |
| PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex3::PEX3-GFP (HIS3MX6) | This study |
| pex29Δ/pex30Δ/PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex29::KanMX4, pex30::KanMX4, pex3::PEX3-GFP (HIS3MX6) | This study |
| rtn1Δ/rtn2Δ/yop1Δ/PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, rtn1::KanMX4, rtn2::KanMX4, yop1::KanMX4, pex3::PEX3-GFP (HIS3MX6) | This study |
| pex19Δ/PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex19::URA3, pex3::PEX3-GFP (HIS3MX6) | This study |
| pex19Δ/pex29Δ/pex30Δ/PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, pex29::KanMX4, pex30::KanMX4, pex3::PEX3-GFP (HIS3MX6), pex19::URA3 | This study |
| pex19Δ/rtn1Δ/rtn2Δ/yop1Δ/PEX3-GFP | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, rtn1::KanMX4, rtn2::KanMX4, yop1::KanMX4, pex3::PEX3-GFP (HIS3MX6), pex19::URA3 | This study |
Yeast Media and Growth Conditions
Strains were grown in YPD (1% yeast extract, 2% peptone, 2% glucose) or YPBO (0.5% KPi, pH 6.0, 0.3% yeast extract, 0.5% peptone, 0.5% Tween 40, 0.15% oleic acid), as indicated. All cultures were grown at 30 °C. When marker selection was required for each strain, defined synthetic medium supplemented with 2% glucose and the necessary amino acid(s) or drug was used. To study peroxisome biogenesis, strains were grown overnight to saturation in YPD containing 5 μm doxycycline, followed by dilution in fresh doxycycline-containing medium to an A600 of 0.2. Cells were then grown to log phase (A600 = 0.7). The log phase cells were harvested by centrifugation, washed five times in YPD to remove doxycycline, resuspended in fresh YPD medium without doxycycline, inoculated into YPD medium at an A600 of 0.2, and cultured at 30 °C in a shaker incubator rotating at 200 rpm. Every 3 h, a sample of the culture was collected for microscopy. Throughout the time course, cells were maintained at an A600 of 0.6–1 by diluting the culture with fresh medium maintained at 30 °C. For all microscopy experiments, cells were harvested from cultures in the exponential phase of growth, fixed in 4% formaldehyde at room temperature for 15 min, washed twice with water, resuspended in water, and imaged within 40 h of sample collection.
Fluorescence Deconvolution Microscopy
Slides were prepared according to Ref. 52 with modifications (53). Essentially, 200 μl of hot 1% agarose in nonfluorescent medium was used to prepare a thin agarose pad on a slide with two 18-mm square wells (Cel-line). 1–2 μl of culture was placed onto the slide, covered with a coverslip, and sealed with Valap (1:1:1 mixture of Vaseline, lanolin, and paraffin). Cells were incubated for 15–30 min at room temperature before image acquisition. Images were acquired as described (54) using a modified LSM 510 META confocal microscope equipped with a ×63/1.3 NA Plan-Neofluar objective (Carl Zeiss). A piezoelectric actuator was used to drive continuous objective movement, allowing for the rapid collection of z-stacks. The sides of each pixel represented 0.057 μm of the sample. Stacks of 41 optical sections spaced 0.16 μm apart were captured. GFP was excited using a 488-nm laser, and its emission was collected using a 488-nm long pass filter (Semrock).
For colocalization experiments, GFP was excited using a 488-nm laser, and its emission was collected using a 514/25-nm bandpass filter (Semrock). mRFP was excited with a 543-nm laser, and its emission was collected with a 629/53-nm bandpass filter (Semrock). Images were captured at room temperature. Transmission images were processed to maximize the fluorescent signal while maintaining cell outlines. Imaris was then used to render the deconvolved three-dimensional data set to the processed transmission image. Final figure assembly was performed in Adobe Photoshop, Adobe Illustrator, or Adobe InDesign.
Images were deconvolved using algorithms provided by Huygens Professional Software (Scientific Volume Imaging BV, The Netherlands). For deconvolution, three-dimensional data sets were processed to remove noise and reassign blur by an iterative Classic Maximum Likelihood Estimation widefield algorithm and confocal algorithms, respectively, with experimentally derived point spread functions. Pearson's correlation coefficient and Manders coefficient were calculated using the JACOP plugin (55) for ImageJ (National Institutes of Health).
Object-based colocalization was performed using Imaris (Bitplane). Fluorescence signal from Pex30-GFP or Pex29-GFP was processed with the “Surface” command function, and fluorescence signal from Mdh2-mRFP or Pot1-mRFP was processed with the “Spots” function. The ImarisXT plugin (Bitplane) was used to compute the distance between all spots and all surfaces and identify all “spots” within 0.28 μm of a “surface.”
For experiments with doxycycline, images were collected with a ×100/NA 1.4 objective on an Olympus IX-71 wide field inverted fluorescence microscope with a 250-watt xenon LED transillumination light source, DeltaVision personalDV (Applied Precision). A GFP filter set was used to filter excitation and emission profiles. Images were deconvolved using the manufacturer's supplied deconvolution software (softWoRx).
Immunoisolation of Protein Complexes from Whole Cell Protein Lysates
To facilitate affinity-based purification, the respective protein of interest was C-terminally tagged with protein A (pA) at the genomic level and expressed as a fusion protein. Cells were grown to an A600 of 1.0–1.2 in YPD medium, harvested by centrifugation, washed twice with cold 20 mm HEPES-KOH, pH 7.4, followed by a wash with cold 20 mm HEPES-KOH, pH 7.4, 1.2% polyvinylpyrrolidone, 1 mm DTT, 1:200 of protease inhibitor mixture (PIC solution) (Sigma) and 1:200 of solution P (90 mg of PMSF and 1 mg of pepstatin A in 5 ml of absolute ethanol). Cells were subjected to centrifugation at 4,000 × g for 20 min, and the pellet was loaded into a plastic syringe and pushed through directly into liquid nitrogen to form “noodles.” Noodles were then cryogenically ground into fine powder using a 25-ml stainless steel grinding jar and ball mill (Retsch PM100 Planetary Ball Mill, Haan, Germany). Each sample was subjected to four runs of grinding (3 min each at 450 rpm with a 1-min immersion in liquid nitrogen between each run). The resulting yeast powder was stored at −80 °C.
Yeast protein lysates were prepared by resuspending 4 g of yeast powder in 14 ml of buffer containing 20 mm HEPES-KOH, pH 7.4, 110 mm potassium acetate, 1 mm DTT, 1 mm MgCl2, 1 mm EDTA, 1:200 protease inhibitor mixture, 0.006% anti-foam B, 1% digitonin. The resuspension was clarified by centrifugation at 4,000 × g for 5 min and then at 50,000 × g for 20 min. The supernatant was then passed through a 2.7-μm pore size glass microfiber, 25-mm diameter syringe filter (Whatman) and collected into a clean 15-ml falcon tube. 20 mg equivalent (140-μl volume) of IgG-conjugated magnetic beads prepared as per the manufacturer's instructions and stored in PBS, pH 7.4, at 4 °C was added to the protein lysate and incubated with gentle agitation on a rocking platform for 40 min at 4 °C. Magnetic beads were collected with a magnet, transferred to a 1.5-ml microcentrifuge tube, and gently washed four times at room temperature (∼25 °C) with 500 μl of resuspension buffer containing 0.1% digitonin. To release protein complexes, the IgG-pA interactions were disrupted by adding 50 μl of 0.2% SDS and incubated at room temperature for 15 min. The beads were separated by a magnet, and the eluate was collected in a fresh microcentrifuge tube. The elution steps were repeated, and the samples pooled (total volume = 100 μl). 30 μl of this sample were aliquoted for separation by SDS-PAGE (NuPAGE Bis-Tris Pre-Cast Gels, Invitrogen) followed by silver staining (Invitrogen) to visualize protein bands. The remaining sample was loaded onto a detergent removal spin column (Thermo Fisher Scientific) to remove SDS following the manufacturer's protocol. The detergent-free flow-through was dried under vacuum and resuspended in 75 μl of 100 mm Tris-HCl, pH 8.3, 1 mm EDTA, 6 m urea. The protein sample was reduced in 5 mm tris(2-carboxyethyl)phosphine at 37 °C for 30 min and alkylated with 25 mm iodoacetamide at room temperature in the dark for 30 min. The alkylation reaction was quenched by adding DTT to a final concentration of 50 mm. The sample was diluted 4-fold to decrease the concentration of urea to 1.5 m. 1 mm CaCl2 was added to the sample, which was then digested with 2 μg of sequencing grade modified porcine trypsin (Promega) overnight at 37 °C. Digested samples were desalted using Vydac C18 Silica MicroSpin columns (The Nest Group) as per the manufacturer's instructions. Purified peptides were resuspended in 10 μl of 0.1% trifluoroacetic acid (v/v) and 1% acetonitrile (v/v), and 6 μl of the sample were analyzed by LTQ-MS/MS (Thermo Fisher Scientific). The resulting peptide finger prints were identified by searching against a yeast peptide database using the Trans Proteomic Pipeline (56). Search result validation was done using Peptide Prophet (version 3.0) (57), and a probability of 1 was used to confidently identify a peptide. We report proteins for which at least two unique peptides were observed. Similar to earlier arguments, in all experiments, ribosomal proteins were excluded from the list of identification as common coeluting contaminants (58). For Pex29-pA and Pex30-pA samples, proteins were identified by both in-gel digestion (59) and the in-solution method. Rtn1-pA and Yop1-pA were analyzed by the in-solution method.
Subcellular Fractionation and ER Shift Assay
Subcellular fractionation of S. cerevisiae cells was performed as in Ref. 30, with modifications. Cells grown in YEPD or YPBO were harvested by centrifugation at 2,000 × g in a Beckman JA10 rotor at room temperature and washed twice with water. Cells were resuspended in 10 mm DTT, 100 mm Tris-HCl, pH 9.4, at a concentration of 10 ml per g of wet cells and incubated at 30 °C for 35 min with gentle agitation to loosen the outer mannoprotein layer. Cells were collected by centrifugation at 2,000 × g in a Beckman JS13.1 rotor for 7 min at room temperature and washed once with Zymolyase buffer (50 mm potassium phosphate, pH 7.5, 1.2 m sorbitol, 1 mm EDTA). Cells were resuspended in Zymolyase buffer containing 0.125 mg of Zymolyase 100T/ml at a concentration of 8 ml per g of wet cells and incubated at 30 °C for 45 min to 1 h with gentle agitation to convert cells to spheroplasts. Spheroplasts were harvested by centrifugation at 2,000 × g in a Beckman JS13.1 rotor for 8 min at room temperature and washed once with 1.2 m sorbitol, 2.5 mm MES, pH 6.0, 1 mm EDTA and harvested by centrifugation at 6,000 × g at 4 °C. They were then resuspended in buffer H (0.6 m sorbitol, 2.5 mm MES, pH 6.0, 1 × cOmplete protease inhibitor mixture (Roche Applied Science)) at a concentration of 2 ml per g of wet cells. Resuspended spheroplasts were transferred to a homogenization mortar and disrupted by 10–20 strokes of a Teflon pestle driven at 1,000 rpm by a stirrer motor (model 4376-00, Cole-Parmer). Cell debris, unbroken cells, and nuclei were pelleted by centrifugation at 1,000 × g in a Beckman JS13.1 rotor for 8 min at 4 °C. The postnuclear supernatant (PNS) was subjected to four additional centrifugations at 1,000 × g in a Beckman JS13.1 rotor for 8 min at 4 °C.
For cells grown in YEPD, 300 μl of PNS was mixed with 700 μl of 11% (w/v) NycoDenz in buffer H and loaded onto the top of a discontinuous NycoDenz gradient (6.6 ml of 17%, 16.5 ml of 25%, 4.5 ml of 35%, and 3 ml of 50% (w/v) NycoDenz in buffer H). Organelles were separated by ultracentrifugation at 100,000 × g for 90 min at 4 °C in a Beckman VTi50 rotor. 18 fractions of 2 ml each were collected from the bottom of the gradient. To shift the ER, buffer H was prepared in 1 mm MgCl2, 0.5 mm EDTA, or 5 mm EDTA.
For cells grown in YPBO, the PNS was fractionated by centrifugation at 20,000 × g in a Beckman JS13.1 rotor for 35 min at 4 °C into pellet and supernatant fractions. The 20,000 × g pellet was resuspended in 11% (w/v) NycoDenz in buffer H and loaded onto the top of a discontinuous NycoDenz gradient (6.6 ml of 17%, 16.5 ml of 25%, 4.5 ml of 35%, and 3 ml of 50% (w/v) NycoDenz in buffer H). Organelles were separated by ultracentrifugation at 100,000 × g for 90 min at 4 °C in a Beckman VTi50 rotor. 18 fractions of 2 ml each were collected from the bottom of the gradient. To shift the ER, buffer H was prepared in 0.5 mm EDTA or 5 mm EDTA.
In Vitro Vesicle Budding Assay
The in vitro cell-free vesicle budding assay was performed according to methods published previously (18, 19). Yeast strains are grown overnight in 1 liter of YEPD to an A600 of 4.0. The cells were collected by centrifugation at 2,000 × g for 7 min at room temperature in a Beckman JA-10 rotor, resuspended in low glucose medium (YEPD but with 0.1% glucose final concentration), and incubated for 30 min at 25 °C with vigorous shaking. Cells were harvested by centrifugation and resuspended in spheroplast medium (1% yeast extract, 2% yeast peptone, 1 g/liter glucose, 1.4 m sorbitol, 50 mm potassium phosphate, pH 7.5, 50 mm 2-mercaptoethanol) supplemented with Zymolase® 100T at 1 mg/ml, to a final concentration of 8 ml/g of wet cells and incubated for 40 min at 37 °C with gentle agitation. Spheroplasts were recovered by centrifugation, resuspended in recovery medium (1% yeast extract, 2% yeast peptone, 1 g/liter glucose, 1 m sorbitol), and incubated at 37 °C for 90 min to allow partial regeneration of the cell wall. The regenerated spheroplasts were subsequently used for generating permeabilized yeast cells and yeast cytosol. For permeabilized yeast cells, regenerated spheroplasts were resuspended in ice-cold spheroplast lysis buffer (100 mm potassium acetate, 200 mm sorbitol, 20 mm HEPES-KOH, pH 7.2, 2 mm MgCl2) at a concentration of 5 ml/75 A600 unit cell equivalents. For cytosol, regenerated spheroplasts were lysed in ice-cold 20 mm HEPES-KOH, pH 7.4, at a concentration of 210 μl/75 A600 unit cell equivalents. The slurry was pipetted 30 times with a 1-ml pipette to ensure efficient lysis. Cell debris was pelleted by centrifugation at 3,000 × g for 5 min at 4 °C, and the supernatant was collected (∼360 μl). To the concentrated cytosol, 50 μl of 10× transport buffer (250 mm HEPES-KOH, pH 7.2, 1.15 m potassium acetate, 25 mm MgCl2), cOmplete protease inhibitor mixture (Roche Applied Science) to 1× concentration, and 50 μl of 2 m sorbitol was added.
Permeabilized yeast cells (PYCs) prepared from pex19Δ, pex19Δ/pex29Δ/pex30Δ, and pex19/rtn1Δ/rtn2Δ/yop1Δ strains were washed twice with TBPS (115 mm potassium acetate, 2.5 mm magnesium acetate, 0.25 m sorbitol, 1× cOmplete protease inhibitor mixture, 25 mm HEPES, pH 7.2) and then resuspended in TBPS at a concentration of 25 μl/5 A600 unit cell equivalents. Pex3 was endogenously tagged with GFP in these strains to differentiate between Pex3 from PYCs versus a potential contamination of Pex3 from cytosol. Reaction conditions were as follows: 25 μl of PYCs, 25 μl of wild-type cytosol, 25 μl of a 4× ATP-regenerating system (4 mm ATP, 0.4 mm GTP, 80 mm creatine phosphate, 0.8 mg/ml creatine phosphate kinase), and 25 μl of 2× TBPS were mixed on ice. The reaction proceeded by incubation at room temperature for 90 min, and chilling the samples on ice terminated the reaction. After the reaction was terminated, the PYCs were pelleted by centrifugation at 13,000 × g for 5 min at 4 °C. The supernatant of two identical reactions was pooled and spun at 200,000 × g for 1 h at 4 °C. The pellet, containing preperoxisomal vesicles, was resuspended in 2× sample buffer (4% SDS, 0.15 m Tris-HCl, pH 6.8, 4 mm EDTA, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue) before being resolved by SDS-PAGE. The presence of Pex3-GFP was detected by immunoblotting with affinity purified Pex3 antibodies (17). Reactions carried out with PYCs alone, in the absence of exogenous ATP, and at 4 °C were included as controls.
Results
Pex30 Localizes to a Punctate Compartment That Is Distinct from Other Organelle Compartments
To investigate the subcellular localization of Pex30, we imaged a genomically integrated chimeric protein, Pex30-GFP, under control of its native promoter. For three-dimensional confocal microscopy of living cells, we employed a fast acquisition protocol with continuous capture along the z axis driven by a piezoelectric actuator (54), which minimizes acquisition time and the introduction of small vibrations from the stepwise progression of a typical three-dimensional acquisition. Use of a multi-immersion objective with a correction collar allowed us to reduce the degrading effects of spherical aberration to image contrast by compensating for mismatches in the refractive indices of the imaging medium and living cells, as well as to correct for the necessary insertion of a coverslip into the light path (60). To remove noise and reassign blur, deconvolution algorithms were applied to the post-acquisition dataset, which also corrects for artifacts introduced by the digital discretization of the analogue fluorescence signal. The resulting three-dimensional reconstruction shows remarkable contrast for weak fluorescence signals, such as those collected by imaging Pex30-GFP, with resolution on three axes approaching the diffraction limits of the confocal microscope (Fig. 1A).
FIGURE 1.

Pex30 localizes to numerous puncta distributed throughout the cytoplasm. Wild-type cells with genomically integrated PEX30-GFP were incubated in YPD medium and imaged by three-dimensional confocal microscopy. A, maximum intensity projection of the z-stacks and a sectional view showing cross-sections in the xy, xz, and yz axes are shown for a small budded cell and a large budded cell. Bar, 1 μm. B, numerical density of peroxisomes and Pex30-GFP puncta. The numerical density was calculated from the number of spots identified with Imaris (Bitplane) software per cell from Mdh2-GFP- and Pex30-GFP-labeled strains. Cell volume was determined by measuring the short and long axes of mother cells and buds and calculating the volume of an oblong spheroid for each cell. Interquartile box and whisker plots for Mdh2 and Pex30 show the results of three independent experiments.
Pex30-GFP localizes to numerous puncta distributed throughout the cell with both cortical and, what appear to be, perinuclear distribution patterns. This localization was independent of cell size or progression through the cell cycle, although an enrichment of Pex30-GFP signal was often seen in small to mid-sized buds (Fig. 1A). Quantification of the number of puncta per cell revealed ∼10-fold more Pex30-GFP puncta as compared with peroxisomes labeled with Mdh2-GFP (Fig. 1B).
To determine the identity of the compartment(s) in which Pex30-GFP resides, we performed quantitative colocalization experiments against a battery of proteins that give a “punctate-composite” localization by light microscopy. These proteins were previously used in a global analysis of protein localization in yeast (61). A wild-type strain expressing Pex30-GFP was mated to strains expressing chimeras of each of the punctate-composite proteins and mRFP. The resulting diploid cells were then imaged by three-dimensional confocal microscopy, and an estimate of the strength of association between the two fluorophores was made by comparing Pearson's correlation coefficients calculated from the different three-dimensional image datasets (Fig. 2A) (62). As a positive control, Pearson's correlation coefficients were calculated for images acquired for Pex3-mRFP and Mdh2-GFP (Fig. 2A and data not shown).
FIGURE 2.

Quantitative three-dimensional colocalization microscopy punctate composite screen reveals partial colocalization between Pex30-GFP and Pex3-mRFP and Sac6-mRFP and Sec13-mRFP. A, diploid cells expressing the indicated fluorescent proteins were grown in YPD medium and imaged by three-dimensional confocal microscopy. Pearson's coefficients were calculated for the deconvolved data sets using the JACOP plugin for ImageJ (National Institutes of Health), and the data are shown by interquartile box and whisker plots. B, maximum intensity projections and middle slice of the median image from A are displayed for the three punctate composite markers showing partial colocalization with Pex30-GFP. Bar, 1 μm. C, intensity correlation analysis of the images shown in B. The intensity correlation analysis plot displays the covariance of the Pex30-GFP signal with a colocalization marker (Pex3-mRFP, Sac6-mRFP, or Sec13-mRFP) as a function of the relative intensity of Pex30-GFP. Intensity correlation analysis plots were calculated using the JACOP plugin for ImageJ. D, cross-correlation function analysis of the images shown in B. The cross-correlation function is a plot of a series of Pearson's coefficients calculated by shifting one image with respect to the other ±20 pixels. The cross-correlation function for each image pair in B was calculated using the JACOP plugin for ImageJ.
Pearson's correlation analysis revealed stronger correlations between Pex30-GFP and Pex3-mRFP, Sac6-mRFP, and Sec13-mRFP, as compared with correlations between Pex30-GFP and Chc1-mRFP, Cop1-mRFP, Erg6-mRFP, and Nic96-mRFP, and Snf7-mRFP (Fig. 2A). Sac6 is yeast fimbrin, an actin bundling and plus-end binding protein (63), whereas Sec13 is a structural component of COPII vesicles and the nuclear pore complex (64, 65). Pearson's correlation coefficients between the ranges of 0.3 and 0.6 could result from random, spurious, or coincident associations or could be the result of a partial colocalization between subsets of the two populations of proteins (55). To distinguish between these possibilities, we performed additional statistical analyses as shown for representative images of Pex30-GFP with Pex3-mRFP, Sac6-mRFP, or Sec13-mRFP (Fig. 2, B and C). Intensity correlation analysis (66) showed high intensity fluorescence from Pex30-GFP covaried with Pex3-mRFP and to a lesser extent with Sec13-mRFP but not with Sac6-mRFP, with which low intensity covariance dominated (Fig. 2C). Furthermore, computation of a cross-correlation function (67) for each image dataset picked up coincident colocalization between Pex30-GFP and Sac6-mRFP, which had an unequal right tail skew likely due to the propensity of both proteins to show enriched signal in the yeast bud (Fig. 2, B and D). We therefore conclude that the partial colocalization between Pex30-GFP and Pex3-mRFP is the result of specific and discrete associations between a subset of each protein population. Partial colocalization between Pex30-GFP and Sec13-mRFP likely results from a broad coincident colocalization of the two proteins residing in the same subcellular compartment, i.e. the ER, whereas colocalization between Pex30-GFP and Sac6-mRFP likely results from spurious and coincident associations. This interpretation comes with the caveat that Pearson's coefficients are strongly impacted by the microscopy setup and its resolution/contrast limits. Although not explored here, we expect that other membrane peroxins would show similar distributions.
We next performed quantitative colocalization experiments with Pex30-GFP and Kar2-mRFP(HDEL) (Fig. 3A). Kar2 is the yeast homologue of binding protein, BiP, an abundant ER lumenal chaperone protein (68). Haploid cells with genomically integrated Pex30-GFP and Kar2-mRFP(HDEL) were grown in the presence of glucose and imaged using three-dimensional confocal microscopy (Fig. 3A). The punctate localization of Pex30-GFP showed a high degree of overlap with the reticular Kar2-mRFP(HDEL) signal, as confirmed by the Pearson's correlation coefficient (Fig. 3D). Surprisingly, Pex30-GFP did not colocalize with a peroxisomal matrix reporter, mRFP(SKL), but did show partial colocalization with Pex3-mRFP (Fig. 3, C and D).
FIGURE 3.
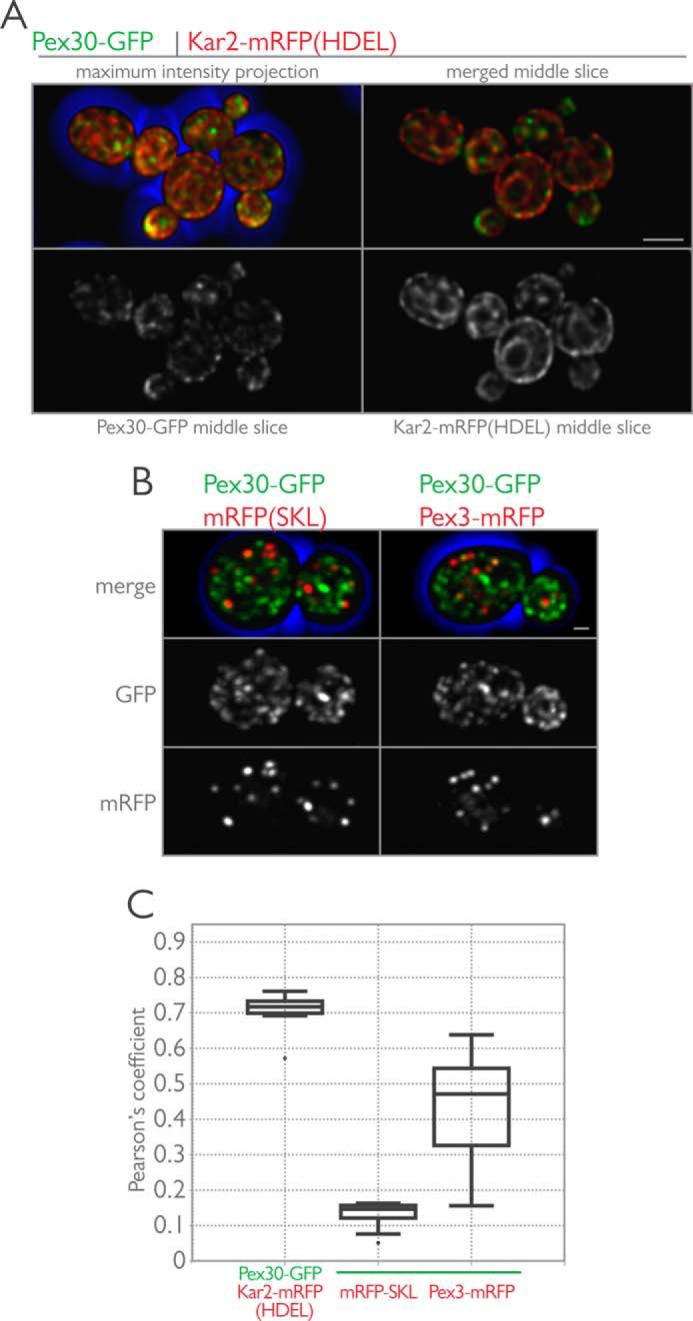
Pex30-GFP localizes to the ER. A, Pex30 localizes to the ER in glucose-grown cells. Endogenously expressed Pex30-GFP and Kar2-mRFP (HDEL) were imaged in wild-type cells grown in the presence of glucose. Shown are maximum intensity projections of all optical sections and single z-sections through the midplane of cells. Bar, 5 μm. B, Pex30-GFP was imaged in wild-type cells expressing mRFP-SKL (left) or Pex3-mRFP (right) and grown in the presence of glucose. Maximum intensity projections of the merged and individual fluorescence channels are displayed. Bar, 1 μm. C, cells expressing the indicated fluorescent proteins were grown in YPD medium and imaged by three-dimensional confocal microscopy. Pearson's coefficients were calculated for the deconvolved data sets using the JACOP plugin for ImageJ (National Institutes of Health), and the data are shown by interquartile box and whisker plots.
Identification of Pex30-interacting Proteins
To gain a better understanding of Pex30 function, we next sought to identify interacting partners of Pex30 by immunoaffinity isolation followed by identification by mass spectrometry. Pex30 was tagged at its C terminus with protein A, Pex30-pA, and immunoisolation of Pex30-pA-containing complexes was performed with whole cell extracts prepared from cells grown in the presence of glucose. Unlike efforts to immunopurify PMPs, which employed enrichment of peroxisomes by ultracentrifugation prior to immunoisolation of protein complexes (69, 70), we used whole cell lysates with the intention of probing for all potential Pex30-containing protein complexes that would be depleted from enriched peroxisomal fractions. Our approach is similar to that employed by David et al. (44) but with several important differences. First, cells were flash-frozen in liquid nitrogen and ground under cryogenic conditions using a planetary ball mill to maintain native protein complexes. This “solid-phase” cell breakage step maintains native protein-protein interactions and minimizes nonspecific protein interactions. Second, the resulting grindate was used for rapid immunoisolation on IgG-coated magnetic beads, which enable faster and cleaner complex isolation as compared with IgG-Sepharose (71–73). This approach reduces the number of steps prior to protein complex purification, thereby leading to a more confident identification of protein complexes (74–76). Experimental conditions were iteratively optimized to provide appropriate solubilization of integral membrane protein complexes while maintaining protein interactions (48, 77). Prominent bands representing putative Pex30-pA-interacting proteins were excised and identified by mass spectrometry.
The major proteins that copurify with Pex30-pA were identified as Pex29, Rtn1, and Yop1 (Fig. 4A). Identification of Pex29 as a Pex30-pA-interacting protein was expected, as they have been shown to interact by two-hybrid analysis and because pex29Δ and pex30Δ strains share similar peroxisome proliferation phenotypes (39, 40). Similarly to Pex30-pA, isolation of Pex29-pA yielded Pex30, Rtn1, and Yop1 as major copurifying proteins. Although both Rtn1 and Yop1 are reported to interact with the GTPase Sey1, we did not find Sey1 associated with either Pex29 or Pex30. This suggests the presence of different Rtn1 and Yop1 subcomplexes in yeast cells or that this interaction is not maintained in our immunopurification conditions. Additionally, from this initial characterization it remains undetermined whether Pex30 and Pex29 exist in a single complex or multiple distinct complexes.
FIGURE 4.

Affinity purification defines an ER-based protein interactome for Pex30 and Pex29. A, endogenously expressed Pex29 and Pex30 C-terminally tagged with protein A were immunopurified from wild-type yeast whole cell lysates under conditions that promote the capture of integral membrane proteins. Shown are silver-stained SDS-polyacrylamide gels exhibiting the typical patterns of proteins observed to copurify with these protein A-tagged baits. Proteins in eluates were identified by two complementary methods, i.e. in-gel digestion of prominent bands excised from silver-stained gels and global shotgun analysis of eluate fractions. The identity of bands excised from Pex29-pA and Pex30-pA gels are indicated. In both immunopurifications, the resident ER proteins, Rtn1 and Yop1, were found to copurify with Pex29-pA and Pex30-pA under these conditions. In support of this interaction, both Pex29 and Pex30 were identified by shotgun mass spectrometry in reciprocal immunoisolations using Rtn1-pA and Yop1-pA as baits. Asterisk indicates IgG heavy chain. B and C, expression ratio (xpress ratio = light isotope/heavy isotope) for proteins identified by the I-DIRT experiments and shotgun mass spectrometry. An xpress ratio of >2 was used as a set point to define specific Pex30-pA- (B) and Pex29-pA (C)-interacting proteins. D, network level view of the protein-protein interactions identified in this study. Horizontal lines represent protein nodes (as labeled in the figure), and vertical lines represent the protein-protein interactions. Graphical representation of the data was generated using BioFabric (Institute for Systems Biology, Seattle, WA).
To confirm the interactions between Pex30, Pex29, Rtn1, and Yop1, we performed reciprocal immunoisolations, this time using Rtn1-pA and Yop1-pA as baits (Fig. 4A). For these immunopurified eluates, proteins were identified by mass spectrometry using in-solution digests rather than gel excision of prominent bands. As expected, both Pex30 and Pex29 were detected in Rtn1-pA and Yop1-pA immunoisolations, strongly suggesting that these four proteins are bona fide interacting proteins. Several other potential interacting proteins, summarized in Table 1, were identified in these experiments. Of note, all four immunoisolations identified Scs2, Dpm1, and Yet3 as interacting partners. Scs2 is the yeast homologue of VAP and a component of the ER-plasma membrane tethering complex involved in the regulation of the phosphatidylinositol-4-phosphate phosphatase, Sac1 (78, 79). Dpm1 is a dolichol-phosphate mannosyltransferase and an integral membrane protein of the ER, but it also interacts with and has an important role in regulating Sac1 (80, 81). Interestingly, we also detected Pom33, an integral membrane protein recently shown to interact with Rtn1 and to dynamically associate with the yeast nuclear pore complex (82) in both Pex29-pA and Pex30-pA eluates. Unexpectedly, we did not identify Pom33 in Rtn1-pA or Yop1-pA eluates; however, we did identify Sey1 and several additional proteins involved in lipid metabolism in the Rtn1-pA eluate (Table 2).
TABLE 2.
Proteins identified by mass spectrometry of Pex30-pA, Pex29-pA, Rtn1-pA, and Yop1-pA immunoisolations
| Protein | IMPa | Localizationb | Molecular massc | GO annotationd |
|---|---|---|---|---|
| kDa | ||||
| Proteins identified with Pex30-pA, Pex29-pA, Rtn1-pA and Yop1-pA | ||||
| Rtn1 | Yes | ER | 32.9 | ER membrane structure |
| Yop1 | Yes | ER | 20.2 | ER membrane structure |
| Pex29 | Yes | ER/P | 63.5 | Regulates peroxisome abundance |
| Pex30 | Yes | ER/P | 59.4 | Regulates peroxisome abundance |
| Scs2 | Yes | ER | 26.9 | Regulates phospholipid metabolism |
| Dpm1 | Yes | ER | 30.3 | Dolichol mannose phosphate synthase |
| Yet3 | Yes | ER | 22.9 | Invertase secretion decreased |
| Proteins identified only with Pex30-pA and Pex29-pA | ||||
| Pom33 | Yes | ER | 32.2 | Nuclear pore complex dynamics |
| Proteins identified only with Rtn1-pA | ||||
| Erg6 | No | ER | 43.4 | Ergosterol biosynthesis |
| Erg1 | Yes | ER | 55.1 | Ergosterol biosynthesis |
| Erg9 | No | ER | 43.4 | Ergosterol biosynthesis |
| Erg11 | Yes | ER | 60.7 | Ergosterol biosynthesis |
| Cbr1 | Yes | ER/M | 31.4 | Microsomal cytochrome b reductase |
| Ncp1 | No | ER | 76.7 | Ergosterol biosynthesis |
| Yip3 | Yes | ER | 19.4 | ER to Golgi transport |
| Lsp1 | No | E | 38 | Primary component of eisosomes |
| Pil1 | No | E | 38.3 | Primary component of eisosomes |
| Sey1 | Yes | PC | 89.4 | GTPase with a role in ER morphology |
a IMP indicates integral membrane protein prediction based on hydropathy index as catalogued in the Saccharomyces Genome Database (yeastgenome.org).
b Localization indicates subcellular localization of the protein as catalogued in the Saccharomyces Genome Database. Curated from Ref. 58. ER is endoplasmic reticulum; P is peroxisome; M is mitochondrion; E is eisosome; PC is punctate composite.
c Molecular mass is as catalogued in the Saccharomyces Genome Database.
d GO annotation indicates the truncated gene ontology from the Saccharomyces Genome Database.
To assess the stability of the association between these membrane proteins and to identify additional interacting proteins of Pex30 and Pex29, we performed isotopic differentiation of interactions as random or targeted (I-DIRT) experiments (75) with Pex30-pA and Pex29-pA followed by shotgun mass spectrometry. This method differentiates between specific and nonspecific interacting proteins by immunoisolation of affinity-tagged protein complexes from cells grown in the presence of isotopically “light” medium, which is subsequently mixed in a 1:1 ratio with cell lysates grown in the presence of isotopically “heavy” medium, before protein identification by mass spectrometry (75). In vivo stably interacting proteins are enriched as isotopically light, whereas spurious interactions that result from lysis and mixing during immunoisolation have a ratio of light to heavy approximating 1:1.
Rtn1, Yop1, and Pex29 were among the top hits for Pex30-pA, confirming the association of these proteins (Fig. 4B). Additionally, Pex28 and Pex32, two proteins previous identified as Pex30 interactors by two-hybrid experiments (40), were also stably associated with Pex30-pA, further validating our proteomics approach. Interestingly, these experiments identified a stable association between Pex30-pA and Tcb3, another component of the ER-plasma membrane tether (78). Scs2 was also identified as a stable interactor but at a lower ratio of light-to-heavy isotope. In addition to these phosphatidylinositol 4-phosphate (PI4P) regulators, Dpm1 also showed an enriched light-to-heavy ratio. Pex30 was the top hit for Pex29-pA experiments with a ratio of light-to-heavy of just over 20, the highest ratio observed in these experiments (Fig. 4C). Pex29-pA also stably associated with Rtn1, but surprisingly Yop1 was not identified as a stable interactor of Pex29 in these experiments despite the identification of light Yop1 peptides. The strong interconnectivity of Pex30- and Pex29-interacting proteins was visualized in a network map of the protein-protein interaction data (Fig. 4D). This view also reinforced the association of Dpm1, Scs2, and Yet3 as additional interacting components of this “reticulon-peroxin” complex.
Pex30 and Pex29 Are ER-resident Proteins Even When Cells Are Grown in the Presence of Oleic Acid
Because most of the proteins detected as interacting with Pex30 and Pex29 are ER-resident proteins, we readdressed and further explored the subcellular localization of these proteins using an ER mobility shift assay (Fig. 5). This assay employs biochemical fractionation of lysed cells by isopycnic density gradient centrifugation in the presence or absence of magnesium (83). Total cellular levels of Mg2+ are in the millimolar range, complexed to anionic ligands, including phosphate, ATP, RNA, and DNA (84). In the presence of excess Mg2+, the ER has increased buoyant density that is most likely due to the strengthened association of ribosomes with the Sec61 complex (83). Ribosome stability requires Mg2+, and ribosomes fall apart when Mg2+ is stripped away by chelation. This phenomenon has been exploited in classic experiments preparing smooth and rough ER membranes for biochemical assays (85, 86). Immunoblot analysis of the separated fractions using antibodies to detect Pex30, Pex29, Pex3, Sec61, and Sdh2 revealed Mg2+-dependent migration of Pex30 and Pex29 along with the ER marker Sec61 from cells grown in the presence of glucose (Fig. 5A). Interestingly, Pex3 also showed Mg2+-dependent migration, moving from fractions of light density to fractions of heavy density with the increase in Mg2+ concentration. Mitochondria, as represented by the matrix protein Sdh2, were minimally affected by changes in Mg2+ and remained in fractions of light density in these experiments. These data suggest that peroxisomes, as labeled by Pex3, associate with the ER in glucose-grown cells but in regions distinct from mitochondria. ER-mitochondria contact sites are well documented in S. cerevisiae (87) but did not appear to affect the density of mitochondria in our experiments. Interestingly, Pex30 cofractionated with Sec61 under these conditions, whereas Pex29 cofractionated with Pex3. Pex29 also showed a bimodal distribution with a portion enriching in fractions of light density under all conditions (Fig. 5A).
FIGURE 5.

Pex30 and Pex29 cofractionate with Sec61 in an ER shift assay. A, wild-type yeast lysates from cells grown in the presence of glucose were prepared in buffer containing either 5 mm EDTA (no Mg2+), 0.5 mm EDTA, or 1 mm Mg2+, and postnuclear supernatants were subjected to isopycnic centrifugation through discontinuous NycoDenz density gradients. Eighteen fractions of increasing density were collected; 1.25% of the fraction volume was separated by SDS-PAGE, and the indicated proteins were detected by immunoblotting. Cofractionation of Pex30 and Pex29 with the ER marker, Sec61, is observed, and a concomitant shift results from increasing the buoyant density of ER by the presence of Mg2+. Pex3 also cofractionates with Sec61, but Sdh2 remains in the lighter fractions. B, similar to A, but this time cells were grown in the presence of oleic acid before preparation of lysates. Lysates were prepared in buffer containing either 5 mm EDTA or 0.5 mm EDTA. Cofractionation of Pex30 and Pex29 with the ER marker, Sec61, is observed, but this time Pex3 is found exclusively in fractions of heavy density and cofractionates with the peroxisomal matrix protein, Pot1.
When cells were grown in the presence of oleic acid, Pex30, Pex29, and Sec61 again showed Mg2+-dependent migration to fractions of heavy density (Fig. 5B). However, this time Pex3 was unaffected in its migration by Mg2+ concentration and was found exclusively in fractions of heavy density where it cofractionated with the oleic acid-induced peroxisomal matrix protein Pot1, as has been demonstrated previously (88). As demonstrated by Erdmann and Blobel (29), peroxisome density is increased in yeast grown in the presence of oleic acid because of the additional import of peroxisomal matrix proteins involved in β-oxidation. Pex29 once again showed a bimodal distribution and could be found in dense fractions that overlapped with Pex3 and Pot1, but in a Mg2+-dependent manner. In contrast, Pex30 cofractionated with Sec61 under all conditions tested (Fig. 5B).
We also tested whether the distribution of Pex30 or Pex29 was affected by the presence of peroxisomes. ER-shift assays performed using lysates from pex3Δ cells or pex19Δ cells, which lack any vestiges of peroxisomes, grown in the presence of glucose demonstrated that the Mg2+-dependent migration of Pex30 and Pex29 was unaffected by the loss of peroxisomes (Fig. 6, A and B). Interestingly, Pex29 still localized to the same slightly denser fractions that it shared with Pex3 in wild-type cells.
FIGURE 6.

Subcellular distribution Pex30 and Pex29 is unaffected by the presence of peroxisomes. A, pex3Δ yeast lysates from cells grown in the presence of oleic acid were prepared in buffer containing either 5 mm EDTA or 0.5 mm EDTA. Cofractionation of Pex30 and Pex29 with the ER marker, Sec61, is observed. B, similar to A but with pex19Δ yeast lysates.
Dynamic Carbon Source-dependent Association of Pex30 and Pex29 with Peroxisomes
We re-evaluated the association of Pex30 and Pex29 with peroxisomes using quantitative three-dimensional colocalization confocal microscopy. Cells endogenously expressing Pex30-GFP or Pex29-mRFP were mated to cells endogenously expressing Mdh2-mRFP or Pot1-mRFP. The resulting diploid cells expressing Mdh2-mRFP and either Pex30-GFP or Pex29-GFP were grown in the presence of glucose, whereas diploid cells expressing Pot1-mRFP and either Pex30-GFP or Pex29-GFP were grown in the presence of oleic acid.
Pex30-GFP associated with peroxisomes more strongly than Pex29-GFP. Quantification showed that 70% of peroxisomes were in contact with Pex30-GFP puncta using an object-based colocalization algorithm (Fig. 7A). This number increased to nearly 100% when cells were grown in the presence of oleic acid. Under these conditions, Pex30-GFP could be seen to associate with peroxisomes and to enrich at sites between peroxisomes that had clustered after growth in the presence of oleic acid. But Pex30-GFP did not overlap with signal from Pot1-mRFP, consistent with our previous colocalization analysis and ER-shift experiments demonstrating Pex30 to be an ER protein. The association between Pex29-GFP and peroxisomes also increased when cells were grown in the presence of oleic acid so that 75% of peroxisomes were in contact with Pex29-GFP puncta (Fig. 7B). However, Pex29-GFP did not redistribute and associate with peroxisomes to the same extent as Pex30, and the overall appearance of Pex29-GFP in cells grown in oleic acid was similar to that of Pex29-GFP in cells grown in the presence of glucose. Despite this lack of redistribution, Pex29-GFP could also be found associated with peroxisomes and enriched at sites between clustered peroxisomes in cells grown in the presence of oleic acid (Fig. 7B).
FIGURE 7.

Pex30, and to a lesser extent Pex29, associates with peroxisomes under peroxisome proliferating conditions as part of a peroxisome-associated ER membrane. A, endogenously expressed Pex30-GFP was localized in vivo with Mdh2-mRFP (left) and Pot1-mRFP (right) in wild-type diploid cells grown in the presence of glucose or oleate, respectively. Bar, 5 μm. Shown is the maximum intensity projection of all optical sections both merged and shown individually. Magnified insets of the merged image from Pex30-GFP and Pot1-mRFP show the association of Pex30-GFP with peroxisomes. The magnified regions represent a 5 × 5-μm area. Graphical results show the percentage of peroxisomes in contact with Pex30-GFP as calculated using an object-based colocalization algorithm using Imaris (Bitplane). B, similar to A but with Pex29-GFP instead of Pex30-GFP. C, Pex30 dynamically associates with peroxisomes. Bar, 1 μm. Two-dimensional video microscopy of wild-type cells expressing Pex30-GFP and mRFP-SKL is shown. Images of the midplane of cells were captured every 200 ms, and selected frames from the video are displayed with the indicated time stamp acting as a guide. Pex30-GFP can be seen to associate dynamically with static peroxisomes in the mother cell and also with mobile peroxisomes in the bud.
To investigate the association of Pex30-GFP with peroxisomes in further detail, we performed time-lapse microscopy on cells expressing Pex30-GFP and the peroxisomal matrix marker mRFP-SKL (Fig. 7C and supplemental Movie 1). We acquired images at a rate of 200 ms/frame to provide a relatively high temporal resolution. Under these conditions, Pex30-GFP could be seen to interact dynamically with peroxisomes. These associations were frequent enough that individual peroxisomes were visited several times over the course of image capture. The dynamic movement of Pex30-GFP throughout the cytoplasm is consistent with previous reports showing the yeast ER to be a highly dynamic structure (22). The association of Pex30 with peroxisomes was also independent of peroxisome placement in the cell, and the dynamic interactions were visualized for static, anchored peroxisomes in the mother cell as well as for fast moving peroxisomes in the bud (Fig. 7C). Pex29-GFP bleached too quickly to reliably visualize whether it dynamically associated with peroxisomes (data not shown).
We next studied the interactions between members of the reticulon family and Pex30 and Pex29 by quantitative colocalization three-dimensional confocal microscopy. We fused GFP to the C terminus of Pex29 or Pex30 and mRFP to the C terminus of Pex30, Rtn1, or Yop1 and tested for colocalization of Pex29-GFP with Pex30-mRFP, Rtn1-mRFP, or Yop1-mRFP, as well as colocalization of Pex30-GFP with Rtn1-mRFP or Yop1-mRFP in diploid cells grown in the presence of glucose or in the presence of oleic acid (Fig. 8, A–E). For each protein pair, partial colocalization, as assessed by comparison of Pearson's coefficients, was observed. In contrast to Pex30-GFP, which showed stable colocalization with Rtn1-mRFP and Yop1-mRFP under both growth conditions, Pex29-GFP associated more strongly with all of its interacting partners when cells were grown in the presence of oleic acid. These findings are consistent with our immunopurification experiments in which Pex29-GFP association with Yop1 was not stable in immunoisolations from cells grown in the presence of glucose (Fig. 4C). A slope graph of the mean Mander's coefficients calculated for each imaging pair reinforced this view (Fig. 8F). The general trend was for stronger association between Pex30, Pex29, Rtn1, and Yop1 when cells were grown in the presence of oleic acid.
FIGURE 8.

Colocalization analysis among Pex30, Pex29, Rtn1, and Yop1. A, endogenously expressed Pex30-GFP was localized in vivo with Rtn1-mRFP in wild-type diploid cells grown in the presence of glucose (left) or oleate (right). Bar, 5 μm. Shown are maximum intensity projections of all optical sections and single z-sections through the midplane of cells. Pearson's coefficients were calculated for the deconvolved data sets using the JACOP plugin for ImageJ (National Institutes of Health), and the data from a representative experiment of five images per strain are shown by interquartile box and whisker plots. Similar to A but with Pex30-GFP and Yop1-mRFP (B), Pex29-GFP and Pex30-mRFP (C), Pex29-GFP and Rtn1-mRFP (D), and Pex29-GFP and Yop1-mRFP (E). F, slope graph of the median Mander's coefficient from the images in A–E. The mean Mander's coefficients were calculated for the deconvolved data sets using the JACOP plugin for ImageJ.
Peroxisome Biogenesis Is Altered in Cells Lacking RTN1, RTN2, and YOP1
As discussed earlier, yeast cells carrying single or double deletion(s) of PEX29 and PEX30 exhibit an increased number of peroxisomes per cell and a decreased average peroxisome volume when grown in oleic acid-containing medium; however, neither deletion affects the viability of yeast cells during growth in glucose- or oleic acid-containing medium (39, 40). Similarly, no detectable growth defect was observed on oleic acid-containing medium for cells deleted for RTN1, YOP1, and RTN2 in a dilution spot assay (data not shown).
We next asked whether Rtn1, Yop1, and Rtn2 directly, or through the establishment of tubular ER structures, contribute to the de novo biogenesis of peroxisomes. Specifically, we measured the kinetics of de novo peroxisome biogenesis in wild-type and mutant strains. To monitor de novo peroxisome biogenesis, we made strains in which the expression of PEX19, a gene essential for peroxisome biogenesis, was tightly regulated. This system is similar to a previously described system used to investigate de novo peroxisome biogenesis (11), but instead of regulating gene expression by a galactose-inducible promoter, we used the regulatable TetO7 promoter (89, 90). This difference is important, as overexpressing PMPs results in several pleiotropies, including the mistargeting of the PMPs to the mitochondria (see “Discussion”).
To visualize peroxisome biogenesis, we introduced an endogenously expressed Gpd1-GFP into both wild-type and triple null, rtn1Δ/rtn2Δ/yop1Δ, strains in which PEX19 expression is under the control of the TetO7 promoter. The rationale behind using the peroxisomal matrix protein, Gpd1, was to facilitate visualization of mature peroxisomes capable of importing matrix proteins, thereby providing a measurement of functional peroxisomes. With this system, we could assess the temporal dynamics of peroxisome biogenesis in individual cells. After prolonged culturing in the presence of doxycycline, repression of PEX19 expression led to the absence of detectable peroxisomes (Fig. 9). Upon doxycycline removal, PEX19 expression resumed and led to the time-dependent reappearance of detectable peroxisomes. Temporal analysis of the percentage of cells in which peroxisomes were detectable following the release of PEX19 repression showed that peroxisome reappearance occurred more quickly in rtn1Δ/rtn2Δ/yop1Δ cells relative to wild-type cells (Fig. 9B). Furthermore, despite observing a plateau in the percentage of cells with detectable peroxisomes for both strains, peroxisome re-emergence was more penetrant in the rtn1Δ/rtn2Δ/yop1Δ strain, as this triple null mutant reached a higher plateau. We interpret these data to suggest that the dynamics of peroxisome formation are accelerated in the mutant cells compared with wild-type cells and demonstrate a role for the ER-resident proteins Rtn1 and Yop1 in peroxisome biogenesis.
FIGURE 9.

Deletion of RTN1, RTN2, and YOP1 increases peroxisome formation and the prevalence of cells containing peroxisomes. The genomically encoded copy of PEX19 was placed under the control of a tetracycline-repressible TetO7 promoter to allow regulatable peroxisome production in wild-type and rtn1Δ/rtn2Δ/yop1Δ triple null cells expressing Gpd1-GFP as a peroxisomal marker. A, Gpd1-GFP localization is presented as maximum intensity projections of z-stack sections through wild-type (left) and rtn1Δ/rtn2Δ/yop1Δ (right) cells. In both strains, peroxisomes are present when cells are grown in the absence of doxycycline (Untreated) and absent in most cells after overnight culturing in medium containing doxycycline (0 h). Release of PEX19 repression by removal of doxycycline from medium results in a partially penetrant, time-dependent reappearance of peroxisomes in cells of both the wild-type and triple null strains. Scale bar, 5 μm. B, quantification of the percentage of cells in which peroxisome could be observed at the indicated time points after removal of doxycycline for the wild-type (solid line) and rtn1Δ/rtn2Δ/yop1Δ (dashed line) strains. Relative to the wild-type control, triple null cells exhibited a more rapid reappearance of peroxisomes and also an overall increase in the total percentage of cells containing peroxisomes throughout the time course. Error bars represent the standard deviation between two independent biological replicates.
Egress of Preperoxisomal Vesicles Is Enhanced in Deletion Mutants of the Reticulons or Pex29 and Pex30
Our preceding experiments, and those of David et al. (44), suggested to us a role for this reticulon-peroxin complex in regulating preperoxisomal vesicle egression from the ER. Pex29 and Pex30 functioning as regulators of peroxisome-destined vesicular flow from the ER is consistent with their observed ER localization, their physical association with the reticulons, and their dynamic association with peroxisomes. To test this hypothesis, we reconstituted peroxisome biogenesis in vitro. A preperoxisomal vesicle budding assay has been previously shown to require the presence of Pex3 in the ER and Pex19 in the cytosol (18). ATP hydrolysis is required for successful budding to occur, and in addition to Pex3, additional peroxins, such as Pex15 and Pex11, have been shown to be present in the vesicles (18, 19, 91).
Vesicle egression was enhanced in cells lacking PEX29 and PEX30 and also in cells lacking RTN1, RTN2, and YOP1 as compared with the control (Fig. 10A). As reported previously, successful budding was dependent on the presence of ATP and could be slowed by incubation at 4 °C.
FIGURE 10.

Egression of preperoxisomal vesicles is enhanced in reticulon-peroxin deletion strains as shown by a cell-free in vitro budding assay. A, PYCs prepared from pex19Δ, pex19Δ/pex29Δ/pex30Δ, and pex19Δ/rtn1Δ/rtn2Δ/yop1Δ cells expressing Pex3-GFP were incubated with wild-type cytosol for 90 min at room temperature in the presence of an ATP-regenerating system (5th, 7th, and 8th lanes, respectively). Controls included incubating the PYCs alone (1st to 3rd lanes), with cytosol but no ATP (4th lane, pex19Δ PYCs only), or with cytosol and ATP, but at 4 °C (6th lane, pex19Δ PYCs only). At the end of the budding reaction, samples were subjected to centrifugation at 13,000 × g for 2 min to pellet the PYCs. The supernatant was collected and subjected to centrifugation at 200,000 × g for 1 h. The resultant pellet was resuspended in sample buffer and separated by SDS-PAGE. Immunoblotting for the presence of Pex3-GFP was performed with affinity-purified antibodies to Pex3. B, cells from wild-type, pex29Δ/pex30Δ, rtn1Δ/rtn2Δ/yop1Δ, pex19Δ, pex19Δ/pex29Δ/pex30Δ, and pex19Δ/rtn1Δ/rtn2Δ/yop1Δ strains endogenously expressing Pex3-GFP were grown in the presence of glucose and imaged by three-dimensional confocal microscopy. Bar, 5 μm. Shown is a maximum intensity projection of all optical sections.
We also imaged the strains used to prepare donor membranes for the in vitro reactions (Fig. 10B). Surprisingly, Pex3-GFP localized to discrete puncta in pex19Δ/pex29Δ/pex30Δ cells and in pex19Δ/rtn1Δ/rtn2Δ/yop1Δ cells, suggesting that the loss of these proteins altered the sorting and accumulation of Pex3-GFP into putative peroxisomal export sites in the ER.
Discussion
Pex30 and Pex29 are integral membrane proteins that have been shown to regulate peroxisome proliferation in the yeast S. cerevisiae. In this study, we show that these proteins are ER-resident proteins that associate with other ER-resident proteins, including Rtn1, Yop1, Scs2, Dpm1, and Yet3. Both Rtn1 and Yop1 play roles in ER membrane curvature and establish the peripheral ER, a site where de novo peroxisome biogenesis is proposed to occur (17). The absence of Rtn1, Rtn2, and Yop1 results in disruption of the peripheral ER (23), and we demonstrate here that the absence of these proteins also leads to the dysregulation of de novo peroxisome biogenesis.
Many of these conclusions are in agreement with the findings of David et al. (44), who also characterized the interactome of Pex30 and Pex29. The reticulon proteins were shown to be prominent interactors with Pex30 and Pex29, and their deletion was shown to dysregulate peroxisome proliferation. These proteomics experiments also identified Scs2, Dpm1, and several other ER proteins as putative interactors. Our independent confirmation of these findings bolsters our confidence in their robustness, and for the role of the reticulons in regulating peroxisome biogenesis through interaction with Pex30 and Pex29.
However, there are several important differences between our findings and those of David et al. (44). The first is their description of a stable Pex30-mediated ER-Peroxisomal CONtact site (EPCON) as a focal point for peroxisome formation. In their report, David et al. (44) relied on an overexpression system to visualize Pex30 and Pex29 in their respective deletion backgrounds as these proteins are difficult to visualize from their endogenous promoters. In general, peroxins and PMPs are difficult to observe when expressed under their endogenous promoter, and the overexpression of these proteins to better improve tractability results in pleiotropic effects. Many PMPs, when overexpressed, localize to mitochondria, and when localized to the ER, they do not segregate into distinct subregions but are found generally throughout the ER (11). In contrast, we were able to visualize these proteins from their endogenous promoters, and our observations lead us to interpret the data differently. We found Pex30, and Pex29, Rtn1, and Yop1, to dynamically associate with peroxisomes and each other in a carbon source-dependent manner. In particular, Pex30-GFP was shown to be highly dynamic when cells were grown in the presence of glucose and only transiently associated with peroxisomes. The results of David et al. (44) and those from our experiments implicate the reticulons Pex29 and Pex30 as negative regulators of peroxisome proliferation, and we observed enhanced egress of Pex3 from the ER in our in vitro budding assay when these proteins are not present in the ER. Therefore, Pex30 does not physically define the peroxisomal ER-exit site but likely regulates its formation by influencing the trafficking and sorting of PMPs such as Pex3 in the ER. We urge care when drawing conclusions from experiments that lead to the overexpression of peroxins.
In their study, David et al. (44) also found a transient association of Pex30 with members the COPI complex and proposed a role for COPI-formed vesicles in peroxisome biogenesis (44). We did not detect COP components in our pullouts of Pex30-pA, Pex29-pA, Rtn1-pA, or Yop1-pA, possibly because of the enhanced stringency of our isolation conditions. However, COP components have been detected in fractions of purified peroxisomes (70) and have been proposed to play a role in rat liver peroxisome biogenesis (92). Inhibition of COP1 gene expression also affects peroxisome numbers in S. cerevisiae (88), but this effect may be indirect as Pex3 egress from the ER occurs independently of COPI and COPII. A proposed role for COPI in peroxisome biogenesis other than as a retrieval system for ER proteins aberrantly trafficked to the peroxisome or for peroxisomal membrane proteins aberrantly trafficked to the Golgi seems unwarranted at this time.
Importantly, despite identifying a similar protein-protein interactome for Pex30 and Pex29, our interpretation of these results differs from that of David et al. (44). Integrating our interactome data with the results of our quantitative fluorescence colocalization microscopy experiments and subcellular fractionation experiments do not support the idea of a protein complex with defined stoichiometry. Rather, we propose that these ER membrane proteins share a similar localization to distinct subregions of the ER where they can physically interact with each other in pairwise or higher ordered protein assemblies. This physical association is dynamic and stabilized under peroxisome proliferating conditions.
A role for the ER in de novo peroxisome biogenesis explains the long established observation that peroxisomes can form in the apparent absence of pre-existing peroxisomes, albeit very inefficiently, in S. cerevisiae (7). This is observed in yeast strains in which mutations lead to a lack of peroxisomes, yet functional peroxisomes reform upon re-introduction of the appropriate wild-type PEX gene (93). Similarly, heterokaryons formed from peroxisome-deficient fibroblasts isolated from Zellweger patients of different complementation groups rapidly form new peroxisomes (94). Thus, the axiom “omnis membrana e membrana” (every membrane from a membrane), as stated by Blobel (95), is not violated by the peroxisome.
Negatively regulating peroxisome biogenesis at the ER seems to be at least one of the functions of the reticulon-peroxin complex. Cells lacking these proteins, and the second reticulon Rtn2, produce peroxisomes more quickly upon reintroduction of Pex19 in the genetic system we used here (Fig. 9). Deletion mutants also led to enhanced egress of Pex3-containing vesicles (Fig. 10). An important question to address in the future is whether regulatory proteins such as Pex30, Pex29, and the reticulons influence the switch between de novo biogenesis and the growth and division of existing peroxisomes. David et al. (44) also demonstrated that peroxisome proliferation was increased in deletion mutants of these proteins but did not distinguish between the relative rates of de novo peroxisome biogenesis versus growth and division of peroxisomes in these cells. Our in vitro budding assay shows that vesicle-mediated Pex3 egress from the ER is enhanced in these mutants (Fig. 10A). However, whether these preperoxisomal vesicles fuse with existing peroxisomes, when present, or go on to mature into peroxisomes without contributions from mature peroxisomes remains to be explored.
Why would cells need to regulate peroxisome biogenesis at the ER? As noted above, several PMPs transit through the ER, and as with other proteins transiting through the ER, assembly must be controlled (96). One simple mechanism to achieve this control, and to prevent the premature assembly of peroxisomal membrane complexes, is to segregate PMPs within the ER. Distinct membrane protein-containing vesicles might then bud from the ER, and subsequent fusion would lead to the formation of import-competent organelles. In P. pastoris both Pex3-dependent and Pex3-independent sorting events regulate the trafficking of PMPs into biochemically distinct preperoxisomal vesicles (91). Similar models have previously been suggested based on biochemical fractionation of distinct peroxisomal populations from the yeast Y. lipolytica (8). Here, we monitored peroxisome formation using only a limited set of peroxisomal marker proteins, namely Pex3, Mdh2, Pot1, and Gpd1, and thus did not observe heterogeneity of peroxisomes. Thus, although it is clear that peroxisomes in rtn1Δ/rtn2Δ/yop1Δ cells can import Gpd1 with improved kinetics over wild-type peroxisomes, it is not clear whether the peroxisome population is homogeneous. If sorting of PMPs within the ER is dependent upon Pex29, Pex30, Rtn1, and Yop1, as we propose, it would be expected that inappropriate preperoxisomal vesicle release from the ER could lead to an aberrant vesicle population.
Presumably, the function of the reticulon-peroxin complex is integrated into, and receives signals from, the cellular response to external cues, which include changes in carbon source (4). Pex30 from wild-type whole cell lysates migrates as multiple bands in immunoblots (data not shown), which may indicate that it is regulated by post-translational modification. In vitro, Pex30 is a target for Kss1 (97), a mitogen-activated protein kinase that is involved in regulating filamentous growth (98). Our previous global analysis of kinases and phosphatases identified Kss1 as a negative regulator of peroxisome proliferation (99), consistent with a role for Kss1 in regulating Pex30. Furthermore, the PI4P phosphatase Sac1 was also shown to be a negative regulator of peroxisome proliferation. Our immunoprecipitation experiments identified several potential linkages between Sac1 and members of the reticulon-peroxin complex. First, all four components interact with Dpm1, which is required for Sac1-mediated regulation of cell secretion and growth by regulating PI4P levels at the ER (80, 81, 100). The reticulon-peroxin complex also interacts with Scs2, a component of the recently characterized ER-plasma membrane tether and an important regulator of Sac1 activity by transducing environmental cues from the plasma membrane to the ER (78, 79, 101). Pex30 and Pex29 also interact with Tcb3, which is also a component of the ER-plasma membrane tether. It would be interesting to investigate how dysregulation of signaling events through the Kss1 signaling pathway, or through pathways involving Sac1, affects the function of the reticulon-peroxin complex.
Our experiments also suggest the subcellular localization of Pex3 is more dynamic than typically thought. Our subcellular fractionation analyses demonstrated that peroxisomes from glucose-grown cells are associated with the ER, and whereas Pex30 partially colocalized with Pex3 when grown in the presence of glucose, it did not colocalize with mRFP-SKL. These data suggest a dual localization for Pex3 in glucose-grown cells and are in agreement with the proposed role for Pex3 to function as an ER tether for peroxisomes (102). However, the dual-localization dynamic of Pex3 changes when cells are grown in the presence of oleic acid, and Pex3 is found exclusively on peroxisomes (Fig. 7). When cells are switched from growth in the presence of glucose to growth in the presence of oleic acid, Rtn1, Yop1, and Pex19 become phosphorylated. Thus, preperoxisomal vesicle export from the ER may be coupled to cellular demands for peroxisomes.
The dynamic and enhanced association of Pex30 and Pex29 with peroxisomes when cells were grown in the presence of oleic acid also suggests the possibility for their role in facilitating lipid exchange between the ER and peroxisomes. Non-vesicular lipid exchange between the ER and peroxisomes has been demonstrated biochemically (103). It remains to be determined whether Pex30, Pex29, and the reticulons function in this non-vesicular transfer of lipids. Reticulons have previously been demonstrated to mediate non-vesicular exchange of lipids between the ER and mitochondria (104).
These discoveries establish the ER as a master regulator of peroxisome proliferation even when the primary method of peroxisome proliferation is through the growth and division of peroxisomes. Our findings, taken together with the likelihood of a conserved function of Rtn1 and Yop1 as mediators of membrane curvature in organelle formation/biogenesis, have led us to propose a model in which members of the reticulon-peroxin complex control the emergence of preperoxisomal vesicles from the peripheral ER and in which this control demands a functionally and spatially defined ER. Our results confirm a direct role for ER-resident proteins in the de novo synthesis of peroxisomes. Combining comprehensive morphological, proteomic, and genetic analyses with the in vitro biochemical assay that produces peroxisome-like vesicles from microsomal fractions promises to reveal further molecular requirements of this fundamental cellular process.
Author Contributions
F. D. M., R. A. R., and J. D. A. conceived the experiments. F. D. M., A. J., R. A. S., D. J. D., and R. S. R. performed the experiments. F. D. M., R. A. R., and J. D. A. analyzed and interpreted the data and wrote the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants P50 GM076547 and P41 GM109824 (to J. D. A.) and by Canadian Institutes for Health Research Foundation Grant 143289 (to R. A. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Movie S1.
- ER
- endoplasmic reticulum
- PYC
- permeabilized yeast cell
- PMP
- peroxisomal membrane protein
- pA
- protein A
- PNS
- postnuclear supernatant
- PI4P
- phosphatidylinositol 4-phosphate
- mRFP
- monomeric red fluorescent protein.
References
- 1. Dubois-Dalcq M., Feigenbaum V., and Aubourg P. (1999) The neurobiology of X-linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci. 22, 4–12 [DOI] [PubMed] [Google Scholar]
- 2. Wanders R. J., and Waterham H. R. (2005) Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin. Genet. 67, 107–133 [DOI] [PubMed] [Google Scholar]
- 3. Smith J. J., and Aitchison J. D. (2013) Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 14, 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mast F. D., Rachubinski R. A., and Aitchison J. D. (2015) Signaling dynamics and peroxisomes. Curr. Opin. Cell Biol. 35, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Distel B., Erdmann R., Gould S. J., Blobel G., Crane D. I., Cregg J. M., Dodt G., Fujiki Y., Goodman J. M., Just W. W., Kiel J. A., Kunau W. H., Lazarow P. B., Mannaerts G. P., Moser H. W., et al. (1996) A unified nomenclature for peroxisome biogenesis factors. J. Cell Biol. 135, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Motley A. M., Galvin P. C., Ekal L., Nuttall J. M., and Hettema E. H. (2015) Reevaluation of the role of Pex1 and dynamin-related proteins in peroxisome membrane biogenesis. J. Cell Biol. 211, 1041–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motley A. M., and Hettema E. H. (2007) Yeast peroxisomes multiply by growth and division. J. Cell Biol. 178, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Titorenko V. I., Chan H., and Rachubinski R. A. (2000) Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica. J. Cell Biol. 148, 29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Titorenko V. I., Ogrydziak D. M., and Rachubinski R. A. (1997) Four distinct secretory pathways serve protein secretion, cell surface growth, and peroxisome biogenesis in the yeast Yarrowia lipolytica. Mol. Cell. Biol. 17, 5210–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Titorenko V. I., and Rachubinski R. A. (1998) Mutants of the yeast Yarrowia lipolytica defective in protein exit from the endoplasmic reticulum are also defective in peroxisome biogenesis. Mol. Cell. Biol. 18, 2789–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoepfner D., Schildknegt D., Braakman I., Philippsen P., and Tabak H. F. (2005) Contribution of the endoplasmic reticulum to peroxisome formation. Cell 122, 85–95 [DOI] [PubMed] [Google Scholar]
- 12. van der Zand A., Gent J., Braakman I., and Tabak H. F. (2012) Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 149, 397–409 [DOI] [PubMed] [Google Scholar]
- 13. Titorenko V. I., and Rachubinski R. A. (2001) The life cycle of the peroxisome. Nat. Rev. Mol. Cell Biol. 2, 357–368 [DOI] [PubMed] [Google Scholar]
- 14. Ma C., Agrawal G., and Subramani S. (2011) Peroxisome assembly: matrix and membrane protein biogenesis. J. Cell Biol. 193, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Titorenko V. I., and Rachubinski R. A. (2009) Spatiotemporal dynamics of the ER-derived peroxisomal endomembrane system. Int. Rev. Cell Mol. Biol. 272, 191–244 [DOI] [PubMed] [Google Scholar]
- 16. Hettema E. H., Erdmann R., van der Klei I., and Veenhuis M. (2014) Evolving models for peroxisome biogenesis. Curr. Opin. Cell Biol. 29, 25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tam Y. Y., Fagarasanu A., Fagarasanu M., and Rachubinski R. A. (2005) Pex3p initiates the formation of a preperoxisomal compartment from a subdomain of the endoplasmic reticulum in Saccharomyces cerevisiae. J. Biol. Chem. 280, 34933–34939 [DOI] [PubMed] [Google Scholar]
- 18. Lam S. K., Yoda N., and Schekman R. (2010) A vesicle carrier that mediates peroxisome protein traffic from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 107, 21523–21528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agrawal G., Joshi S., and Subramani S. (2011) Cell-free sorting of peroxisomal membrane proteins from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 108, 9113–9118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Titorenko V. I., and Rachubinski R. A. (2000) Peroxisomal membrane fusion requires two AAA family ATPases, Pex1p and Pex6p. J. Cell Biol. 150, 881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Preuss D., Mulholland J., Kaiser C. A., Orlean P., Albright C., Rose M. D., Robbins P. W., and Botstein D. (1991) Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast 7, 891–911 [DOI] [PubMed] [Google Scholar]
- 22. Prinz W. A., Grzyb L., Veenhuis M., Kahana J. A., Silver P. A., and Rapoport T. A. (2000) Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Biol. 150, 461–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., and Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 [DOI] [PubMed] [Google Scholar]
- 24. De Craene J. O., Coleman J., Estrada de Martin P., Pypaert M., Anderson S., Yates J. R. 3rd, Ferro-Novick S., and Novick P. (2006) Rtn1p is involved in structuring the cortical endoplasmic reticulum. Mol. Biol. Cell 17, 3009–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M. M., Rapoport T. A., and Prinz W. A. (2008) Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250 [DOI] [PubMed] [Google Scholar]
- 26. Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., and Blackstone C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A., and Daga A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983 [DOI] [PubMed] [Google Scholar]
- 28. Lynes E. M., and Simmen T. (2011) Urban planning of the endoplasmic reticulum (ER): how diverse mechanisms segregate the many functions of the ER. Biochim. Biophys. Acta 1813, 1893–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erdmann R., and Blobel G. (1995) Giant peroxisomes in oleic acid-induced Saccharomyces cerevisiae lacking the peroxisomal membrane protein Pmp27p. J. Cell Biol. 128, 509–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith J. J., Marelli M., Christmas R. H., Vizeacoumar F. J., Dilworth D. J., Ideker T., Galitski T., Dimitrov K., Rachubinski R. A., and Aitchison J. D. (2002) Transcriptome profiling to identify genes involved in peroxisome assembly and function. J. Cell Biol. 158, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tam Y. Y., Torres-Guzman J. C., Vizeacoumar F. J., Smith J. J., Marelli M., Aitchison J. D., and Rachubinski R. A. (2003) Pex11-related proteins in peroxisome dynamics: a role for the novel peroxin Pex27p in controlling peroxisome size and number in Saccharomyces cerevisiae. Mol. Biol. Cell 14, 4089–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koch J., Pranjic K., Huber A., Ellinger A., Hartig A., Kragler F., and Brocard C. (2010) PEX11 family members are membrane elongation factors that coordinate peroxisome proliferation and maintenance. J. Cell Sci. 123, 3389–3400 [DOI] [PubMed] [Google Scholar]
- 33. Opaliński Ł., Kiel J. A., Williams C., Veenhuis M., and van der Klei I. J. (2011) Membrane curvature during peroxisome fission requires Pex11. EMBO J. 30, 5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knoblach B., and Rachubinski R. A. (2010) Phosphorylation-dependent activation of peroxisome proliferator protein PEX11 controls peroxisome abundance. J. Biol. Chem. 285, 6670–6680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rottensteiner H., Stein K., Sonnenhol E., and Erdmann R. (2003) Conserved function of Pex11p and the novel Pex25p and Pex27p in peroxisome biogenesis. Mol. Biol. Cell 14, 4316–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tower R. J., Fagarasanu A., Aitchison J. D., and Rachubinski R. A. (2011) The peroxin Pex34p functions with the Pex11 family of peroxisomal divisional proteins to regulate the peroxisome population in yeast. Mol. Biol. Cell 22, 1727–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huber A., Koch J., Kragler F., Brocard C., and Hartig A. (2012) A subtle interplay between three Pex11 proteins shapes de novo formation and fission of peroxisomes. Traffic 13, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang J., Klute M. J., Tower R. J., Mast F. D., Dacks J. B., and Rachubinski R. A. (2015) An ancestral role in peroxisome assembly is retained by the divisional peroxin Pex11 in the yeast Yarrowia lipolytica. J. Cell Sci. 128, 1327–1340 [DOI] [PubMed] [Google Scholar]
- 39. Vizeacoumar F. J., Torres-Guzman J. C., Tam Y. Y., Aitchison J. D., and Rachubinski R. A. (2003) YHR150w and YDR479c encode peroxisomal integral membrane proteins involved in the regulation of peroxisome number, size, and distribution in Saccharomyces cerevisiae. J. Cell Biol. 161, 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vizeacoumar F. J., Torres-Guzman J. C., Bouard D., Aitchison J. D., and Rachubinski R. A. (2004) Pex30p, Pex31p, and Pex32p form a family of peroxisomal integral membrane proteins regulating peroxisome size and number in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown T. W., Titorenko V. I., and Rachubinski R. A. (2000) Mutants of the Yarrowia lipolytica PEX23 gene encoding an integral peroxisomal membrane peroxin mislocalize matrix proteins and accumulate vesicles containing peroxisomal matrix and membrane proteins. Mol. Biol. Cell 11, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tam Y. Y., and Rachubinski R. A. (2002) Yarrowia lipolytica cells mutant for the PEX24 gene encoding a peroxisomal membrane peroxin mislocalize peroxisomal proteins and accumulate membrane structures containing both peroxisomal matrix and membrane proteins. Mol. Biol. Cell 13, 2681–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yan M., Rachubinski D. A., Joshi S., Rachubinski R. A., and Subramani S. (2008) Dysferlin domain-containing proteins, Pex30p and Pex31p, localized to two compartments, control the number and size of oleate-induced peroxisomes in Pichia pastoris. Mol. Biol. Cell 19, 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. David C., Koch J., Oeljeklaus S., Laernsack A., Melchior S., Wiese S., Schummer A., Erdmann R., Warscheid B., and Brocard C. (2013) A combined approach of quantitative interaction proteomics and live-cell imaging reveals a regulatory role for endoplasmic reticulum (ER) reticulon homology proteins in peroxisome biogenesis. Mol. Cell. Proteomics 12, 2408–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 46. Campbell R. E., Tour O., Palmer A. E., Steinbach P. A., Baird G. S., Zacharias D. A., and Tsien R. Y. (2002) A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 99, 7877–7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dilworth D. J., Suprapto A., Padovan J. C., Chait B. T., Wozniak R. W., Rout M. P., and Aitchison J. D. (2001) Nup2p dynamically associates with the distal regions of the yeast nuclear pore complex. J. Cell Biol. 153, 1465–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aitchison J. D., Rout M. P., Marelli M., Blobel G., and Wozniak R. W. (1995) Two novel related yeast nucleoporins Nup170p and Nup157p: complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore-membrane protein Pom152p. J. Cell Biol. 131, 1133–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garí E., Piedrafita L., Aldea M., and Herrero E. (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848 [DOI] [PubMed] [Google Scholar]
- 50. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., and Tsien R. Y. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 51. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 52. Adames N. R., Oberle J. R., and Cooper J. A. (2001) The surveillance mechanism of the spindle position checkpoint in yeast. J. Cell Biol. 153, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fagarasanu A., Mast F. D., Knoblach B., Jin Y., Brunner M. J., Logan M. R., Glover J. N., Eitzen G. A., Aitchison J. D., Weisman L. S., and Rachubinski R. A. (2009) Myosin-driven peroxisome partitioning in S. cerevisiae. J. Cell Biol. 186, 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hammond A. T., and Glick B. S. (2000) Raising the speed limits for 4D fluorescence microscopy. Traffic 1, 935–940 [PubMed] [Google Scholar]
- 55. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 56. Keller A., Eng J., Zhang N., Li X. J., and Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 58. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 59. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 60. Pawley J. B. (eds) (2006) Handbook of Biological Confocal Microscopy, 3rd Ed., Springer, New York [Google Scholar]
- 61. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., and O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 62. Manders E. M., Stap J., Brakenhoff G. J., van Driel R., and Aten J. A. (1992) Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 103, 857–862 [DOI] [PubMed] [Google Scholar]
- 63. Adams A. E., Botstein D., and Drubin D. G. (1991) Requirement of yeast fimbrin for actin organization and morphogenesis in vivo. Nature 354, 404–408 [DOI] [PubMed] [Google Scholar]
- 64. Siniossoglou S., Wimmer C., Rieger M., Doye V., Tekotte H., Weise C., Emig S., Segref A., and Hurt E. C. (1996) A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell 84, 265–275 [DOI] [PubMed] [Google Scholar]
- 65. Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., and Schekman R. (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 [DOI] [PubMed] [Google Scholar]
- 66. Li Q., Lau A., Morris T. J., Guo L., Fordyce C. B., and Stanley E. F. (2004) A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 24, 4070–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van Steensel B., van Binnendijk E. P., Hornsby C. D., van der Voort H. T., Krozowski Z. S., de Kloet E. R., and van Driel R. (1996) Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 109, 787–792 [DOI] [PubMed] [Google Scholar]
- 68. Rose M. D., Misra L. M., and Vogel J. P. (1989) KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57, 1211–1221 [DOI] [PubMed] [Google Scholar]
- 69. Agne B., Meindl N. M., Niederhoff K., Einwächter H., Rehling P., Sickmann A., Meyer H. E., Girzalsky W., and Kunau W. H. (2003) Pex8p: an intraperoxisomal organizer of the peroxisomal import machinery. Mol. Cell 11, 635–646 [DOI] [PubMed] [Google Scholar]
- 70. Marelli M., Smith J. J., Jung S., Yi E., Nesvizhskii A. I., Christmas R. H., Saleem R. A., Tam Y. Y., Fagarasanu A., Goodlett D. R., Aebersold R., Rachubinski R. A., and Aitchison J. D. (2004) Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol. 167, 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oeffinger M., Wei K. E., Rogers R., DeGrasse J. A., Chait B. T., Aitchison J. D., and Rout M. P. (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods 4, 951–956 [DOI] [PubMed] [Google Scholar]
- 72. Hakhverdyan Z., Domanski M., Hough L. E., Oroskar A. A., Oroskar A. R., Keegan S., Dilworth D. J., Molloy K. R., Sherman V., Aitchison J. D., Fenyö D., Chait B. T., Jensen T. H., Rout M. P., and LaCava J. (2015) Rapid, optimized interactomic screening. Nat. Methods 12, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cristea I. M., and Chait B. T. (2011) Affinity purification of protein complexes. Cold Spring Harb. Protoc. 2011, prot5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dilworth D. J., Tackett A. J., Rogers R. S., Yi E. C., Christmas R. H., Smith J. J., Siegel A. F., Chait B. T., Wozniak R. W., and Aitchison J. D. (2005) The mobile nucleoporin Nup2p and chromatin-bound Prp20p function in endogenous NPC-mediated transcriptional control. J. Cell Biol. 171, 955–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tackett A. J., DeGrasse J. A., Sekedat M. D., Oeffinger M., Rout M. P., and Chait B. T. (2005) I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J. Proteome Res. 4, 1752–1756 [DOI] [PubMed] [Google Scholar]
- 76. Tackett A. J., Dilworth D. J., Davey M. J., O'Donnell M., Aitchison J. D., Rout M. P., and Chait B. T. (2005) Proteomic and genomic characterization of chromatin complexes at a boundary. J. Cell Biol. 169, 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rout M. P., Blobel G., and Aitchison J. D. (1997) A distinct nuclear import pathway used by ribosomal proteins. Cell 89, 715–725 [DOI] [PubMed] [Google Scholar]
- 78. Manford A. G., Stefan C. J., Yuan H. L., Macgurn J. A., and Emr S. D. (2012) ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 23, 1129–1140 [DOI] [PubMed] [Google Scholar]
- 79. Stefan C. J., Manford A. G., and Emr S. D. (2013) ER-PM connections: sites of information transfer and inter-organelle communication. Curr. Opin. Cell Biol. 25, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Faulhammer F., Kanjilal-Kolar S., Knödler A., Lo J., Lee Y., Konrad G., and Mayinger P. (2007) Growth control of Golgi phosphoinositides by reciprocal localization of Sac1 lipid phosphatase and Pik1 4-kinase. Traffic 8, 1554–1567 [DOI] [PubMed] [Google Scholar]
- 81. Faulhammer F., Konrad G., Brankatschk B., Tahirovic S., Knödler A., and Mayinger P. (2005) Cell growth-dependent coordination of lipid signaling and glycosylation is mediated by interactions between Sac1p and Dpm1p. J. Cell Biol. 168, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chadrin A., Hess B., San Roman M., Gatti X., Lombard B., Loew D., Barral Y., Palancade B., and Doye V. (2010) Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution. J. Cell Biol. 189, 795–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roberg K. J., Rowley N., and Kaiser C. A. (1997) Physiological regulation of membrane protein sorting late in the secretory pathway of Saccharomyces cerevisiae. J. Cell Biol. 137, 1469–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Graschopf A., Stadler J. A., Hoellerer M. K., Eder S., Sieghardt M., Kohlwein S. D., and Schweyen R. J. (2001) The yeast plasma membrane protein Alr1 controls Mg2+ homeostasis and is subject to Mg2+-dependent control of its synthesis and degradation. J. Biol. Chem. 276, 16216–16222 [DOI] [PubMed] [Google Scholar]
- 85. Blobel G., and Dobberstein B. (1975) Transfer of proteins across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 67, 852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jackson R. C., and Blobel G. (1977) Post-translational cleavage of presecretory proteins with an extract of rough microsomes from dog pancreas containing signal peptidase activity. Proc. Natl. Acad. Sci. U.S.A. 74, 5598–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S., and Walter P. (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Perry R. J., Mast F. D., and Rachubinski R. A. (2009) Endoplasmic reticulum-associated secretory proteins Sec20p, Sec39p, and Dsl1p are involved in peroxisome biogenesis. Eukaryot. Cell 8, 830–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hughes T. R., Marton M. J., Jones A. R., Roberts C. J., Stoughton R., Armour C. D., Bennett H. A., Coffey E., Dai H., He Y. D., Kidd M. J., King A. M., Meyer M. R., Slade D., Lum P. Y., et al. (2000) Functional discovery via a compendium of expression profiles. Cell 102, 109–126 [DOI] [PubMed] [Google Scholar]
- 90. Mnaimneh S., Davierwala A. P., Haynes J., Moffat J., Peng W. T., Zhang W., Yang X., Pootoolal J., Chua G., Lopez A., Trochesset M., Morse D., Krogan N. J., Hiley S. L., Li Z., et al. (2004) Exploration of essential gene functions via titratable promoter alleles. Cell 118, 31–44 [DOI] [PubMed] [Google Scholar]
- 91. Agrawal G., Fassas S. N., Xia Z. J., and Subramani S. (2016) Distinct requirements for intra-ER sorting and budding of peroxisomal membrane proteins from the ER. J. Cell Biol. 212, 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Passreiter M., Anton M., Lay D., Frank R., Harter C., Wieland F. T., Gorgas K., and Just W. W. (1998) Peroxisome biogenesis: involvement of ARF and coatomer. J. Cell Biol. 141, 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Matsuzono Y., Kinoshita N., Tamura S., Shimozawa N., Hamasaki M., Ghaedi K., Wanders R. J., Suzuki Y., Kondo N., and Fujiki Y. (1999) Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc. Natl. Acad. Sci. U.S.A. 96, 2116–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brul S., Wiemer E. A., Westerveld A., Strijland A., Wanders R. J., Schram A. W., Heymans H. S., Schutgens R. B., Van den Bosch H., and Tager J. M. (1988) Kinetics of the assembly of peroxisomes after fusion of complementary cell lines from patients with the cerebro-hepato-renal (Zellweger) syndrome and related disorders. Biochem. Biophys. Res. Commun. 152, 1083–1089 [DOI] [PubMed] [Google Scholar]
- 95. Blobel G. (1980) Intracellular protein topogenesis. Proc. Natl. Acad. Sci. U.S.A. 77, 1496–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Braakman I., and Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 97. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R., McCartney R. R., Schmidt M. C., Rachidi N., Lee S. J., Mah A. S., et al. (2005) Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 [DOI] [PubMed] [Google Scholar]
- 98. Cook J. G., Bardwell L., and Thorner J. (1997) Inhibitory and activating functions for MAPK Kss1 in the S. cerevisiae filamentous-growth signalling pathway. Nature 390, 85–88 [DOI] [PubMed] [Google Scholar]
- 99. Saleem R. A., Knoblach B., Mast F. D., Smith J. J., Boyle J., Dobson C. M., Long-O'Donnell R., Rachubinski R. A., and Aitchison J. D. (2008) Genome-wide analysis of signaling networks regulating fatty acid-induced gene expression and organelle biogenesis. J. Cell Biol. 181, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Konrad G., Schlecker T., Faulhammer F., and Mayinger P. (2002) Retention of the yeast Sac1p phosphatase in the endoplasmic reticulum causes distinct changes in cellular phosphoinositide levels and stimulates microsomal ATP transport. J. Biol. Chem. 277, 10547–10554 [DOI] [PubMed] [Google Scholar]
- 101. Stefan C. J., Manford A. G., Baird D., Yamada-Hanff J., Mao Y., and Emr S. D. (2011) Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144, 389–401 [DOI] [PubMed] [Google Scholar]
- 102. Knoblach B., Sun X., Coquelle N., Fagarasanu A., Poirier R. L., and Rachubinski R. A. (2013) An ER-peroxisome tether exerts peroxisome population control in yeast. EMBO J. 32, 2439–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Raychaudhuri S., and Prinz W. A. (2008) Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 105, 15785–15790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Voss C., Lahiri S., Young B. P., Loewen C. J., and Prinz W. A. (2012) ER-shaping proteins facilitate lipid exchange between the ER and mitochondria in S. cerevisiae. J. Cell Sci. 125, 4791–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.