

Abstract
Protein misfolding results in devastating degenerative diseases and cancer. Among the culprits involved in these illnesses are prions and prion-like proteins, which can propagate by converting normal proteins to the wrong conformation. For spongiform encephalopathies, a real prion can be transmitted among individuals. In other disorders, the bona fide prion characteristics are still under investigation. Besides inducing misfolding of native proteins, prions bind nucleic acids and other polyanions. Here, we discuss how nucleic acid binding might influence protein misfolding for both disease-related and benign, functional prions and why the line between bad and good amyloids might be more subtle than previously thought.
Keywords: cancer, neurodegenerative disease, nucleic acid, prion, protein aggregation, protein misfolding, functional amyloid, prions, p53, functional amyloids
Introduction
Several degenerative human diseases are triggered by protein misfolding. Prions are proteins that misfold and can cause normal cellular proteins to aggregate and misfold, thus amplifying the conversion process and triggering disease propagation. Additionally, true prions can propagate from cell to cell and between individuals; they are transmissible. The true transmissibility of these diseases has been fully attributed to the prion protein (PrP),2 which is involved in transmissible spongiform encephalopathies and other prion diseases. However, recent evidence suggests that other degenerative disorders may share the same molecular mechanisms (1–4). It has been shown that both amyloid and amorphous aggregates formed by proteins implicated in human degenerative diseases, such as amyloid β (Aβ), α-synuclein, and p53 in Alzheimer disease, Parkinson disease, and cancer, respectively, can trigger protein misfolding that can spread among cells and/or tissues (4–6). Although these disorders may not be absolutely transmissible, they are characterized as prion-like diseases.
In the past decade, more attention has been directed toward the topic of sustained protein aggregation leading to physiological cellular processes. The type of protein involved in this benign aggregation is known as a functional amyloid, or prion. Although it undergoes conformational changes and organized polymerization, it is an important player in many processes, such as memory consolidation (CPEB, CPEB3, and Orb2), the immune response to viral infection (mitochondrial antiviral signaling protein (MAVS)), and hormone transport (Pmel17) (7–10). Yeast prions have such characteristics, and there are several important reviews on this subject (11, 12). Amyloids in prokaryotes also have similarities to those found in eukaryotic amyloid diseases. Examples include the curli proteins (13), an Escherichia coli protein that aggregates upon nucleic acid binding (14), and the formation of biofilms in Staphylococcus aureus, in which extracellular DNA stimulates the formation of amyloid fibers (15). In this review, we will focus mainly on disease-related and functional prions or amyloids in multicellular organisms and their involvement with nucleic acids.
A common characteristic of these proteins (disease-related or functional prions) is that they bind polyanions, such as nucleic acids, glycosaminoglycans, and lipids (16–19). Nucleic acid binding has been described for PrP, α-synuclein, amyloid-β, and huntingtin, which are involved in transmissible spongiform encephalopathies, Parkinson, Alzheimer, and Huntington diseases, respectively (18, 20). Proteins involved in amyotrophic lateral sclerosis (ALS), such as superoxide dismutase, TDP43, and FUS/TLS proteins, also bind DNA and RNA sequences. These binding patterns are related to their cellular functions (21). In the case of the prion proteins, Aβ and α-synuclein, binding occurs, but no related functional impact has been discovered. Several recent studies have expanded the prion-like amyloid aggregation research to cancer because of the aggregation of p53 and other tumor suppressors (6, 22–24). This correlation between misfolding/aggregation and nucleic acid binding is not restricted to pathological events and serves some routine functions, including the persistence of long-term memory, as in the case of CPEB in Aplysia and CPEB3 in mammals (10), as well as the amyloid-like aggregation of the RNA-binding protein Rim4, involved in gametogenesis (25). Recently, it was shown that several different amyloid fibrils induce the release of extracellular traps of chromatin from neutrophils (26).
On the pathological side, there are no effective therapies against diseases involving the prion-like aggregation of proteins. An approach that focuses on protein-nucleic acid interactions, which are the key characteristic of these diseases, might reveal new therapeutic targets. Nucleic acids can have opposing effects on protein aggregation, depending on the specific cellular context, either in function or in pathology. Thus, this relationship suggests a molecular personification of Dr. Jekyll and Mr. Hyde.
PrP Requires Polyanions, Such as RNA or DNA, as Partners during Pathological Conversion
Prion diseases are attributed to the conversion of the α-helical cellular form of PrP (PrPC) into a β-sheet-rich conformation, PrPSc, which is prone to aggregation (27). The nucleic acid binding properties of PrP have been known for more than 15 years (16). Interestingly, even before the first experiments describing the aggregation of recombinant PrP upon nucleic acid binding were published (28, 29), it was hypothesized that a cofactor triggers PrP aggregation in vivo (30, 31). The initial studies on the interaction of PrP with nucleic acids began with simple in vitro assays that used different plasmids, small nucleic acid molecules, and recombinant purified PrP (28, 29, 32). In some studies, PrP aggregation and polymerization in amyloid-like fibers were observed upon binding to DNA (32). In 2001, we proposed that nucleic acids act as catalysts in the conversion of PrP, either by helping PrPC and PrPSc encounter one another or by accelerating PrP misfolding into a species prone to aggregation that could further recruit new PrP molecules (29, 33) (Fig. 1).
FIGURE 1.
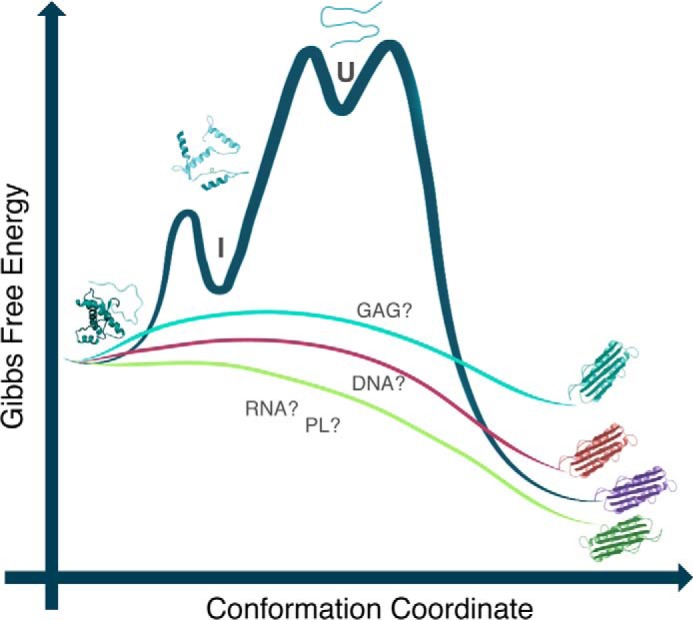
Free energy diagram representing the role of polyanions in the conversion reaction from PrPC to PrPSc. DNA, RNA, phospholipid (PL), and glycosaminoglycan (GAG) molecules may bind PrPC and lower the energy barrier that prevents its conversion to the pathogenic, misfolded form, PrPSc. PrPC binding to different cofactor molecules may change the efficiency of the conversion to PrPSc and/or result in the generation of PrPSc with varying conformations (i.e. prion strains, depending on the nature of the cofactor). Modified from Refs. 5 and 18.
Over the next few years, our group and others identified three putative DNA-binding regions along PrP: the octarepeat portion and the extreme N terminus (KKRPK) in the N-terminal region (34, 35) and the C-terminal domain (34). The interaction of PrP with RNA has also been investigated. It was shown that RNA induced the aggregation of PrP into a proteinase K-resistant species that can be toxic to cultured cells (36). RNA was also shown to be needed for the in vitro conversion of PrP into a PrPSc-like species using a modified protein misfolding cyclic amplification (PMCA) protocol (37). PMCA was originally developed to mimic prion replication under in vitro conditions using biological samples containing minimal traces of infectious PrPSc (38). The work by Suppatapone and co-workers (37) was crucial to corroborate the need for nucleic acid molecules in generating scrapie prions. Later, it became clear that other polyanions also induce the conversion of PrP in vitro, as shown by PMCA and other in vitro conversion assays (16, 19, 37, 39). The resultant PrPSc was shown to be infectious to transgenic mice, even when recombinant PrP was used as the seed (40). Although it became clear that an as yet unidentified negatively charged accessory molecule is important to drive the conversion process, this hypothesis was also strongly questioned because of the lack of knowledge of how and where this encounter would occur in vivo. A recent study describes the aggregation of PrPC into a β-sheet-rich species induced by shaking recombinant PrP under appropriate conditions without the addition of any cofactor (41). However, the infectivity of the aggregated species was not investigated in vivo. It has also been shown that phospholipids can act as cofactors in prion conversion (19, 42) (Fig. 1).
Although a specific nucleic acid consensus sequence recognized by PrP has not been defined, there are some sequences to which PrP prefers to bind (43, 44). The minimal size of the nucleic acid molecule that can convert PrP into PrPSc-like species is also arguable (18, 36, 45). The particular structure of the nucleic acid also seems to be important for the binding affinity and specificity. Recent studies have shown that both DNA and RNA quadruplex structures bind PrP with a high affinity (44, 46) and can modify the PrP conformation. It was shown that PrP can bind its own mRNA, which contains G4 motifs, causing it to form a quadruplex structure under particular conditions (47). It is becoming more and more obvious that PrP binds to structured nucleic acids, and both the nucleic acid and the protein undergo structural changes when this interaction occurs (29, 43, 44, 48, 49). Indeed, different prion strains display different interactions with RNA molecules when added as conversion cofactors under cell-free conditions (50). One might speculate that different conformational changes can take place in the PrPSc pool depending on the nucleic acid/cofactor present, and the changes can be reflected in the amplification of a specific prion strain (Fig. 1).
Although no single RNA or DNA molecule has been found to be associated with PrP infectivity in vivo, recent work provides strong evidence that small RNA sequences (27- and 55-mers) associated with brain scrapie material can trigger the conversion of the recombinant protein to a disease-causing form when incubated with recombinant PrPC (45). Although not all preparations induced prion-like disease in recipient mice, these results indicate that a sustained RNA population is present in scrapie-associated fibrils, in partial disagreement with the protein-only hypothesis (27). Recently, when two different prion strains were treated with a detergent-resistant nuclease, a significant reduction in the prion infectivity toward hypothalamic neuronal GT1 cells was observed (51). All these data indicate that nucleic acids (and other polyanions) are good candidates for catalysts of the formation of PrPSc in vivo (Fig. 1).
The binding of PrP to polyanions has also been explored for the development of anti-scrapie therapeutics. Modified nucleic acids, such as thioaptameric DNAs (small sequences), have been shown to inhibit prion conversion (52, 53). Glycosaminoglycans have been investigated as candidates for treating prion and prion-like diseases (54). Recent studies have demonstrated that the interaction of low-molecular-weight heparin (LMWHep) with the PrP protein affects the extent of PrP fibrillization and its kinetics (55, 56). The protective effects of low-molecular-weight heparin provide the groundwork for the development of therapeutic strategies based on glycosaminoglycans against prion and prion-like diseases.
Binding of Nucleic Acids to Other Proteins Involved in Prion-like Diseases
In the previous section, we discussed the ability of PrP to bind nucleic acids and other polyanions. In fact, most prion-like proteins do bind nucleic acids, but to different extents (18, 57). Along with nucleic acid binding, the proteins discussed below share other common characteristics with PrP.
It was recently demonstrated that Aβ, α-synuclein, and tau protein have strain-like properties. Thus, they can exist in different conformations that have different seeding properties and different levels of neurotoxicity and could contribute to the heterogeneity of neurodegenerative diseases (4, 58–60). There are several in vivo and in vitro studies that show the prion-like propagation of α-synuclein in Parkinson disease (PD), indicating that this protein can seed its own aggregation and transmission from cell to cell and likely between individuals (4). α-Synuclein has been found to bind both double-stranded and single-stranded DNA, which can then either trigger or prevent α-synuclein fibrillation (61, 62). This information, along with the fact that α-synuclein can localize to the nucleus, supports the role of nucleic acids in the function of α-synuclein and in the pathogenesis of PD. Recently, Prusiner et al. (3) reported that multiple system atrophy can be caused by a prion version of the α-synuclein protein. In contrast, brain extracts from patients with Parkinson disease could not transmit the disease to engineered cells or mice (3), which supports the possibility of different strains or dependence on a cofactor to explain the difference in behavior between multiple system atrophy and PD.
Tau protein also has prion-like properties, and its propagation in human tissues and in cell and animal models has been described (4, 63, 64). Another prion-like feature of tau is that it can bind dsDNA and RNA with no apparent sequence specificity (18, 65). Both Aβ and huntingtin were reported to directly interact with DNA, and this interaction changes the nucleic acid conformation (66, 67).
In particular, several proteins containing RNA recognition motifs (RRM) are involved in neurodegenerative diseases, such as ALS and frontotemporal lobar degeneration (FTLD) (68). These proteins have a modular structure that contains, in addition to the RRM, a prion-like domain (PrLD) that essentially consists of an intrinsically disordered region (IDR), also referred to as a low-complexity domain (LCD) (69). Just recently, we started to learn about the three-dimensional structure of these proteins. Because the intact protein does not form crystals, NMR is usually employed to obtain site-specific structural information.
Most of the information about these proteins containing RRMs and PrLDs was obtained because of their involvement in human pathologies, especially neurodegenerative diseases such as ALS, FTLD, Huntington disease, and tauopathies (68, 70). Proteins such as FUS, TDP-43, ataxin2, and hnRNPA1 have clear RRM and IDR segments (Fig. 2). Recently, several of these proteins were found to participate in non-membranous assemblies, such as those found in nuclear bodies (nucleoli, Cajal bodies, promyelocytic leukemia bodies, and speckles), as well as processing P bodies and stress granules in the cytoplasm (68, 71). Toretsky and Wright (70) coined the term “assemblages” for these structures, and their principal constituent proteins contain IDRs. The formation of these structures occurs by means of liquid-liquid phase separation, and RNA molecules appear to have a crucial role. Like the characters in Robert Louis Stevenson's The Strange Case of Dr Jekyll and Mr. Hyde, the association of these proteins and RNA in liquid droplets can transform one of the components from “good” to “evil” and result in fibrillogenesis and devastating diseases (Fig. 2). We know little about these molecular Jekyll and Hyde partners, and it is urgent to investigate these associations as we search for preventive and therapeutic strategies to combat neurodegenerative diseases.
FIGURE 2.

Cellular representation of functional (blue, right) and misfunctional (red, left) interactions of proteins involved in neurodegenerative disorders with nucleic acids. Three RNA-binding proteins were selected as examples: PrP, FUS/TLS, and TDP-43. FUS/TLS and TDP-43 have RRMs and glycine-rich, prion-like domains. Both FUS-TLS and TDP-43 are found to be functional in the nucleus, where they bind RNA and can associate to form stress granules with RNA and other proteins (physiological stress response). Mutations in TDP-43 and FUS/TLS can cause ALS and FTLD that relate to dysfunction or dysregulation of neuronal RNA granules and SGs in the cytoplasm. It is hypothesized that the prolonged presence of aggregated TDP-43 (mutation, aging) in the cytoplasm contributes to degeneration by interfering with mRNA homeostasis (pathological stress granules). Besides, both native and mutant FUS/TLS can form reversible (blue, right) and irreversible (red, left) hydrogels, respectively, that trap RNA-binding proteins and RNA. PrP is in general found anchored to the cell membrane (upper right side), but it also interacts with nucleic acids (both DNA and RNA) with a yet unknown function. It aggregates upon interaction with RNA, and these aggregates can be toxic to cells. RGG, Arg-Gly-Gly repeat.
Several recent studies have provided strong evidence that RNA regulates the biophysical properties of the liquid droplets formed by prion-like proteins containing RRM and LCD (72, 73). Using Whi3, a fungal protein that has an RRM and an IDR, Zhang et al. (72) showed that different mRNAs result in droplets with dissimilar characteristics. Their work also suggested that RNA prevents fibrillogenesis. In the case of FUS and hnRNPA1, which are involved in neurodegenerative diseases such as ALS, FTLD, and multisystem proteinopathy, both RNA and mutations in these proteins affect the transition into pathological aggregates (73–75).
Patel et al. (74) demonstrated that FUS forms liquid compartments at sites of DNA damage and cytoplasmic stress. The droplets were found to convert from a liquid to an aggregated state during aging (74). In this context, mutations in FUS could accelerate its conversion into amyloid-like structures (75). In this latter study, the authors showed that physiological liquid droplets and hydrogel-like structures can turn into insoluble fibrillar hydrogels (unlike conventional amyloids) when FUS is mutated (75). How different RNA sequences might affect the conversion needs to be further explored. In addition to RNA, FUS binds the polyanion poly ADP-ribose, which boosts the formation of liquid droplets, especially at the sites that require DNA repair (74).
Taking into account that several of these proteins can localize to the nucleus (functionally or abnormally), bind nucleic acids and other polyanions, and have intrinsically unfolded, prion-like or low-complexity domains, one can speculate that liquid droplet formation occurs in vivo, under either physiological or pathological conditions (Fig. 2). As represented in Fig. 2, nuclear proteins such as TDP-43 and FUS would go to the cytoplasm associating with stress granules (SGs). An imbalance in the nuclear-cytoplasmic shuttling of these proteins would lead into increased formation of SGs at pathological conditions (77).
Prion-like Conversion Is a Physiological Process Involving Nucleic Acid Binding
As pointed out above, many macromolecular assemblies lacking membrane envelopes have been described at the molecular level as involving protein-protein and protein-nucleic acid interactions. The disordered regions of RNA-binding proteins can undergo phase separation, resulting in liquid droplets (73). The structure inside these droplets converts into amyloid-like fibrils. It is likely that the tendency to form amyloid-like structures was a requirement for protein in RNP granules and was under evolutionary pressure. We anticipate that we are starting to open a Pandora's box of information concerning the cells that encase physiological assemblages, which are crucial to cell function, and the culprit structures related to several diseases (Fig. 2).
In parallel to the findings related to proteins involved in diseases, there have been several studies that describe “functional” amyloids, such as the CPEB family of proteins, which is involved in long-term memory in Aplysia and mammals, and the Rim4 protein, which is involved in gametogenesis. These proteins share the properties of amyloid aggregation and RNA binding, and they also act as translational effectors. One of the main differences between functional and pathological amyloids may be the prompt reversibility of the functional prions.
CPEB was first described to have prion-like behavior in the marine snail Aplysia (7). The persistence of long-term memory and synaptic plasticity are regulated by a transition from the soluble to the aggregated state. CPEB and the Drosophila homolog Orb2 act as translation regulators. In the soluble state, their binding represses the translation of mRNA. Upon signaling, the protein aggregates; this results in the polyadenylation of target RNAs, which leads to the translation into proteins involved in synaptic plasticity and memory storage (7, 76). Recently, Kandel and co-workers (77) demonstrated that the prion-like mechanism also operates in mammals. They provided strong evidence that CPEB3 aggregation is a crucial mediator of consolidation and the persistence of memory in mice. Other important factors that help to keep the protein soluble, such as SUMOylation, also participate (78). Upon neuronal stimulation, CPEB3 is deSUMOylated and undergoes aggregation.
More recently, Si and co-workers (79) observed that monomers of Drosophila Orb2 repress translation and remove the mRNA poly(a) tails, whereas the amyloid-like oligomeric form activates translation, elongates the poly(A) tails, and converts the monomers. In another study, they found that Orb2 aggregation is similar to the aggregation of other proteins related to diseases (80). For example, they found the formation of toxic amyloid oligomers. The main difference is that the toxic intermediates of Orb2 are transient. Interestingly, an anti-amyloidogenic peptide interferes with long-term memory in Drosophila. Thus, the division between bad and good amyloids might be more subtle than previously thought (Fig. 2).
Another remarkable recent example of a “Dr. Jekyll” amyloid is Rim4. The prion-like aggregate of Rim4 acts as a translation repressor regulating gametogenesis in yeast (25) in a similar way to CPEB. In a beautifully controlled process, when Rim4 adopts the aggregated state, translation is repressed, and the degradation of Rim4 amyloid aggregates during meiosis II releases this repression. Starvation leads to the conversion of monomeric Rim4 into amyloid-like aggregates, which represses translation. Rim4 amyloid aggregates are dissociated at the onset of meiosis II, resulting in translation. This amyloid-like aggregation of Rim4 during gametogenesis is conserved (25).
Prion-like Aggregation of Tumor Suppressors in Cancer
“Mr. Hyde” amyloids are also involved in other diseases in addition to neurodegeneration. The most intriguing example is the amyloid-like aggregation of the tumor suppressor p53, a DNA-binding protein (6). p53 mutations are the most common mutations in cancers and occur in more than 50% of all tumors, most of them in the DNA-binding domain of the protein (81). p53, a tetrameric protein, is the main controller of cell homeostasis and DNA stability. p53 works as a transcription factor, binding to specific sequences and inducing the transcription of genes involved in cell cycle control, apoptosis, and senescence, among other processes (81). The p53 mutations cause a significant decrease in structural stability and/or modify transcriptional activity (82). In addition to the loss of function caused by mutations, gain-of-function effects, such as increased migration, invasion, and metastasis, are also observed (81). Another important feature related to p53 mutations is the dominant-negative effect exerted by mutant p53 on wild-type p53, which originates from different alleles in the same cell (83). Our group has previously demonstrated that this phenomenon appears to be related to a prion-like effect exerted by mutp53 on WT p53 (22, 23, 84, 85).
The groundwork for the dominant-negative effect caused by the mutation is that amyloid-like mutant p53 converts WT p53 into a more aggregated species (Fig. 3). The co-aggregation of mutant p53 with other proteins may also lead to the gain-of-function phenotype. Mutant p53 aggregation appears to occur with its paralogs p63 and p73 (6, 24, 86). Amyloid aggregates of mutant p53 have been discovered in breast cancer (23, 84), malignant skin tumors (87), and ovarian cancer (88). Although the three functional domains of p53 have the potential to form amyloid-like aggregates (22, 89, 90), the DNA-binding domain has the highest tendency to form amyloid oligomers and fibrils.
FIGURE 3.

Free energy landscape showing native, misfolded, amyloid oligomers and fibrils of p53 along the folding funnel. Aggregation of mutant p53 is an important therapeutic target. Researchers have been trying to use peptides, aptameric nucleic acids, and small molecules (SMs) to prevent the formation of aggregates, responsible for the dominant-negative effects and gain-of-function effects of mutant p53. Modified from Refs. 5 and 6.
Whether the prion-like effects of mutant p53 could be extended to other cells is another open question. A recent study showed that aggregates of p53C are internalized by cells and result in co-aggregation with the endogenous p53 protein (91). The prion-like behavior of oncogenic p53 mutants provides an explanation for its dominant-negative and gain-of-function properties, including the potential to cause metastasis. The blockage of p53 aggregation into oligomeric and fibrillar amyloids has been considered a promising target for therapeutic intervention in cancer (6, 23, 88, 92). In silico analyses corroborate the experimental data that p53 and its paralogous proteins (p63 and p73) have several hot spots for aggregation, similar to PrP and other proteins involved in prion-like diseases (85). Intriguingly, p63 and p73 have a lower propensity than p53 to aggregate when tested in vitro (86).
Another piece of evidence for the possibility of spreading of the misfolded conformation from one cell to another is the fact that different aggregates of p53 are toxic to different cell lines (22, 23, 87). Toxicity is typical of amyloid oligomers. As discussed above for Orb2, the toxic effect might depend on the kinetics of the formation of the intermediate and its evolution into fibrils (80). The toxicity of mutant p53 oligomers would not kill the cell (similar to Orb2) but would perturb protein homeostasis, likely resulting in the release of misfolded proteins, which could interact with proteins from different compartments in the cell or even move into other cells in the tissue. The hypothesis of cell-to-cell transmission of mutant p53 oligomers still demands additional experimental evidence.
Gain-of-function p53 Mutants and Anti-amyloid Therapeutic Intervention
In addition to providing an explanation for the dominant-negative effect, the prion-like behavior of oncogenic p53 mutants may explain the gain-of-function properties of several mutations (6, 23). Different types of mechanisms operate in the gain-of-function activity of p53 mutants (81). The prion-like aggregation of mutant p53 with WT p53, p63, p73, and other transcription factors is likely the most important one (6, 24). The co-localization of p53 with small amyloid oligomers in breast cancer tissues (23, 84) and in basal cell carcinoma (87) underpins the prion-like aggregation hypothesis for the gain of function of some p53 mutants.
Quite intriguingly, p53 aggregation in tumor cells, such as those in breast cancer, appears predominantly in the form of amyloid oligomers (23, 84). In neurodegenerative diseases, there is evidence that oligomers affect cellular homeostasis more than fibrils (93). The cell likely has mechanisms to attenuate the toxic effect of misfolded oligomers. However, it seems that with age, the oligomers win the battle, causing cellular death and spreading to other cells in a prion-like fashion. In cancer, mutant p53 oligomers could confer this “Dr. Jekyll and Mr. Hyde” effect to the cell: the oligomers would kill some cells but would guarantee the immortality of the surviving cells with the loss of tumor suppression.
The amyloid aggregation of mutant p53 has emerged as a new therapeutic treatment for cancer (6). The blockage of cell proliferation due to a dominant-negative effect or a gain of function caused by the prion-like behavior of mutant p53 appears to be a novel target (Fig. 3). Most therapeutic strategies have focused on identifying small molecules that can reactivate mutant p53. For example, PRIMA-1 is converted to compounds that form adducts with the thiols in mutant p53, inducing apoptosis in tumor cells (94). PRIMA-1 has shown positive results in stage-I/II clinical trials (95). We have recently found that the main molecular mechanism of PRIMA-1 is to inhibit the prion-like aggregation of mutant p53 (96). In the case of the Y220C p53 mutant, compounds that bind to the cavity where the mutation is located are good candidates for blocking aggregation (97). A protein assembly modulator (CLR01) was recently demonstrated to induce the rapid formation of p53 aggregates of intermediate sizes and inhibit additional p53 aggregation, decreasing the cytotoxicity of the amyloid aggregates (98).
Another potential therapeutic strategy is the use of aptameric nucleic acids to prevent aggregation and prion-like conversion (92). Our group showed that small, cognate double-stranded DNA stabilizes both the p53 DNA binding domain and the full-length p53, preventing amyloid formation (Fig. 3). Therefore, such DNA sequences might be useful as part of a new approach to cancer therapy (92).
The design of specific peptides to interact with the protein segment, which has a propensity to aggregate, is another strategy that has been explored by Eisenberg and co-workers (88). They developed a peptide (ReACp53) that binds to the amyloidogenic domain of mutant p53, preventing its amyloid aggregation. The peptide was able to rescue p53 function in high-grade serous ovarian carcinomas with p53 mutations. Fig. 3 summarizes the different strategies that can be used to prevent the aggregation of p53 and promote important anti-tumoral effects.
Conclusions and Perspectives
We are witnessing a paradigm shift in the protein folding field. Proteins that were long believed to function in solution appear to acquire different states: soluble, amyloid precursor oligomers, liquid droplets, hydrogels, and fibrils. We know little about the structures that occur between the soluble and fibrillar states, especially oligomers. It is generally believed that oligomers are the villains in toxic gain-of-function mutations in neurodegenerative diseases. In their functional states, similar multimeric structures are now thought of as “good” amyloids (Dr. Jekyll forms), such as the CPEB family of proteins and Rim4. The distinction between good and evil might be more subtle, and nucleic acids are key players in the conversion between the two forms. It is quite likely that the nature of the interaction between a given nucleic acid and an amyloid precursor will determine the final outcome. The appropriate liquid-liquid phase separation inside the nucleus and the cytoplasm is the key to maintaining homeostasis in the cell. The deterioration of several regulatory processes occurs during aging and eventually ensues in most of the neurodegenerative diseases. However, it is likely present in other situations as well, such as chronic traumatic encephalopathy, which is caused by severe shock to the head and is often found in players of American football and other sports (ice hockey, boxing, etc.) (99).
In the case of cancer, p53 aggregation appears to sustain the proliferative nature of tumors (6), whereas aggregation in neurodegenerative diseases leads to cell death, although the proteins appear to share the same mechanisms for prion-like conversion. Interestingly, a recent study demonstrated that p53 gain-of-function mutants bind to chromatin regulatory genes by interacting with the transcription factor ETS2, resulting in histone methylation and acetylation (100). Likely, this occurs by formation of heteroligomers containing mutp53 and ETS2 with altered DNA binding activity.
In conclusion, the prions and prion-like proteins have a Jekyll and Hyde behavior that is highly dependent on the interaction with cellular partners, especially with nucleic acids
Acknowledgment
We thank Dr. Martha M. Sorenson for critical reading of the manuscript.
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq awards and the Instituto Nacional de Ciência e Tecnologia (INCT) Program), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The authors declare that they have no conflicts of interest with the contents of this article.
- PrP
- prion protein
- Aβ
- amyloid-β peptide
- ALS
- amyotrophic lateral sclerosis
- PD
- Parkinson disease
- FTLD
- frontotemporal dementia
- CPEB
- cytoplasmic polyadenylation element-binding protein
- FUS/TLS
- fused in sarcoma/translocated in liposarcoma protein
- PMCA
- protein misfolding cyclic amplification
- TDP-43
- transactive response DNA binding protein 43 kDa
- IDR
- intrinsically disordered region
- RRM
- RNA recognition motifs
- SG
- stress granule
- RNP
- ribonucleoprotein.
References
- 1. Jaunmuktane Z., Mead S., Ellis M., Wadsworth J. D., Nicoll A. J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A. S., Rudge P., Collinge J., and Brandner S. (2015) Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 [DOI] [PubMed] [Google Scholar]
- 2. Morales R., Bravo-Alegria J., Duran-Aniotz C., and Soto C. (2015) Titration of biologically active amyloid-β seeds in a transgenic mouse model of Alzheimer's disease. Sci. Rep. 5, 9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prusiner S. B., Woerman A. L., Mordes D. A., Watts J. C., Rampersaud R., Berry D. B., Patel S., Oehler A., Lowe J. K., Kravitz S. N., Geschwind D. H., Glidden D. V., Halliday G. M., Middleton L. T., Gentleman S. M., et al. (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brettschneider J., Del Tredici K., Lee V. M.-Y. Y., and Trojanowski J. Q. (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silva J. L., Vieira T. C. R. G., Gomes M. P. B., Ano Bom A. P., Lima L. M. T. R., Freitas M. S., Ishimaru D., Cordeiro Y., and Foguel D. (2010) Ligand binding and hydration in protein misfolding: insights from studies of prion and p53 tumor suppressor proteins. Acc. Chem. Res. 43, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silva J. L., De Moura Gallo C. V., Costa D. C. F., and Rangel L. P. (2014) Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 39, 260–267 [DOI] [PubMed] [Google Scholar]
- 7. Si K., Lindquist S., and Kandel E. R. (2003) A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell 115, 879–891 [DOI] [PubMed] [Google Scholar]
- 8. Fowler D. M., Koulov A. V., Alory-Jost C., Marks M. S., Balch W. E., and Kelly J. W. (2006) Functional amyloid formation within mammalian tissue. PLoS Biol. 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hou F., Sun L., Zheng H., Skaug B., Jiang Q. X., and Chen Z. J. (2011) MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Si K. (2015) Prions: what are they good for? Annu. Rev. Cell Dev. Biol. 31, 149–169 [DOI] [PubMed] [Google Scholar]
- 11. Wickner R. B., Shewmaker F. P., Bateman D. A., Edskes H. K., Gorkovskiy A., Dayani Y., and Bezsonov E. E. (2015) Yeast prions: structure, biology, and prion-handling systems. Microbiol. Mol. Biol. Rev. 79, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liebman S. W., and Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., and Hultgren S. J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giraldo R. (2007) Defined DNA sequences promote the assembly of a bacterial protein into distinct amyloid nanostructures. Proc. Natl. Acad. Sci. U.S.A. 104, 17388–17393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz K., Ganesan M., Payne D. E., Solomon M. J., and Boles B. R. (2016) Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol. Microbiol. 99, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Silva J. L., Lima L. M. T. R., Foguel D., and Cordeiro Y. (2008) Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem. Sci. 33, 132–140 [DOI] [PubMed] [Google Scholar]
- 17. Silva J. L., Gomes M. P., Vieira T. C., and Cordeiro Y. (2010) PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. (Landmark Ed.) 15, 132–150 [DOI] [PubMed] [Google Scholar]
- 18. Cordeiro Y., Macedo B., Silva J. L., and Gomes M. P. B. (2014) Pathological implications of nucleic acid interactions with proteins associated with neurodegenerative diseases. Biophys. Rev. 6, 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., and Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu C., and Zhang Y. (2011) Nucleic acid-mediated protein aggregation and assembly. Adv. Protein Chem. Struct. Biol. 84, 1–40 [DOI] [PubMed] [Google Scholar]
- 21. Colombrita C., Onesto E., Megiorni F., Pizzuti A., Baralle F. E., Buratti E., Silani V., and Ratti A. (2012) TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 287, 15635–15647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishimaru D., Andrade L. R., Teixeira L. S. P., Quesado P. A., Maiolino L. M., Lopez P. M., Cordeiro Y., Costa L. T., Heckl W. M., Weissmüller G., Foguel D., and Silva J. L. (2003) Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 42, 9022–9027 [DOI] [PubMed] [Google Scholar]
- 23. Ano Bom A. P. D., Rangel L. P., Costa D. C. F., de Oliveira G. A. P., Sanches D., Braga C. A., Gava L. M., Ramos C. H. I., Cepeda A. O. T., Stumbo A. C., De Moura Gallo C. V., Cordeiro Y., and Silva J. L. (2012) Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J. Biol. Chem. 287, 28152–28162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu J., Reumers J., Couceiro J. R., De Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.-C., Lambrechts D., Suh Y.-A., Rousseau F., and Schymkowitz J. (2011) Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295 [DOI] [PubMed] [Google Scholar]
- 25. Berchowitz L. E., Kabachinski G., Walker M. R., Carlile T. M., Gilbert W. V., Schwartz T. U., and Amon A. (2015) Regulated formation of an amyloid-like translational repressor governs gametogenesis. Cell 163, 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Azevedo E. P. C., Guimarães-Costa A. B., Torezani G. S., Braga C. A., Palhano F. L., Kelly J. W., Saraiva E. M., and Foguel D. (2012) Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J. Biol. Chem. 287, 37206–37218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nandi P. K. (1997) Interaction of prion peptide HuPrP106–126 with nucleic acid: brief report. Arch. Virol. 142, 2537–2545 [DOI] [PubMed] [Google Scholar]
- 29. Cordeiro Y., Machado F., Juliano L., Juliano M. A., Brentani R. R., Foguel D., and Silva J. L. (2001) DNA converts cellular prion protein into the β-sheet conformation and inhibits prion peptide aggregation. J. Biol. Chem. 276, 49400–49409 [DOI] [PubMed] [Google Scholar]
- 30. Weissmann C. (1991) A “unified theory” of prion propagation. Nature 352, 679–683 [DOI] [PubMed] [Google Scholar]
- 31. Radulescu R. T., and Korth C. (1996) Prion function and dysfunction: a structure-based scenario. Med. Hypotheses 46, 225–228 [DOI] [PubMed] [Google Scholar]
- 32. Nandi P. K. (1998) Polymerization of human prion peptide HuPrP 106–126 to amyloid in nucleic acid solution. Arch. Virol. 143, 1251–1263 [DOI] [PubMed] [Google Scholar]
- 33. Cordeiro Y., and Silva J. L. (2005) The hypothesis of the catalytic action of nucleic acid on the conversion of prion protein. Protein Pept. Lett. 12, 251–255 [DOI] [PubMed] [Google Scholar]
- 34. Lima L. M. T. R., Cordeiro Y., Tinoco L. W., Marques A. F., Oliveira C. L. P., Sampath S., Kodali R., Choi G., Foguel D., Torriani I., Caughey B., and Silva J. L. (2006) Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 45, 9180–9187 [DOI] [PubMed] [Google Scholar]
- 35. Yin S., Fan X., Yu S., Li C., and Sy M. S. (2008) Binding of recombinant but not endogenous prion protein to DNA causes DNA internalization and expression in mammalian cells. J. Biol. Chem. 283, 25446–25454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gomes M. P. B., Millen T. A., Ferreira P. S., Cunha E Silva N. L., Vieira T. C. R. G., Almeida M. S., Silva J. L., and Cordeiro Y. (2008) Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J. Biol. Chem. 283, 19616–19625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deleault N. R., Harris B. T., Rees J. R., and Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saborio G. P., Permanne B., and Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 39. Abid K., Morales R., and Soto C. (2010) Cellular factors implicated in prion replication. FEBS Lett. 584, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang F., Wang X., Yuan C.-G., and Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ladner-Keay C. L., Griffith B. J., and Wishart D. S. (2014) Shaking alone induces de novo conversion of recombinant prion proteins to β-sheet rich oligomers and fibrils. PLoS ONE 9, e98753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Srivastava S., and Baskakov I. V (2015) Contrasting effects of two lipid cofactors of prion replication on the conformation of the prion protein. PLoS ONE 10, e0130283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Macedo B., Millen T. A., Braga C. A. C. A., Gomes M. P. B., Ferreira P. S., Kraineva J., Winter R., Silva J. L., and Cordeiro Y. (2012) Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 51, 5402–5413 [DOI] [PubMed] [Google Scholar]
- 44. Cavaliere P., Pagano B., Granata V., Prigent S., Rezaei H., Giancola C., and Zagari A. (2013) Cross-talk between prion protein and quadruplex-forming nucleic acids: a dynamic complex formation. Nucleic Acids Res. 41, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simoneau S., Thomzig A., Ruchoux M.-M., Vignier N., Daus M. L., Poleggi A., Lebon P., Freire S., Durand V., Graziano S., Galeno R., Cardone F., Comoy E., Pocchiari M., Beekes M., et al. (2015) Synthetic scrapie infectivity: interaction between recombinant PrP and scrapie brain-derived RNA. Virulence 6, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mashima T., Matsugami A., Nishikawa F., Nishikawa S., and Katahira M. (2009) Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 37, 6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olsthoorn R. C. L. (2014) G-quadruplexes within prion mRNA: the missing link in prion disease? Nucleic Acids Res. 42, 9327–9333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bera A., and Nandi P. K. (2007) Biological polyamines inhibit nucleic-acid-induced polymerisation of prion protein. Arch. Virol. 152, 655–668 [DOI] [PubMed] [Google Scholar]
- 49. Gomes M. P. B., Vieira T. C. R. G., Cordeiro Y., and Silva J. L. (2012) The role of RNA in mammalian prion protein conversion. Wiley Interdiscip. Rev. RNA. 3, 415–428 [DOI] [PubMed] [Google Scholar]
- 50. Saá P., Sferrazza G. F., Ottenberg G., Oelschlegel A. M., Dorsey K., and Lasmézas C. I. (2012) Strain-specific role of RNAs in prion replication. J. Virol. 86, 10494–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Botsios S., and Manuelidis L. (2016) CJD and scrapie require agent-associated nucleic acids for infection. J. Cell Biochem. 117, 1947–1958 [DOI] [PubMed] [Google Scholar]
- 52. Kocisko D. A., Vaillant A., Lee K. S., Arnold K. M., Bertholet N., Race R. E., Olsen E. A., Juteau J. M., and Caughey B. (2006) Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 50, 1034–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. King D. J., Safar J. G., Legname G., and Prusiner S. B. (2007) Thioaptamer interactions with prion proteins: sequence-specific and non-specific binding sites. J. Mol. Biol. 369, 1001–1014 [DOI] [PubMed] [Google Scholar]
- 54. Rainov N. G., Tsuboi Y., Krolak-Salmon P., Vighetto A., and Doh-Ura K. (2007) Experimental treatments for human transmissible spongiform encephalopathies: is there a role for pentosan polysulfate? Expert Opin. Biol. Ther. 7, 713–726 [DOI] [PubMed] [Google Scholar]
- 55. Vieira T. C. R. G., Reynaldo D. P., Gomes M. P. B., Almeida M. S., Cordeiro Y., and Silva J. L. (2011) Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of RNA-resistant species. J. Am. Chem. Soc. 133, 334–344 [DOI] [PubMed] [Google Scholar]
- 56. Vieira T. C. R. G., Cordeiro Y., Caughey B., and Silva J. L. (2014) Heparin binding confers prion stability and impairs its aggregation. FASEB J. 28, 2667–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vanderweyde T., Youmans K., Liu-Yesucevitz L., and Wolozin B. (2013) Role of stress granules and RNA-binding proteins in neurodegeneration: a mini-review. Gerontology 59, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guo J. L., and Lee V. M. Y. (2011) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Watts J. C., Condello C., Stöhr J., Oehler A., Lee J., DeArmond S. J., Lannfelt L., Ingelsson M., Giles K., and Prusiner S. B. (2014) Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc. Natl. Acad. Sci. U.S.A. 111, 10323–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cherny D., Hoyer W., Subramaniam V., and Jovin T. M. (2004) Double-stranded DNA stimulates the fibrillation of α-synuclein in vitro and is associated with the mature fibrils: an electron microscopy study. J. Mol. Biol. 344, 929–938 [DOI] [PubMed] [Google Scholar]
- 62. Hegde M. L., Vasudevaraju P., and Rao K. J. (2010) DNA induced folding/fibrillation of α-synuclein: new insights in Parkinson's disease. Front. Biosci. (Landmark Ed.) 15, 418–436 [DOI] [PubMed] [Google Scholar]
- 63. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., and Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kfoury N., Holmes B. B., Jiang H., Holtzman D. M., and Diamond M. I. (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hua Q., and He R. Q. (2003) Tau could protect DNA double helix structure. Biochim. Biophys. Acta 1645, 205–211 [DOI] [PubMed] [Google Scholar]
- 66. Benn C. L., Sun T., Sadri-Vakili G., McFarland K. N., DiRocco D. P., Yohrling G. J., Clark T. W., Bouzou B., and Cha J. H. (2008) Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 28, 10720–10733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yu H., Ren J., and Qu X. (2007) Time-dependent DNA condensation induced by amyloid β-peptide. Biophys. J. 92, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li Y. R., King O. D., Shorter J., and Gitler A. D. (2013) Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xiang S., Kato M., Wu L. C., Lin Y., Ding M., Zhang Y., Yu Y., and McKnight S. L. (2015) The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell 163, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Toretsky J. A., and Wright P. E. (2014) Assemblages: functional units formed by cellular phase separation. J. Cell Biol. 206, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Meng F., Na I., Kurgan L., and Uversky V. N. (2016) Compartmentalization and functionality of nuclear disorder: intrinsic disorder and protein-protein interactions in intra-nuclear compartments. Int. J. Mol. Sci. 17, E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang H., Elbaum-Garfinkle S., Langdon E. M., Taylor N., Occhipinti P., Bridges A. A., Brangwynne C. P., and Gladfelter A. S. (2015) RNA controls PolyQ protein phase transitions. Mol. Cell 60, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lin Y., Protter D. S. W., Rosen M. K., Parker R., Lin Y., Protter D. S. W., Rosen M. K., and Parker R. (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins article formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Patel A., Lee H. O., Jawerth L., Maharana S., Jahnel M., Hein M. Y., Stoynov S., Mahamid J., Saha S., Franzmann T. M., Pozniakovski A., Poser I., Maghelli N., Royer L. A., Weigert M., et al. (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 [DOI] [PubMed] [Google Scholar]
- 75. Murakami T., Qamar S., Lin J. Q., Schierle G. S. K., Rees E., Miyashita A., Costa A. R., Dodd R. B., Chan F. T. S., Michel C. H., Kronenberg-Versteeg D., Li Y., Yang S. P., Wakutani Y., Meadows W., et al. (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Si K., Choi Y. B., White-Grindley E., Majumdar A., and Kandel E. R. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140, 421–435 [DOI] [PubMed] [Google Scholar]
- 77. Fioriti L., Myers C., Huang Y. Y., Li X., Stephan J. S., Trifilieff P., Colnaghi L., Kosmidis S., Drisaldi B., Pavlopoulos E., and Kandel E. R. (2015) The persistence of hippocampal-based memory requires protein synthesis mediated by the prion-like protein CPEB3. Neuron 86, 1433–1448 [DOI] [PubMed] [Google Scholar]
- 78. Drisaldi B., Colnaghi L., Fioriti L., Rao N., Myers C., Snyder A. M., Metzger D. J., Tarasoff J., Konstantinov E., Fraser P. E., Manley J. L., and Kandel E. R. (2015) SUMOylation is an inhibitory constraint that regulates the prion-like aggregation and activity of CPEB3. Cell Rep. 11, 1694–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Khan M. R., Li L., Pérez-Sánchez C., Saraf A., Florens L., Slaughter B. D., Unruh J. R., and Si K. (2015) Amyloidogenic oligomerization transforms Drosophila Orb2 from a translation repressor to an activator. Cell 163, 1468–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hervás R., Li L., Majumdar A., Fernández-Ramírez M. del C., Unruh J. R., Slaughter B. D., Galera-Prat A., Santana E., Suzuki M., Nagai Y., Bruix M., Casas-Tintó S., Menéndez M., Laurents D. V., Si K., and Carrión-Vázquez M. (2016) Molecular basis of Orb2 amyloidogenesis and blockade of memory consolidation. PLOS Biol. 14, e1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Muller P. A., and Vousden K. H. (2013) p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 [DOI] [PubMed] [Google Scholar]
- 82. Joerger A. C., Ang H. C., and Fersht A. R. (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U.S.A. 103, 15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Freed-Pastor W. A., and Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Levy C. B., Stumbo A. C., Ano Bom A. P. D., Portari E. A., Cordeiro Y., Silva J. L., and De Moura-Gallo C. V. (2011) Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 43, 60–64 [DOI] [PubMed] [Google Scholar]
- 85. Rangel L. P., Costa D. C. F., Vieira T. C. R. G., and Silva J. L. (2014) The aggregation of mutant p53 produces prion-like properties in cancer. Prion. 8, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang G., and Fersht A. R. (2015) Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. U.S.A. 112, 2443–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lasagna-Reeves C. A., Clos A. L., Castillo-Carranza D., Sengupta U., Guerrero-Muñoz M., Kelly B., Wagner R., and Kayed R. (2013) Dual role of p53 amyloid formation in cancer: loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 430, 963–968 [DOI] [PubMed] [Google Scholar]
- 88. Soragni A., Janzen D. M., Johnson L. M., Lindgren A. G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A. B., Lu J., Jiang L., Faull K. F., Pellegrini M., Memarzadeh S., and Eisenberg D. S. (2016) A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee A. S., Galea C., DiGiammarino E. L., Jun B., Murti G., Ribeiro R. C., Zambetti G., Schultz C. P., and Kriwacki R. W. (2003) Reversible amyloid formation by the p53 tetramerization domain and a cancer-associated mutant. J. Mol. Biol. 327, 699–709 [DOI] [PubMed] [Google Scholar]
- 90. Rigacci S., Bucciantini M., Relini A., Pesce A., Gliozzi A., Berti A., and Stefani M. (2008) The (1–63) region of the p53 transactivation domain aggregates in vitro into cytotoxic amyloid assemblies. Biophys. J. 94, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Forget K. J., Tremblay G., and Roucou X. (2013) p53 aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS ONE 8, e69242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ishimaru D., Ano Bom A. P. D., Lima L. M. T. R., Quesado P. A., Oyama M. F. C., de Moura Gallo C. V., Cordeiro Y., and Silva J. L. (2009) Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 48, 6126–6135 [DOI] [PubMed] [Google Scholar]
- 93. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 94. Lambert J. M. R., Gorzov P., Veprintsev D. B., Söderqvist M., Segerbäck D., Bergman J., Fersht A. R., Hainaut P., Wiman K. G., and Bykov V. J. N. (2009) PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 [DOI] [PubMed] [Google Scholar]
- 95. Bykov V. J. N., and Wiman K. G. (2014) Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 588, 2622–2627 [DOI] [PubMed] [Google Scholar]
- 96. Rangel L., da Costa C., da Costa D., and da Silva J. (2014) p53 aggregation and prionoid effect and the modulation of this process by PRIMA-1 and its active metabolite, 2-methylene-3-quinuclidinone hydrate (754.4). FASEB J. 28, Supplement 754.4 [Google Scholar]
- 97. Wilcken R., Wang G., Boeckler F. M., and Fersht A. R. (2012) Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 109, 13584–13589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Herzog G., Shmueli M. D., Levy L., Engel L., Gazit E., Klärner F. G., Schrader T., Bitan G., and Segal D. (2015) The Lys-specific molecular tweezer, CLR01, modulates aggregation of the mutant p53 DNA binding domain and inhibits its toxicity. Biochemistry 54, 3729–3738 [DOI] [PubMed] [Google Scholar]
- 99. McKee A. C., Stein T. D., Kiernan P. T., and Alvarez V. E. (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 25, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhu J., Sammons M. A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B. W., Shilatifard A., Huang J., Hua X., Arrowsmith C. H., and Berger S. L. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]