

Abstract
Glycyl radical enzymes (GREs) represent a diverse superfamily of enzymes that utilize a radical mechanism to catalyze difficult, but often essential, chemical reactions. In this work we present the first biochemical and structural data for a GRE-type diol dehydratase from the organism Roseburia inulinivorans (RiDD). Despite high sequence (48% identity) and structural similarity to the GRE-type glycerol dehydratase from Clostridium butyricum, we demonstrate that the RiDD is in fact a diol dehydratase. In addition, the RiDD will utilize both (S)-1,2-propanediol and (R)-1,2-propanediol as a substrate, with an observed preference for the S enantiomer. Based on the new structural information we developed and successfully tested a hypothesis that explains the functional differences we observe.
Keywords: chemical biology, enzyme structure, intestinal metabolism, metabolic engineering, radical, diol dehydratase, glycerol dehydratase, glycyl radical enzyme, radical dehydration
Introduction
In contrast to the well characterized B12-dependent dehydratases (1, 2), only one glycyl radical enzyme (GRE)2 or GRE-type dehydratase has been characterized to date (3). Although pyruvate formate lyase (PFL) and anaerobic ribonucleotide reductase are among the most comprehensively studied GREs (4–9), recent work provides evidence that the catalytic diversity of GREs is considerably greater than originally anticipated (10). It is now recognized that GREs can catalyze the formation of C-C bonds (benzyl succinate synthase) (11), C-C bond cleavage (PFL and 4-hydroxyphenylacetate decarboxylase) (5, 12), anaerobic ribonucleotide reduction (8), dehydration reactions (glycerol dehydratase) (3), and C-N bond cleavage (choline trimethylamine-lyase) (13–15). Taken together, these observations underscore a superfamily of enzymes that utilize a conserved structural core to perform vastly different radical-catalyzed reactions. Many of these reactions are important to human health or represent difficult chemical conversions that may be significant to the development of future biocatalysts.
At the present time the only structural information available for any GRE-type dehydratase is that of the glycerol dehydratase from Clostridium butyricum (CbGD) (3, 16). The presence of a homologous 1,2-propandiol dehydratase in Roseburia inulinivorans was proposed based on genomic and metabolic analyses of the organism (17). However, the enzyme was never isolated, and only propanol production was observed when R. inulinivorans was cultured under anaerobic conditions (17). In addition to the presence of the proposed GRE-type dehydratase gene, the operon containing the diol dehydratase (WP_007885173) also contained another gene that was consistent with that of an “activating enzyme” (ABC25540). These observations are significant because all of the GREs that have been investigated to date are expressed in an inactive form that is post-translationally activated by an activating enzyme (AE). The generally accepted mechanism for the activation of all GREs involves a “radical SAM” activating enzyme that is specific to the GRE (10). The activation process that is catalyzed by the AE involves a uniquely coordinated [4Fe-4S] cluster and S-adenosylmethionine (SAM) as a co-substrate. The [4Fe-4S] cluster (formerly in the 1+ oxidation state) catalyzes the reductive cleavage of SAM to generate a highly reactive 5′-deoxyadenosyl radical (5′-dA·) that is positioned to facilitate the abstraction of a hydrogen atom from a conserved glycine residue on the GRE (10). The activation reaction and subsequent dehydration investigated herein are highly sensitive to molecular oxygen and must be performed under strictly anaerobic conditions. In vitro, the catalytic [4Fe-4S] can be reduced from the 2+ to the 1+ oxidation state using a biological reducing system, such as a NADPH flavodoxin/oxidoreductase and flavodoxin system (18). In addition, chemical reductants such as sodium dithionite or a light-driven reducing system are also effective electron donors (19, 20). The activation process, or generation of the catalytic glycyl radical, is further complicated by recent observations that demonstrate the substrate for the GRE accelerates the activation rate (21). Regardless of the electron source, once the glycyl radical is generated, all GREs will catalyze their primary metabolic reaction, regenerating the radical with each turnover. Precisely how or when GREs are inactivated in vivo remains an outstanding question in the field.
In this work we have expressed and purified the predicted diol dehydratase from R. inulinivorans (RiDD) as well as the RiDD activating enzyme (RiDD-AE) (homologously expressed in Escherichia coli). In addition to assay data, we have determined the crystal structures for the ethanediol- and propanediol-bound RiDD. Structural alignment of the crystallographic models with our previously published models for the CbGD reveals a similar overall-fold and a highly conserved active site structure. However, subtle variations in the active site amino acid residues provide a viable explanation for the observed functional differences. This hypothesis is tested through site directed mutagenesis herein.
Results
Overall Structure of the RiDD
All of the GRE structures published to date represent a model of the inactive enzyme. This is not surprising considering that maintaining the glycyl radical during crystal growth is simply not feasible. Despite this technical detail, the crystal structures for GREs have provided significant insight into enzyme mechanism (3, 22–28). Numerous computational predictions have also been made based on this structural information (29–31). In this work we were able to crystallize and model the RiDD with ethanediol or 1,2-propanediol bound in the active site (Table 1).
TABLE 1.
Data collection and refinement statistics
| RiDD |
||
|---|---|---|
| Ethanediol-bound | 1,2-Propanediol-bound | |
| PDB ID code | 5I2A | 5I2G |
| Space group | C2221 | C2 |
| Wavelength | 0.98 | 0.98 |
| Resolution range (Å) | 50.0–2.1 | 50.0–2.4 |
| Unit cell dimensions (Å) | a = 138.2, b = 183.2, c = 228.3 | a = 204.3, b = 83.2, c = 138.2 |
| Outer shell | 2.18-2.1 | 2.45-2.4 |
| Unique observations | 166,139 | 74,266 |
| Completeness (%) | 98.9 (90.3)a | 98.8 (88.9) |
| Rmerge (%)b | 0.06 (0.29) | 0.12 (0.33) |
| Redundancy | 6.9 (3.4) | 7.2 (3.5) |
| I/σ | 22.2 (2.8) | 22.1 (4.9) |
| Protein atoms | 13,127 | 12,512 |
| Solvent atoms | 1,153 | 370 |
| Rcryst (%) | 15.2 | 14.5 |
| Rfree (%) | 19.0 | 20.3 |
| r.m.s.d. bonds (Å) | 0.007 | 0.008 |
| r.m.s.d. angles ( ° ) | 1.01 | 1.04 |
| Average B factor (2) | 30.2 | 36.4 |
| Ramachandrian outliers (%) | 0.3 | 0.4 |
| Rotamer outliers (%) | 0.6 | 0.7 |
a Numbers in parentheses denote values for the outermost resolution shell.
b Rsym = Σhkl[ΣI( Ihkl,I − 〈Ihkl〉 )]/Σhkl,I〈Ihkl〉, where Ihkl is the intensity of an individual measurement of the reflection with indices hkl, and 〈Ihkl〉 is the mean intensity of that reflection.
The overall fold of the RiDD is most similar to the CbGD. Fig. 1, panel a, shows an overlay comparing the model of the RiDD reported herein, aligned with the structure previously reported for the GRE-type CbGD (3). Fig. 1, panel b, also highlights the location of the conserved glycine residues in both enzymes and the high degree of structural homology found in the C-terminal domain. With a small exception, discussed below, we did not observe any significant changes in the overall position of the backbone atoms when the ethanediol- and propanediol-bound models were compared with each other or the CbGD structures. However, there is a 48-amino acid insert that is present in the RiDD (Fig. 1, panel a). Excluding this insertion and the first 20 N-terminal amino acids of the RiDD, the structural alignment of monomer A from the CbGD model (PDB ID 1R9D) with the A monomer of the RiDD model resulted in a root mean square deviation (r.m.s.d.) of 0.6 Å for the backbone α carbon atoms. The additional 48-amino acid insert that is found in the RiDD extends an existing helix and forms an additional helix, loop, and helix motif. This region of the RiDD structure is highlighted in dark blue in Fig. 1, panel a, and forms an important crystallographic contact by interacting with the identical domain in a symmetry-related molecule (data not shown). Interestingly, three hydrophobic amino acids (Ile-623, Ile-627, and Leu-631) form a largely hydrophobic interface between the helical domains of this insert with a buried surface area of ∼470 Å2. The precise function of the additional domain is not clear; however, much of this region could not be modeled for the (S)-1,2-propanediol-bound model due to a lack of electron density. This observation is consistent with the change we observed in the space group for the propanediol-treated crystals (Table 1), suggesting that substrate binding may be communicated to this region of the enzyme. A recent report on the GRE choline lyase (CutC) also demonstrated that substrate binding can influence the overall structure for that enzyme (15).
FIGURE 1.

A schematic representation of the RiDD model aligned on the CbGD model (panel a) and a structural alignment of the C-terminal domain for both models (panel b). The CbGD model is colored green, and the RiDD model is colored magenta with the exception of a 48-amino acid insert that is colored blue. The conserved glycine residue in both structures is represented by red space-filling atoms in panel B.
The RiDD Forms a Dimer in Solution
Sedimentation velocity analysis of the RiDD in solution, without substrate, shows a sedimentation coefficient (c(s)) distribution dominated by a 9.2S species (70.5%), which is close to the predicted values of 8.8S and 8.7S for the dimers produced by non-crystallographic and crystallographic symmetry operations, respectively (Fig. 2, panel a). The dimer with the largest surface area is shown in Fig. 2, panel b. The difference in the observed and expected Svedberg values suggests that the substrate-free dimer may adopt a slightly more compact conformation than that observed in the crystal lattice of substrate bound RiDD. We also observe a 5.6S (16.7%) and 13.4S (5.5%) species that closely match the predicted values for the monomer (5.8S) and tetramer (13.6S). Finally, there is a minor 3.6S species (6.0%) in the c(s) distribution that likely corresponds to misfolded monomer. These results suggest that the RiDD may undergo a concentration-dependent association to form a dimer and a relatively unstable tetramer. The asymmetric unit we observe for the RiDD is a dimer; however, precedence in the literature for PFL (26–28) and the CbGD (3) indicate that the dimer in our asymmetric unit is not functionally relevant. In fact, the buried surface area for the interface of the dimer in our asymmetric unit is only ∼350 Å2. The dimer that is shown in Fig. 2, panel b, has an interface that is substantially larger consisting of ∼800 Å2 of buried surface area based on PISA analysis (32). Interestingly, in the unit cell of both RiDD crystal forms it is possible to construct a tetramer out of two of the dimers shown in Fig. 2 and two of the smaller interfaces.
FIGURE 2.

Sedimentation velocity study for the RiDD (panel a) and a schematic representation (panel b) of the proposed RiDD dimer overlaid on top of the asymmetric unit observed for the model of the CbGD (PDB ID 1R9D). The c(s) sedimentation distribution reveals species corresponding to monomer (M, 5.6 S), dimer (D, 9.2 S), and tetramer (T, 13.4 S). The peak at 3.6 S likely represents a small amount of misfolded monomer. The color scheme for the schematic representation of the RiDD and CbGD models is the same as shown in Fig. 1 except that the atoms of the conserved glycine in the RiDD model are highlighted as cyan space-filling spheres.
Enzyme Activation and Dehydratase Assays
All GREs are activated by their corresponding AE in a SAM-dependent manner (10). The activation reaction is not trivial as all aspects of the procedure, including the glycyl radical, are sensitive to molecular oxygen. Previous work from this laboratory has demonstrated the SAM-dependent activation of the CbGD by the CbGD-AE (3, 33), but the activation of the RiDD by the RiDD-AE has not been reported. The protein sequence of the RiDD-AE indicates that this activating enzyme contains only a single [4Fe-4S] cluster. Consistent with this observation and unlike what we observe for the CbGD-AE (33), iron analysis of the anaerobically expressed, purified, and reconstituted RiDD-AE used in this investigation revealed 3.73 ± 0.26 iron atoms/mol of protein. In this regard, the RiDD-AE is more similar to PFL-AE (20) in that both activating enzymes (PFL-AE and RiDD-AE) contain only the catalytic [4Fe-4S] cluster. To further address whether a [4Fe-4S] cluster was present in the RiDD-AE and functioned in the activation of the RiDD, we applied electron paramagnetic resonance (EPR) spectroscopy to probe the electronic environment of the [4Fe-4S] cluster. The EPR spectrum of the purified RiDD-AE in the presence of 1.0 mm sodium dithionite is consistent with that of a reduced [4Fe-4S]1+ cluster (Fig. 3, panel a). Specifically, we observe a predominantly axial signal with g‖ of 2.016 and g⊥ of 1.923 at 10 K. The addition of excess SAM to the purified RiDD-AE resulted in a shift in the signal to give a g‖ of 2.029 and g⊥ of 1.863 at 10 K (Fig. 3, panel b). These observations are typical of what has been extensively observed for the [4Fe-4S] cluster of other radical SAM AEs and has been recently reviewed elsewhere (10).
FIGURE 3.

EPR spectra monitoring the SAM-dependent generation of a catalytic radical on the RiDD by the RiDD-AE. EPR spectra of 0.1 mm RiDD-AE alone (panel a), in the presence of 1.25 mm SAM (panel b), and after the addition of an equimolar amount of the RiDD (panel c). The spectra presented in panel a and b were recorded at 10 K with a microwave power of 0.25 milliwatt, whereas the data presented in panel c were recorded at 70 K with a microwave power of 0.05 milliwatt. All of the spectra represent the sum of three scans and were recorded with a microwave frequency of 9.582 GHz, a modulation amplitude of 6.477 G, and the modulation frequency was 100 kHz.
When the RiDD is added to the reduced RiDD-AE in the presence of SAM, the Fe-S cluster signal was lost, and a radical signal, centered at g = 2.006, was observed. (Fig. 3, panel c). All of the glycyl radicals that have been characterized to date have been centered at g = 2.00; therefore, the slightly shifted value we observed for the g-tensor of the RiDD radical is slightly different. In addition to the g-tensor, the line shape and splitting of glycyl radicals is typically indicative of 2-fold splitting due to the remaining proton on the glycine residue (14, 20). Additional results are described below that further address the differences in the radical signal we observed for the activated RiDD, and we expanded upon this in the discussion. However, it is becoming increasingly apparent that it is important to utilize the physiological electron donor when studying radical SAM enzymes (35), and we are utilizing sodium dithionite as an electron source in this work. Regardless, we do not observe any dehydration activity unless we perform the SAM- and RiDD-AE-dependent activation of the RiDD. The complete loss of the [4Fe-4S] signal was also somewhat surprising; however, spin integration of this signal against a Cu-EDTA standard indicated that sodium dithionite is incapable of fully reducing this cluster. Specifically, although iron analysis indicates that each monomer of the RiDD-AE contains an intact [4Fe-4S] cluster, spin integration against a Cu-EDTA standard resulted in 0.3 spins per monomer of RiDD. This confirms that <30% of the [4Fe-4S] clusters are in the reduced, or 1+, oxidation state. This also explains the complete loss of the EPR signal for the cluster at 10 K after SAM is added and the radical species has been generated, as all of the spin density has been converted to the radical signal. Given the difficulty working with these enzymes and evidence that the physiological electron donor is important for complete reduction of the [4Fe-4S] cluster in other radical SAM enzymes (35), future efforts will be made to isolate the appropriate donor(s) from R. inulinivorans and optimize reduction of the catalytic cluster.
“Preactivation” of the RiDD
Given the observations above, it was important to establish a linear range for RiDD turnover using the partially activated enzyme before investigating the dehydration of 1,2-propanediol. Various concentrations of preactivated RiDD were investigated using a previously reported coupled assay (3) with a small modification. Specifically, we preactivated a batch of RiDD before adding known amounts of this activated RiDD to the dehydration assay. The decrease in the absorbance at 340 nm (Fig. 4) is due to the NADH-dependent reduction of propanal to propanol by the coupling enzyme, yeast alcohol dehydrogenase (YADH). Based on the rate of NADH consumption by the coupling enzyme at the various RiDD concentrations investigated (Fig. 4, inset), the average specific activity under these experimental conditions is calculated to be 5.5 ± 0.4 μmol min−1 mg−1. If the RiDD was not preactivated, then a significant lag time was observed before the production of propanal, and subsequent NADH-dependent reduction to propanol by YADH was observed (data not shown). However, the specific activity calculated from the maximum slope observed at the highest concentrations of substrate (S)-1,2-propanediol was very similar to what was observed for the preactivated enzyme (∼5.0 compared with 5.5 μmol min−1 mg−1). Notably, increasing the 1,2-propanediol concentration resulted in a decrease in the lag time required to achieve maximum turnover, suggesting a faster activation rate in the presence of substrate, a proposal that is consistent with recent observations for PFL (21). Moreover, as shown in Fig. 5, the RiDD catalyzes the dehydration of (S)-1,2-propanediol considerably faster than (R)-1,2-propanediol. However, due to the degree of error and technical challenges associated with these experiments it is impossible to determine a reliable Km for the two substrates at this time. In addition, as we demonstrate below, there may be additional factors that complicate the kinetic analysis. Regardless, what can be interpreted from these data is that the RiDD is in fact a 1,2-propanediol dehydratase with a preference for the S enantiomer.
FIGURE 4.
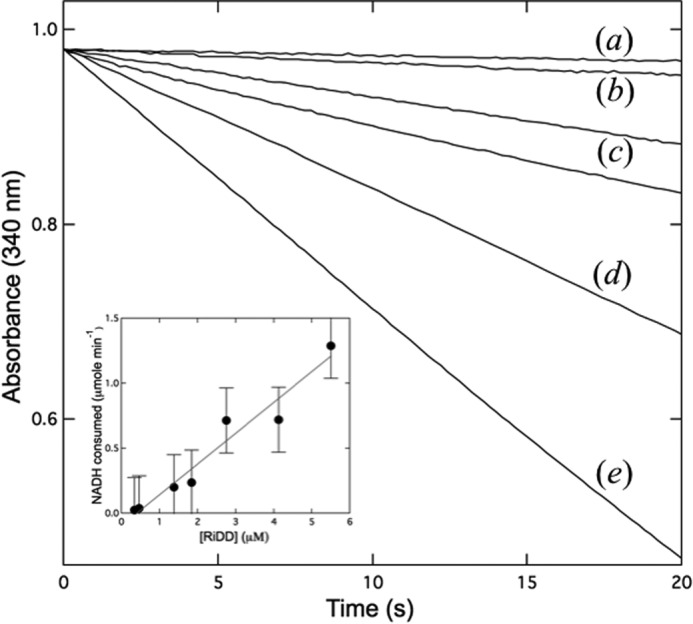
Monitoring RiDD activity using preactivated enzyme. The RiDD was preactivated as described under “Experimental Procedures” and then added to a coupled assay where the NADH-dependent reduction of propanal by YADH (1 mg/ml) was monitored in real time at 340 nm using the extinction coefficient of 6220 m−1 cm−1 to follow the change in NADH concentration. Raw data for assays that were performed with 0 (a), 0.3 μm (b), 0.5 μm (c), 1.4 μm (d), 2.7 μm (e) and 5.5 μm concentrations of the RiDD. The inset shows a plot of the rate of NADH consumption versus the concentration of RiDD (molecular mass ∼94 kDa) for multiple experiments. Assays were performed in triplicate, and error bars represent the S.D. across the data set.
FIGURE 5.
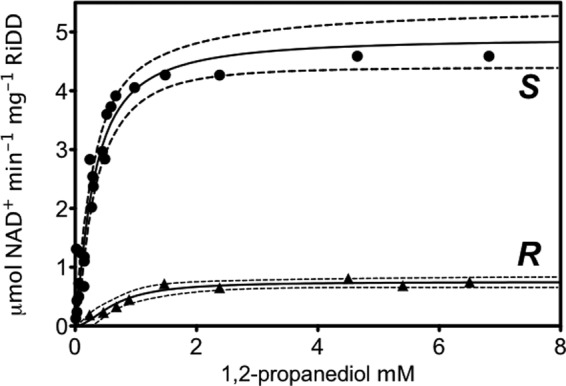
Substrate saturation plot showing the specific activity, calculated from the maximum rates of NADH consumption in the coupled assay, which were observed for the RiDD at different concentrations of (S)-1,2-propanediol (S) or (R)-1,2-propanediol (R). Data points represent the average value for three experiments and were fit to the Hill equation with a 95% confidence range displayed by the dotted lines.
Direct Detection of Dehydration Products by Gas Chromatography
Gas chromatography (GC) was also employed to look directly at the products of the RiDD-catalyzed dehydration reaction. As previously reported, it was possible to obtain good separation of the compounds 1,2-propanediol, acetone, and propionaldehyde (36). It is important to note that this experiment is performed on a significantly larger scale and at a much higher concentration of 1,2-propanediol (5% v/v). No coupling enzyme was included, thus, allowing the products of the dehydration reaction to accumulate. Samples were analyzed by GC/flame ionization detector analysis to directly quantitate the production of propanal. In particular, when glycerol was used as a substrate for the RiDD, no activity was observed when up to 8 mm glycerol was tried as the substrate. Surprisingly, when 1,2-propanediol was the substrate, we observed acetone production in addition to propanal production with both enantiomers of 1,2-propanediol (Fig. 6). An explanation for acetone production is not immediately apparent; however, these observations further confirm that the RiDD is in fact selective for 1,2-propanediol. These data may have mechanistic implications as we do not observe any acetone production when 1,2-propanediol is the substrate for the CbGD.
FIGURE 6.
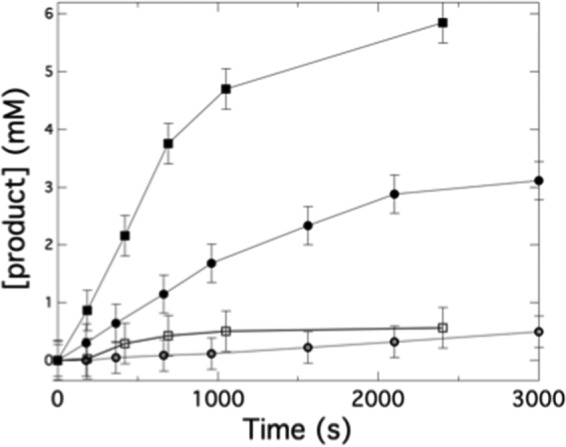
GC data monitoring the conversion of 1,2-propanediol to propionaldehyde and acetone by the RiDD. Assays were performed, and samples for GC analysis were prepared as described under “Experimental Procedures.” Samples were taken and analyzed at the indicted time points. The data show the amount of propionaldehyde (■) and acetone (□) produced when S-1,2-propanediol was used as substrate as well as the propionaldehyde produced (●) and acetone (○) produced when R-1,2-propanediol was used as the substrate for the RiDD in the assay. All experiments were performed in triplicate, and the error bars represent the S.D. across all data points for a given data set.
Active Site Structure of the RiDD Compared with the CbGD
The active site structure of the RiDD may provide an explanation for the functional observations described above. Fig. 7 shows the models for the RiDD determined in this work. Ethanediol, presumably a contaminant of the low molecular weight PEG we used to crystallize the RiDD, was observed in the active site of crystals obtained for the “as-isolated” enzyme (Fig. 7, panel a). Fortunately, it was possible to transfer these crystals in a stepwise fashion to mother liquor containing 5% 1,2-propanediol (racemic mixture). The diffraction quality did decreased to 2.4 Å (Table 1), but the 1,2-propanediol was clearly observed in the active site (Fig. 7, panel b). Refinement with either enantiomer, visual inspection of the difference density, and slightly lower R-values indicated that (S)-1,2-propanediol is bound and, therefore, has been modeled as such. Notably, the hydroxyl groups of the ethanediol and 1,2-propandiol are similarly positioned in the active site of the RiDD models (compare panel a with panel b in Fig. 7). The binding mode of the ethanediol further supports modeling (S)-1,2-propanediol in the active site as alignment of the backbone carbon atoms of the R enantiomer for 1,2-propanediol would cause a steric clash with the side chain of Phe-344. Furthermore, when the 1,2-propanediol-bound RiDD model is aligned with the glycerol-bound model of the CbGD, both substrates are in similar positions within the active site (Fig. 8). However, there is a very distinct difference between the active site structure of the RiDD and the CbGD. Specifically, amino acids Tyr-339 and Ser-642 are within hydrogen bonding distance of each other, and Ser-642 is also within hydrogen bonding distance of the third hydroxyl group of glycerol. The equivalent residues in the RiDD are Phe-344 and Val-696. In fact, based on the alignment shown in Fig. 8, the side chain of Val-696 occupies the space that would be required to accommodate glycerol.
FIGURE 7.

Wall-eyed stereo view showing a stick representation of the active site model determined for the RiDD with ethanediol (panel a) or S-1,2-propanediol bound (panel b). In both cases, the 2Fo − Fc composite omit map (Green cage) was generated using the simulated annealing protocol with 7% of the model omitted per cycle. The electron density is contoured at 1.3 σ, and the carbon, oxygen, nitrogen, and sulfur atoms are colored tan, red, blue, and cyan, respectively. Ethanediol and S-1,2-propanediol are labeled EDO and PGO, respectively. The dashed lines highlight atoms within hydrogen bonding distance of the hydroxyl groups of ethanediol (panel A) or propanediol (panel B).
FIGURE 8.

Wall-eyed stereo view of a stick diagram showing an overlay of the RiDD model with ethanediol bound (this work, tan carbon atoms), the RiDD model with S-1,2-propanediol bound (this work, yellow carbon atoms), and the CbGD model with glycerol bound (green carbon atoms). The amino acid residues for the RiDD and CbGD models are labeled in black and brown text, respectively. The dashed lines indicate atoms that are within 3.7 Å of one another.
Site-directed Mutagenesis of the RiDD and Glycerol Dehydration
To test the hypothesis that residues Phe-344 and Val-696 are responsible for the preventing glycerol binding and turnover in the wild-type RiDD, we constructed the F344Y/V696S RiDD variant. This variant was also useful in re-examining the radical signal we observe for the wild-type RiDD. Specifically, before performing a coupled assay using glycerol as the substrate, we first investigated the SAM-dependent activation of the F344Y/V696S RiDD by the RiDD-AE in a side-by-side activation experiment with the wild-type CbGD and RiDD. These data are shown in Fig. 9. Similar to what we observed for the wild-type RiDD, the g-tensor for the activated F344Y/V696S RiDD signal is also somewhat shifted by comparison to the radical signal observed for the CbGD. Both the wild-type RiDD and F344Y/V696S RiDD are weaker than the radical signal observed for the activated CbGD, consistent with the partial reduction we observed for the dithionite-treated RiDD-AE. Interestingly, the line shape for the activated F344Y/V696S RiDD is more similar to what we observed for the CbGD. In the absence of access to the physiological reduction partners for the RiDD-AE, we are hesitant to read too much into these observations. However, although this is a minor difference, it clearly implies spin-orbit coupling in the RiDD that is different from the CbGD. If the unpaired electron in the activated RiDD is coupled to a different atom relative to the typical glycyl radical observed for GREs, this might provide new mechanistic insight and is being investigated further. Regardless, in all cases we did not observe any dehydration activity for any of these enzymes unless the enzymes were activated first.
FIGURE 9.
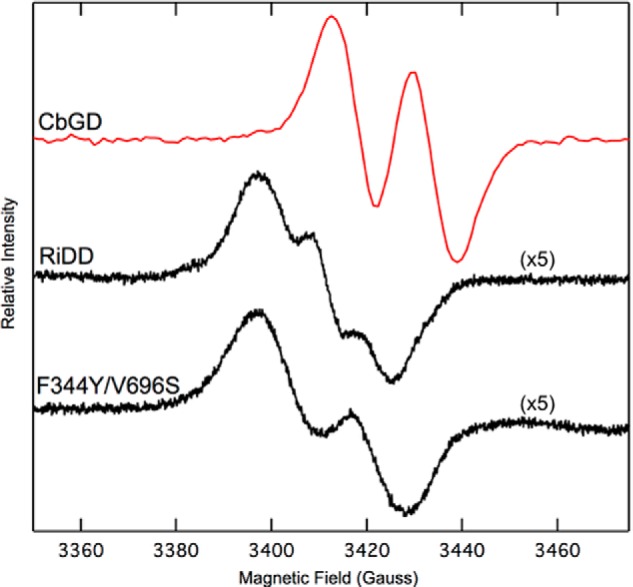
EPR spectra recorded at 70 K for the activated CbGD, RiDD, and the RiDD double mutant F344Y/V696S. All of the spectra represent the sum of three scans and were recorded with a microwave power of 0.05 milliwatt, microwave frequency of 9.582 GHz, modulation amplitude of 6.477 G, and a modulation frequency of 100 kHz. The intensity of the RiDD and the F344Y/V696S variant was considerably less that what was observed for the CbGD and was increased 5-fold for a better comparison.
Given that the F344Y/V696S RiDD could be activated, we asked whether or not the enzyme could dehydrate glycerol. We did in fact observe NADH consumption in a coupled assay when glycerol was the substrate for either the CbGD or the F344Y/V696S RiDD variant (Fig. 10). However, the specific activity for glycerol dehydration by the F344Y/V696S RiDD variant was considerably less than what is observed for the activated CbGD. The F344Y/V696S RiDD did purify with slightly lower yields, suggesting it might be less stable than the wild-type enzyme, but nonetheless this variant catalyzed glycerol dehydration at maximum rate of 0.81 ± 0.02 μmol min−1 mg−1. Although this rate was almost 5-fold slower when compared with the CbGD, these observations suggest that these two amino acids play a role in the substrate selectivity. The slower rate of activity suggests that the conformational dynamics of the active site and dehydration mechanism are considerably more complex, and in both cases (CbGD and RiDD) it may be more appropriate to utilize a physiological electron donor in all future investigations.
FIGURE 10.
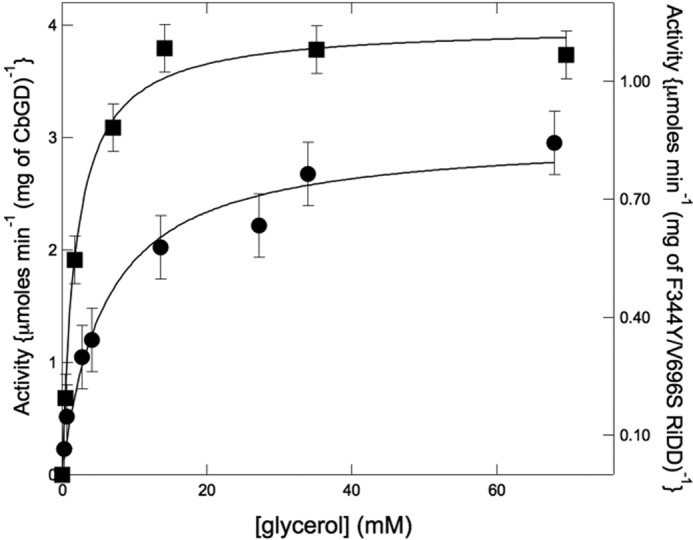
Substrate saturation plot showing the specific activity, calculated from the maximum rates of NADH consumption in the coupled assay, for the CbGD (left axis, solid squares) and the F344Y/V696S variant of the RiDD (right axis, solid circles) when glycerol was used as the substrate. The coupled assay was performed as described under “Experimental Procedures” with the notable exception that purified 1,3-propanediol dehydrogenase was used as the coupling enzyme instead of YADH. Experiments were performed in triplicate, and error bars represent the S.D. across all data points.
Discussion
The functional diversity of glycyl radical enzymes is impressive (9, 37, 38) and has important implications for many aspects of human health and general quality of life. For example, the inhibition of some of these reactions will prevent anaerobic growth by certain pathogenic microorganisms (39). In addition, many of the reactions are industrially significant, making GREs of interest as potential biocatalysts. However, there are a number of unanswered questions regarding the activation, mechanism, and deactivation of GREs that remain to be addressed.
Overall Structure and the C-terminal Glycyl Radical Domain
The RiDD model exhibits the anticipated 10-α/β-barrel fold and aligns best with the structure reported for the CbGD. The enzyme exists predominantly as a dimer in solution. The presence of some tetrameric species in solution was unexpected and potentially interesting in light of the observation of two additional protein-protein interfaces in the unit cell of the RiDD crystals. One of these interfaces is facilitated by an additional stretch of amino acids unique to the RiDD, although the function of this domain and interaction is unclear. Regardless, the conserved glycyl radical domain is found in the C-terminal domain of the 10-α/β-barrel fold. The general consensus is that significant conformational changes are required to move the glycyl radical loop out of the core of the GRE structure and into the active site of the activating enzyme. Recent work with CutC has provided evidence that the substrate itself may help induce some of the conformational changes required for activation (15). In addition to the CbGD, the overall fold of the RiDD is also more similar to the large subunits of benzyl succinate synthase and 4-hydroxyphenyl acetate decarboxylase (24, 40). However, both benzyl succinate synthase and 4-hydroxyphenyl acetate decarboxylase have additional peptide subunits, presenting an additional challenge when moving the glycyl radical loop out of the C-terminal domain and into the active site of their respective activating enzymes (25). Funk et al. (25) noticed that contacts between the C-terminal glycyl radical domain and “loop 2” of the N-terminal domain in the CbGD resulted in burying an additional 100 Å. They speculated that loop 2 would have to move to facilitate the opening of the glycyl radical domain in the CbGD. We observed a similar interaction in the RiDD structures reported here; however, we do see a subtle difference in the positioning of loop 2 when the RiDD structures are aligned with the CbGD model. Specifically, with the exception of backbone residues 106–109 in the RiDD, the N-terminal and C-terminal glycyl radical domain align with an r.m.s.d. of <0.5 Å. Interestingly, the backbone α carbon atoms of residues 106–109, found within loop 2, are 2–3 Å away from the equivalent atoms in the CbGD model. This observation provides further support for the hypothesis put forth by Funk et al. (25) regarding the activation of the CbGD. This interaction is not present in the structural models of PFL. However, PFL remains somewhat unique in that it has also been shown to exhibit “half-sites” reactivity (19, 20, 41) and appears to have a “repair peptide,” YfiD, which can be post-translationally activated by PFL-AE. The activated YfiD can replace any glycyl radical domains in PFL that have been oxygen damaged (42). These data indicate significantly greater freedom of movement for the C-terminal domain of PFL.
Generation of the Glycyl Radical
At the present time, there is no evidence to suggest that there is any allosteric communication between active sites in the physiological dimer we observed for the RiDD. In addition, there is no evidence that the glycyl radical must be generated at both active sites to observe activity in any GRE. However, one common feature that all of the GREs have been shown (or predicted) to contain is a catalytic dyad consisting of a conserved glycine and cysteine residue. The thiol group of the conserved cysteine is positioned close to the α carbon atom of the glycine residue (typically within ∼3.5 Å). The location and position of the catalytic dyad facilitates radical transfer from the α carbon atom of the glycine residue to the sulfur atom of the cysteine residue via hydrogen atom abstraction. The latter radical is positioned to abstract a hydrogen atom from the primary metabolic substrate. For the RiDD reported here, the sulfur atom of Cys-438 is within 3.6 Å of the C1 carbon atom of 1,2-propanediol (Fig. 7, panel B). At the present time, we have not identified or isolated the physiological electron donor for the RiDD-AE, and therefore, we are hesitant to speculate on the precise identity of the radical signals we observed for the wild-type and F344Y/V696S variant of the RiDD at 70 K. However, chemical reductants can lead to artifacts, and the importance of the electron source for radical SAM enzymes has been underscored in recent work (35). Interestingly, the line shape of the EPR spectra we observed for the activated F344Y/V696S variant looks more similar to the radical signal we observed for the activated CbGD, suggesting that these amino acids may have some influence on the generation and/or environment of the glycyl radical in the RiDD. Future work will focus on identification of the physiological electron donor to the RiDD-AE in R. inulinivorans.
Mechanism of GRE-dependent Dehydration
At least two mechanisms have been proposed for the dehydration of glycerol as catalyzed by the CbGD (3, 31). Both mechanisms are also relevant to 1,2-propanediol dehydration, and both begin with the radical on the catalytic cysteine residue (Cys-433 or Cys-438 in the CbGD or RiDD, respectively). This radical is positioned to abstract a hydrogen atom from substrate (either 1,2-propanediol or glycerol). The former mechanism, proposed by O'Brien et al. (3), involves abstraction of the pro-S hydrogen atom from the primary carbon atom followed by the migration of the new substrate radical and hydroxyl group in opposite directions. The substrate/intermediate radical re-abstracts a hydrogen atom from the catalytic cysteine to produce a hydrated aldehyde that spontaneously decomposes to water and 3-hydroxy propionaldehyde. In the O'Brien (3) proposal the migrating hydroxyl is stabilized by a pair of conserved and appropriately protonated histidine side chains (His-164 and His-281 of the CbGD). In a purely computational investigation, Feliks et al. (31) proposed that these histidine residues, His-164 in particular, could facilitate the direct donation of a proton and loss of water. In such a “direct dehydration” mechanism, only the substrate/intermediate radical migrates from the primary to secondary carbon atom of substrate before re-abstracting a hydrogen atom from Cys-433 and resetting the enzyme for another round of turnover.
Prior to this work there was no evidence to suggest that the mechanism of glycerol dehydration would be significantly different from 1,2-propanediol dehydration in the GRE-dependent dehydratases. In fact, as both mechanisms for the CbGD had predicted, it was reasonable to expect that a proton on the C1 carbon atom of 1,2-propanediol would be abstracted first in the mechanism of the RiDD. However, the production of some acetone with either enantiomer of 1,2-propanediol by the RiDD calls this into question. Although both the RiDD and the CbGD prefer the S enantiomer of 1,2-propanediol, the CbGD will not utilize the R enantiomer at all and did not produce any acetone. This suggests that the two dehydratases have subtly different molecular dynamics. In particular, the RiDD may be a “sloppy” enzyme by comparison to the CbGD with a slightly more flexible active site. In theory, the F344Y/V696S RiDD should have identical interactions with glycerol and catalytic activity when compared with the CbGD or wild-type RiDD; clearly this is not the case. In light of this, acetone production by the RiDD may simply reflect this flexibility and may be the result of some improperly oriented substrate leading to hydrogen atom abstraction from the C2 atom of 1,2-propanediol and direct removal of the hydroxyl group on the C1 carbon atom of 1,2-propanediol.
Taken together, these data also indicate that both dehydration mechanisms (direct dehydration versus hydroxyl/radical co-migration) may be valid depending on the dynamics of the active site and precisely how the substrate is positioned. In fact, multiple binding modes have been proposed to explain the mechanism-based inactivation by glycerol that is observed for the B12-dependent diol dehydratase (43). Specifically, although glycerol is not a chiral compound, glycerol can bind to the active site of the B12-dependent diol dehydratase in two different orientations. One orientation mimics the geometry and binding mode of S-1,2-propanediol (GS), whereas the other binding mode is similar to that of R-1,2-propanediol (GR). Because of the different geometry, the hydrogen bond strength varies between the GS or GR orientation. The starting orientation is important because during the dehydration reaction, and specifically the migration of the middle hydroxyl group of glycerol, there is an energetic penalty associated with the stronger hydrogen bonding of the GS orientation. Hence, it was proposed that undesirable side reactions leading to the suicide inhibition of the B12-dependent diol dehydratase occur with a much higher probability for glycerol that is bound in the GS orientation (43). Consistent with this proposal, the B12-dependent diol dehydratase will utilize both enantiomers of 1,2-propanediol but has a preference for the R enantiomer (44). Interestingly, the results reported here are consistent with the CbGD having a more selective binding site with glycerol or S-1,2-propanediol bound in a single orientation, whereas the RiDD has slightly more conformational flexibility.
Conclusions
The catalytic power of GREs is of significant interest, especially in light of anaerobic metabolism in the intestinal microbiome (15, 22, 23) and growing demand for biocatalysts capable of performing difficult chemical rearrangements. Although B12-dependent glycerol and diol dehydratases have been well studied, this is the first report of a GRE-dependent diol dehydratase. Interestingly, a similar mechanistic parallel is observed when comparing the activity of the GRE-dependent glycerol dehydratase, CbGD, with the GRE-dependent diol dehydratase, RiDD. Specifically, similar to what has been reported for the B12-dependent enzymes, the diol dehydratase may be more amendable to re-engineering (43). However, to address this thoroughly as well as the mechanism of all GREs, it will be imperative to utilize the physiological donors (i.e. flavodoxin or ferredoxin from the native organism) when providing reducing equivalents for the respective activating enzymes.
Experimental Procedures
Enzyme Expression, Purification, and Crystallization
Unless explicitly stated, all experimental procedures were carried out under strictly anaerobic conditions, and all solutions were made anaerobic by purging with oxygen-free argon on a vacuum manifold. In addition, all sample preparation for spectroscopic studies or enzyme assays was carried out in a Coy™ anaerobic chamber containing an atmosphere of 5% hydrogen with the balance being nitrogen and <1 ppm oxygen. The genes for the RiDD, the RiDD-AE, and the 3-hydroxyprionaldehyde reductase (a.k.a. 1,3-propanediol dehydrogenase) from C. butyricum (NCBI accession numbers WP_007885173, ABC25540, and AAM54730.1, respectively) were all codon-optimized for expression in E. coli and cloned into the commercially available pTRCHis™ vector introducing a 6× polyhistidine tail at the N terminus (specifically MSHHHHHHSGS) before the first amino acid of the native sequence. The RiDD, the RiDD-AE, and 1,3-propanediol dehydrogenase were purified using the same affinity column procedures reported for the CbGD and CbGD-AE (3). Briefly, this involves affinity purification using the cobalt-loaded Talon™ affinity resin followed by gel filtration using an S-200 column equilibrated with 50 mm Tris, pH 8.0, and 500 mm KCl. Identical buffer was used to further purify the RiDD using gel filtration Superdex S-200 resin (GE Healthcare). In the case of the RiDD-AE, a chemical reconstitution of the [4Fe-4S] cluster was performed as previously described (45), and the enzyme was subjected to further purification using anion exchange chromatography. Specifically, sodium sulfide (0.2 mm), 2-mercapoethanol (5 mm), and ferrous ammonium sulfate (0.25 mm) were added to the solution, and the entire mix was gently stirred overnight at 4 °C. Precipitate was removed by centrifugation, and the supernatant was loaded onto a 5-ml Sepharose QFF column. The protein was then eluted from the column by applying a linear salt gradient (from 0 to 0.5 m KCl) in 20 mm Tris, pH 8.1. Elution typically occurred around 150 mm KCl. Essentially, the reconstituted activating enzymes were subjected to anion exchange chromatography to remove adventitiously bound iron and sulfide. All of the purified enzymes were concentrated and kept in liquid nitrogen. Protein concentrations were determined using the modified Biuret method (46), and all proteins were judged homogeneous by SDS-PAGE analysis.
EPR Spectroscopy
EPR spectroscopy was employed to monitor SAM-dependent changes in the spectrum of the [4Fe-4S]1+ cluster in the RiDD-AE as well as the SAM-dependent generation of a catalytic radical on the RiDD. All EPR samples contained 1 mm sodium dithionite and were recorded using a microwave frequency of 9.602 GHz, modulation amplitude of 6.3 G, and a modulation frequency of 100 kHz as previously described (3). The concentrations of enzyme or SAM added were as noted in the figure legend. Spectra were recorded at 10 K as well as 70 K for all samples unless stated otherwise.
Coupled Enzyme Assay for the CbGD and RiDD
The coupled enzyme assay for the CbGD and the RiDD were performed in anaerobic quartz cuvettes essentially as previously described (3) at 25 °C except that where indicated the RiDD was preactivated before the addition of the substrate as described below. Similar assays utilizing a coupling enzyme have been reported by those studying the B12-dependent glycerol and diol dehydratases (47). Essentially, an aldehyde reductase, which utilizes NADH or NADPH to reduce the aldehyde to the corresponding alcohol, is used as a coupling enzyme. Depending on the specific dehydration reaction being investigated, several coupling enzymes have already been well characterized (16, 48–52). Unless stated otherwise, the assay contained 1 mm SAM, 1 mm sodium dithionite, 100 mm KCl, and 200 μm NADH in 50 mm HEPES, pH 7.5. The assay also contained 28 μm YADH. The total volume of the coupled assay was 0.5 ml, and the substrate as well as the enzymes (RiDD and RiDD-AE) were all added to the concentrations indicated in the appropriate figures. Before initiating the assay the spectrophotometer was blanked against assay buffer. To preactivate the RiDD, this enzyme was incubated with the RiDD-AE (2:1 RiDD-AE:RiDD) in the presence of 2 mm SAM and 2 mm sodium dithionite for 10 min to allow for generation of the glycyl radical, a variety of enzyme concentrations were tried in these experiments, and the assay was initiated by the addition of the substrate. In general, the coupled assay takes advantage of a catalytic excess of the reductase YADH to rapidly reduce the propionaldehyde product of the dehydratase reaction. We independently investigated the specific activity of commercially available YADH (Sigma) by measuring the NADH-dependent reduction of propionaldehyde in the assay buffer described above and found that it was >400 μmol min−1 mg−1. To address glycerol dehydration by the CbGD and the F344Y/V696S variant of the RiDD, the YADH was replaced with purified 1,3-propanediol dehydrogenase in the coupled assay.
Direct Monitoring of 1,2-Propanediol Dehydration by GC
The direct detection of 1,2-propanediol, propionaldehyde, and acetone was performed using a GC equipped with a 30-m × 0.329-mm Carbowax 20-m column. Due to the sensitivity of the GC/flame ionization detector measurement and the sample collection protocol, the assay had to be performed at a much larger scale. Specifically, these assays were performed in 20 ml of assay solution with a concentration of RiDD of 1 μm. Yeast alcohol dehydrogenase as well as NADH were omitted from this assay to allow the propionaldehyde to accumulate. A stir bar was also included in the experiment and provided stirring at a rate of ∼75 rpm. In all these experiments the reaction was initiated by the addition of RiDD-AE to 2 μm in the presence of 2 mm SAM and 2 mm sodium dithionite. Propanediol (either S-1,2-propanediol or R-1,2-propanediol) was added to 5% (v/v) at the beginning of the assay. At the time points indicated, 1-ml samples were extracted anaerobically and immediately treated with 250 μl of 1 m formic acid. The acid-treated samples were all centrifuged in a microcentrifuge at 14,000 rpm for 10 min to remove any precipitated material. The supernatant was then injected into the GC and separated using a temperature gradient that ramped from 60 °C to 120 °C over a 20-min period. The GC was equipped with a flame ionization detector, and a series of standards for 1,2-propanediol, acetone, and propionaldehyde was used to calibrate the integrated peak areas.
Crystallization, Data Collection, and Structure Determination
Initial crystallization conditions for the RiDD were identified in sitting drop experiments and then optimized in a hanging-drop tray. The final conditions for diffraction quality crystals of the RiDD were 0.125 m sodium acetate, 42% PEG 400 (w/v), and 0.025 m HEPES, pH 7.5. Crystallization was performed in hanging drop experiments with 800 μl of precipitating solution in the well. Protein (40 mg/ml) and precipitation solution were mixed (2 μl each) to initiate the reaction, and then the tray was placed at 4 °C for 12 h before being moved to an incubator at 18 °C. Crystals of the RiDD took approximately 1 week to form, and no further cryo-protection was required for freezing. Ethanediol was serendipitously observed in the active site of the RiDD for the native crystals, but it was possible to soak 1,2-propanediol (racemic mixture) into the crystals by simply transferring the crystals to mother liquor containing 5% 1,2-propanediol in a stepwise manner.
Data were collected at the Advanced Photon Source through SER-CAT on beam line 22BM at 0.98 Å. The program PHENIX (53) was used to solve the initial phase problem by using a polyalanine model of the CbGD (PDB ID 1R9D) and molecular replacement. Iterative rounds of model building and refinement using COOT (54) and PHENIX were performed with experimental phase restraints and a 5.0% Rfree test set (55) that was generated by PHENIX and used throughout all stages of model building and refinement. A summary of the data collection and refinement statistics for the ethanediol- and 1,2-propanediol-bound models is shown in Table 1.
Site-directed Mutagenesis
The F344Y/V696S variant of the RiDD was generated using the QuikChange II kit (Agilent Technologies) by following the manufacturer's instructions. Mutagenic oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA), and both mutations were verified by sequencing at the Georgia Genomics Facility.
Sedimentation Velocity Experiments
The RiDD was dialyzed into 10 mm HEPES, pH 7.5 and 150 mm KCl and then diluted to a final concentration of 5 μm for analysis. RiDD (400 μl) or reference buffer (410 μl) was loaded into 12-mm double-sector Epon centerpieces equipped with quartz windows and equilibrated at 20 °C in an AN60 Ti rotor for 1 h. Sedimentation velocity data were collected in an Optima XLA analytical ultracentrifuge using a rotor speed of 50,000 rpm at 20 °C. Data were recorded at a wavelength of 280 nm using a radial step size of 0.003 cm. The partial specific volume of RiDD (0.73478 ml/g) was calculated from the amino acid sequence. The program SEDNTERP was used to calculate the density (1.00609 g/ml) and viscosity (0.01009 P) of the buffer (56). SEDFIT was used to model and analyze the sedimentation data. Modeled data were fit as a continuous c(s) distribution using the baseline, meniscus, frictional coefficient, systematic time-invariant noise, and radial-invariant noise (57). The r.m.s.d. value for the experiment was <0.004 absorbance. Theoretical sedimentation coefficient (s) values were calculated from the atomic coordinates of RiDD using HYDROPRO (34).
Author Contributions
W. N. L. conceived and coordinated the study and wrote the paper. J. W. L., S. K., and P. R. contributed substantially to conception and design and acquisition of data. N. D. K. assisted with the sedimentation experiments exclusively. All authors contributed to analysis and interpretation of the data presented in this work. F. G., D. J. K., and I. E. G. provided technical assistance and contributed to the preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We to thank Sarah Ann Lee for performing the GC/HPLC analysis and Dr. Mark Eiteman in the engineering department for the use of that equipment. We also thank Dr. Zac Wood for assistance with the analytical ultracentrifugation experiments and helpful discussions in the preparation of the revised manuscript.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5I2A and 5I2G) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GRE
- glycyl radical enzyme
- PFL
- pyruvate formate lyase
- CbGD
- C. butyricum glycerol dehydratase
- AE
- activating enzyme
- SAM
- S-adenosylmethionine
- RiDD
- R. inulinivorans diol dehydratase
- r.m.s.d.
- root mean square deviation
- YADH
- yeast alcohol dehydrogenase
- c(s)
- sedimentation coefficient.
References
- 1. Toraya T. (2002) Enzymatic radical catalysis: coenzyme B12-dependent diol dehydratase. Chem. Rec. 2, 352–366 [DOI] [PubMed] [Google Scholar]
- 2. Toraya T. (2002) Radical-catalyzed enzymatic reactions: structure and mechanism of B12 enzymes. Seikagaku 74, 87–102 [PubMed] [Google Scholar]
- 3. O'Brien J. R., Raynaud C., Croux C., Girbal L., Soucaille P., and Lanzilotta W. N. (2004) Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry 43, 4635–4645 [DOI] [PubMed] [Google Scholar]
- 4. Knappe J., and Wagner A. F. (1995) Glycyl free radical in pyruvate formate-lyase: synthesis, structure characteristics, and involvement in catalysis. Methods Enzymol. 258, 343–362 [DOI] [PubMed] [Google Scholar]
- 5. Knappe J., Elbert S., Frey M., and Wagner A. F. (1993) Pyruvate formate-lyase mechanism involving the protein-based glycyl radical. Biochem. Soc. Trans. 21, 731–734 [DOI] [PubMed] [Google Scholar]
- 6. Frey M., Rothe M., Wagner A. F., and Knappe J. (1994) Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. Studies of [2H]glycine-substituted enzyme and peptides homologous to the glycine 734 site. J. Biol. Chem. 269, 12432–12437 [PubMed] [Google Scholar]
- 7. Sun X., Ollagnier S., Schmidt P. P., Atta M., Mulliez E., Lepape L., Eliasson R., Gräslund A., Fontecave M., Reichard P., and Sjöberg B. M. (1996) The free radical of the anaerobic ribonucleotide reductase from Escherichia coli is at glycine 681. J. Biol. Chem. 271, 6827–6831 [PubMed] [Google Scholar]
- 8. Sun X., Eliasson R., Pontis E., Andersson J., Buist G., Sjöberg B. M., and Reichard P. (1995) Generation of the glycyl radical of the anaerobic Escherichia coli ribonucleotide reductase requires a specific activating enzyme. J. Biol. Chem. 270, 2443–2446 [DOI] [PubMed] [Google Scholar]
- 9. Selmer T., Pierik A. J., and Heider J. (2005) New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol. Chem. 386, 981–988 [DOI] [PubMed] [Google Scholar]
- 10. Shisler K. A., and Broderick J. B. (2014) Glycyl radical activating enzymes: structure, mechanism, and substrate interactions. Arch. Biochem. Biophys. 546, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krieger C. J., Roseboom W., Albracht S. P., and Spormann A. M. (2001) A stable organic free radical in anaerobic benzylsuccinate synthase of Azoarcus sp. strain T. J. Biol. Chem. 276, 12924–12927 [DOI] [PubMed] [Google Scholar]
- 12. Selmer T., and Andrei P. I. (2001) p-Hydroxyphenylacetate decarboxylase from Clostridium difficile: a novel glycyl radical enzyme catalysing the formation of p-cresol. Eur. J. Biochem. 268, 1363–1372 [DOI] [PubMed] [Google Scholar]
- 13. Craciun S., and Balskus E. P. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Craciun S., Marks J. A., and Balskus E. P. (2014) Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem. Biol. 9, 1408–1413 [DOI] [PubMed] [Google Scholar]
- 15. Kalnins G., Kuka J., Grinberga S., Makrecka-Kuka M., Liepinsh E., Dambrova M., and Tars K. (2015) Structure and function of CutC choline lyase from human microbiota bacterium Klebsiella pneumoniae. J. Biol. Chem. 290, 21732–21740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raynaud C., Sarçabal P., Meynial-Salles I., Croux C., and Soucaille P. (2003) Molecular characterization of the 1,3-propanediol (1,3-PD) operon of Clostridium butyricum. Proc. Natl. Acad. Sci. U.S.A. 100, 5010–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scott K. P., Martin J. C., Campbell G., Mayer C. D., and Flint H. J. (2006) Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium Roseburia inulinivorans. J. Bacteriol. 188, 4340–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grove T. L., Lee K. H., St Clair J., Krebs C., and Booker S. J. (2008) In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry 47, 7523–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng Y., Veneziano S. E., Gillispie G. D., and Broderick J. B. (2010) Pyruvate formate-lyase, evidence for an open conformation favored in the presence of its activating enzyme. J. Biol. Chem. 285, 27224–27231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henshaw T. F., Cheek J., and Broderick J. B. (2000) The [4Fe-4S]1+ cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover. J. Am. Chem. Soc. 122, 8331–8332 [Google Scholar]
- 21. Crain A. V., and Broderick J. B. (2014) Pyruvate formate-lyase and its activation by pyruvate formate-lyase activating enzyme. J. Biol. Chem. 289, 5723–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Becker A., Fritz-Wolf K., Kabsch W., Knappe J., Schultz S., and Volker Wagner A. F. (1999) Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Biol. 6, 969–975 [DOI] [PubMed] [Google Scholar]
- 23. Logan D. T., Andersson J., Sjöberg B. M., and Nordlund P. (1999) A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science 283, 1499–1504 [DOI] [PubMed] [Google Scholar]
- 24. Martins B. M., Blaser M., Feliks M., Ullmann G. M., Buckel W., and Selmer T. (2011) Structural basis for a Kolbe-type decarboxylation catalyzed by a glycyl radical enzyme. J. Am. Chem. Soc. 133, 14666–14674 [DOI] [PubMed] [Google Scholar]
- 25. Funk M. A., Judd E. T., Marsh E. N., Elliott S. J., and Drennan C. L. (2014) Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc. Natl. Acad. Sci. U.S.A. 111, 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lehtiö L., Leppänen V. M., Kozarich J. W., and Goldman A. (2002) Structure of Escherichia coli pyruvate formate-lyase with pyruvate. Acta Crystallogr. D Biol. Crystallogr. 58, 2209–2212 [DOI] [PubMed] [Google Scholar]
- 27. Leppänen V. M., Merckel M. C., Ollis D. L., Wong K. K., Kozarich J. W., and Goldman A. (1999) Pyruvate formate lyase is structurally homologous to type I ribonucleotide reductase. Structure 7, 733–744 [DOI] [PubMed] [Google Scholar]
- 28. Leppänen V. M., Parast C. V., Wong K. K., Kozarich J. W., and Goldman A. (1999) Purification and crystallization of a proteolytic fragment of Escherichia coli pyruvate formate-lyase. Acta Crystallogr. D Biol. Crystallogr. 55, 531–533 [DOI] [PubMed] [Google Scholar]
- 29. Bharadwaj V. S., Dean A. M., and Maupin C. M. (2013) Insights into the glycyl radical enzyme active site of benzylsuccinate synthase: a computational study. J. Am. Chem. Soc. 135, 12279–12288 [DOI] [PubMed] [Google Scholar]
- 30. Feliks M., Martins B. M., and Ullmann G. M. (2013) Catalytic mechanism of the glycyl radical enzyme 4-hydroxyphenylacetate decarboxylase from continuum electrostatic and QC/MM calculations. J. Am. Chem. Soc. 135, 14574–14585 [DOI] [PubMed] [Google Scholar]
- 31. Feliks M., and Ullmann G. M. (2012) Glycerol dehydratation by the B12-independent enzyme may not involve the migration of a hydroxyl group: a computational study. J. Phys. Chem. B 116, 7076–7087 [DOI] [PubMed] [Google Scholar]
- 32. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 33. Demick J. M., and Lanzilotta W. N. (2011) Radical SAM activation of the B12-independent glycerol dehydratase results in formation of 5′-deoxy-5′-(methylthio)adenosine and not 5′-deoxyadenosine. Biochemistry 50, 440–442 [DOI] [PubMed] [Google Scholar]
- 34. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bruender N. A., Young A. P., and Bandarian V. (2015) Chemical and biological reduction of the radical SAM enzyme CPH4 synthase. Biochemistry 54, 2903–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pontes H., Guedes de Pinho P., Casal S., Carmo H., Santos A., Magalhães T., Remião F., Carvalho F., and Lourdes Bastos M. (2009) GC determination of acetone, acetaldehyde, ethanol, and methanol in biological matrices and cell culture. J. Chromatogr. Sci. 47, 272–278 [DOI] [PubMed] [Google Scholar]
- 37. Hioe J., Savasci G., Brand H., and Zipse H. (2011) The stability of Cα peptide radicals: why glycyl radical enzymes? Chemistry 17, 3781–3789 [DOI] [PubMed] [Google Scholar]
- 38. Frey P. A. (2001) Radical mechanisms of enzymatic catalysis. Ann. Rev. Biochem. 70, 121–148 [DOI] [PubMed] [Google Scholar]
- 39. Ofer A., Kreft J., Logan D. T., Cohen G., Borovok I., and Aharonowitz Y. (2011) Implications of the inability of Listeria monocytogenes EGD-e to grow anaerobically due to a deletion in the class III NrdD ribonucleotide reductase for its use as a model laboratory strain. J. Bacteriol. 193, 2931–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L., Patterson D. P., Fox C. C., Lin B., Coschigano P. W., and Marsh E. N. (2009) Subunit structure of benzylsuccinate synthase. Biochemistry 48, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Knappe J., and Sawers G. (1990) A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol. Rev. 6, 383–398 [DOI] [PubMed] [Google Scholar]
- 42. Nnyepi M. R., Peng Y., and Broderick J. B. (2007) Inactivation of E. coli pyruvate formate-lyase: role of AdhE and small molecules. Arch. Biochem. Biophys. 459, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamanishi M., Kinoshita K., Fukuoka M., Saito T., Tanokuchi A., Ikeda Y., Obayashi H., Mori K., Shibata N., Tobimatsu T., and Toraya T. (2012) Redesign of coenzyme B12 dependent diol dehydratase to be resistant to the mechanism-based inactivation by glycerol and act on longer chain 1,2-diols. FEBS J. 279, 793–804 [DOI] [PubMed] [Google Scholar]
- 44. Shibata N., Nakanishi Y., Fukuoka M., Yamanishi M., Yasuoka N., and Toraya T. (2003) Structural rationalization for the lack of stereospecificity in coenzyme B12-dependent diol dehydratase. J. Biol. Chem. 278, 22717–22725 [DOI] [PubMed] [Google Scholar]
- 45. Külzer R., Pils T., Kappl R., Hüttermann J., and Knappe J. (1998) Reconstitution and characterization of the polynuclear iron-sulfur cluster in pyruvate formate-lyase-activating enzyme. Molecular properties of the holoenzyme form. J. Biol. Chem. 273, 4897–4903 [DOI] [PubMed] [Google Scholar]
- 46. Chromý V., Fischer J., and Kulhánek V. (1974) Re-evaluation of EDTA-chelated biuret reagent. Clin. Chem. 20, 1362–1363 [PubMed] [Google Scholar]
- 47. Sottocasa G. L., Stagni N., and De Bernard B. (1971) A spectrophotometric method for the direct recording of dioldehydrase activity. Experientia 27, 1247–1248 [DOI] [PubMed] [Google Scholar]
- 48. Trivić S., Leskovac V., Zeremski J., Stancić B., and Anderson B. M. (1998) Influence of Tris(hydroxymethyl)aminomethane on kinetic mechanism of yeast alcohol dehydrogenase. J. Enzyme Inhib. 13, 57–68 [DOI] [PubMed] [Google Scholar]
- 49. Zhuge B., Zhang C., Fang H., Zhuge J., and Permaul K. (2010) Expression of 1,3-propanediol oxidoreductase and its isoenzyme in Klebsiella pneumoniae for bioconversion of glycerol into 1,3-propanediol. Appl. Microbiol. Biotechnol. 87, 2177–2184 [DOI] [PubMed] [Google Scholar]
- 50. Daniel R., Boenigk R., and Gottschalk G. (1995) Purification of 1,3-propanediol dehydrogenase from Citrobacter freundii and cloning, sequencing, and overexpression of the corresponding gene in Escherichia coli. J. Bacteriol. 177, 2151–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jarboe L. R. (2011) YqhD: a broad-substrate range aldehyde reductase with various applications in production of biorenewable fuels and chemicals. Appl. Microbiol. Biotechnol. 89, 249–257 [DOI] [PubMed] [Google Scholar]
- 52. Sulzenbacher G., Alvarez K., Van Den Heuvel R. H., Versluis C., Spinelli S., Campanacci V., Valencia C., Cambillau C., Eklund H., and Tegoni M. (2004) Crystal structure of E. coli alcohol dehydrogenase YqhD: evidence of a covalently modified NADP coenzyme. J. Mol. Biol. 342, 489–502 [DOI] [PubMed] [Google Scholar]
- 53. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 55. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 56. Laue T. M., and Pelletier S. L. (1986) Interactive computer-aided interpretation of sedimentation data. Biophys. J. 49, A274–A274 [Google Scholar]
- 57. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]