

Abstract
Transport of secretory proteins from the endoplasmic reticulum to the Golgi is mediated by the coat protein II (COPII) complex comprising a Sec23-Sec24 heterodimer and a Sec13-Sec31 heterotetramer. The mechanisms underlying COPII-mediated protein trafficking have been well defined, but the extent of regulation of this secretory machinery by cellular signaling pathways remains poorly understood. Here, we report that CRK1, a G1 cyclin-dependent kinase in Trypanosoma brucei, regulates anterograde protein trafficking by phosphorylating Sec31. Depletion of CRK1 abolished anterograde transport of the secretory protein and disrupted the localization of multiple Golgi proteins, reminiscent of Sec31 depletion. CRK1 phosphorylates Sec31 at multiple serine/threonine sites, and mutation of these phosphosites to alanine recapitulates the protein trafficking defects caused by Sec31 depletion. Mutation of these CRK1 phosphosites to aspartate restored Sec31 function. Taken together, these results uncover a novel function of CRK1 in anterograde protein trafficking and elucidate the mechanistic role of CRK1 in protein trafficking through regulation of the COPII subunit Sec31.
Keywords: chemical biology, COPII, cyclin, cyclin-dependent kinase (CDK), trafficking, Trypanosoma brucei
Introduction
Secretory proteins are synthesized in the endoplasmic reticulum (ER),3 transported to the Golgi, and then delivered to their final destination (1, 2). The secretory pathway consists of the rough ER, ER exit site (ERES), the ER-to-Golgi intermediate compartment, the Golgi apparatus, and post-Golgi carriers (3). Secretory proteins first accumulate into the ERES, which is mediated by the coat protein II (COPII) complex (4, 5). Assembly of the COPII complex is initiated by recruiting the Sar1 GTPase to the cytoplasmic face of the ER (6–8). Subsequently, Sar1, through binding to Sec23, recruits the Sec23-Sec24 heterodimer complex (9, 10), which further recruits cargo proteins to the forming vesicle (11–13). The complex formed by Sar1-Sec23-Sec24 and cargo proteins is further captured by the Sec13-Sec31 heterotetramer, which consists of two each of Sec13 and Sec31 and forms the outer coat layer of COPII (14). The Sec13-Sec31 heterotetramer complex promotes vesicle scission and release from the ER membrane (10, 15, 16).
Protein phosphorylation has been reported to play important roles in regulating a variety of membrane trafficking processes, including the regulation of Golgi fragmentation by Cdc2-mediated phosphorylation of the cis-Golgi matrix protein GM130 during mitosis (17), the regulation of ERES disassembly by Cdc2-mediated phosphorylation of the AAA-ATPase p97 cofactor p47 during mitosis (18), the modulation of secretory cargo transport by the PCTAIRE kinase and its potential regulation of Sec23 (19), and the regulation of ER-to-Golgi trafficking by Akt kinase-mediated phosphorylation of Sec24 (20) and by casein kinase II (CK2)-mediated phosphorylation of Sec31 (21). These findings suggest the extensive regulation of protein trafficking by diverse cellular signaling pathways.
Cyclin-dependent kinases (CDKs) are crucial regulators of the cell division cycle in eukaryotes and are activated by their cyclin partners whose levels oscillate during the cell cycle. In the early branching parasitic protozoan Trypanosoma brucei, 11 CDK-related kinases, designated CRK1–CRK4 and CRK6–CRK12, have been identified (22, 23). CRK1 and CRK3 are involved in controlling the G1/S transition and the G2/M transition, respectively (24–26). CRK1 associates with four PHO80-like cyclins, CYC2, CYC4, CYC5, and CYC7, that appear to play distinct cellular roles in addition to driving the G1/S transition in the procyclic form of T. brucei (27). CYC2 and CYC7 cooperate with CRK1 to regulate posterior cytoskeletal morphogenesis, whereas the CYC4-CRK1 complex appears to regulate certain unidentified nuclear processes required for the G1/S transition (27). Despite the essential functions of CRK1, however, its mechanistic roles remain poorly understood, mainly because its downstream substrates have not been identified.
We have set out to identify the substrates of CRK1 by chemical genetic approach, which utilizes an analog-sensitive CRK1 mutant and an engineered ATP analog to thiophosphorylate native CRK1 substrates in crude cell lysate. This allowed us to identify novel CRK1 substrates, including the Sec31 homolog, which was characterized in detail in this report. We demonstrated that CRK1 co-localizes with Sec31 at the ERES and phosphorylates Sec31 at multiple serine/threonine sites, thereby regulating anterograde protein transport. These findings elucidated the mechanistic role of CRK1 in ER-to-Golgi protein trafficking through regulating Sec31.
Results
Identification of CRK1 Substrates Using Analog-sensitive CRK1 Mutant and Bulky ATP Analog
To identify the substrates of CRK1, we employed the chemical genetic approach, which utilizes an engineered protein kinase with an enlarged ATP binding pocket to accept bulky ATP analogs (28). The CRK1 analog-sensitive (CRK1as) mutant, generated by mutating the conserved bulky gatekeeper residue phenylalanine 81 to alanine, was capable of utilizing the N6-(benzyl)ATPγS to thiophosphorylate its substrates (Fig. 1, A and B), as confirmed by in vitro kinase assay with purified CRK1as in complex with CYC2 and with recombinant histone H1 as the substrate (Fig. 1C). Thiophosphorylated histone H1 was alkylated with p-nitrobenzyl mesylate (PNBM), which yields a specific thiophosphate ester (29), and then detected by Western blotting with an anti-thiophosphate ester antibody (α-thioP). In contrast, the wild-type CRK1 was not able to utilize the ATP analog (Fig. 1C). It should be noted that CRK1as appeared to be more active than wild-type CRK1 in the presence of ATPγS (Fig. 1C), presumably because enlargement of the ATP binding pocket enabled more efficient access of ATPγS.
FIGURE 1.

Identification of CRK1 substrates by chemical genetic approach. A, procedures of the chemical genetic approach used for CRK1 substrate detection and identification. Thiophosphorylated CRK1 substrates were either detected by Western blotting after PNBM alkylation, which yields thiophosphate ester and can be detected by a thiophosphate ester-specific rabbit monoclonal antibody (anti-thioP), or trypsin-digested, affinity-purified, and analyzed by LC-MS/MS. B, alignment of the sequence surrounding the gatekeeper residue of CRK1 with its orthologs from human, budding yeast, and fission yeast. The gatekeeper residue is highlighted in red. C, in vitro kinase assays to confirm that the CRK1as, but not the wild-type CRK1, is capable of utilizing the bulky ATP analog to thiophosphorylate histone H1. CRK1 (wild-type and analog-sensitive mutant) and CYC2 were co-expressed in E. coli and co-purified. Thiophosphorylated histone H1 was detected by Western blotting with a thiophosphate ester monoclonal antibody (α-thioP). CRK1-His6 and CRK1as-His6 were detected with anti-His antibody, whereas histone H1 was detected by Coomassie Blue staining. D, in vitro thiophosphorylation of T. brucei total proteins by CRK1as. Crude trypanosome cell lysate was incubated with purified CRK1-CYC2 complex and CRK1as-CYC2 complex in the presence of N6-(benzyl)ATPγS. GTP (0.5 or 3 mm) was added to the kinase reaction to act as a competitor of nonspecific labeling. Thiophosphorylated proteins were detected by Western blotting with α-thioP antibody. Total T. brucei proteins used for in vitro kinase assay were stained with Coomassie Blue. IB, immunoblot.
To examine whether the purified CRK1as-CYC2 complex was able to thiophosphorylate native trypanosome proteins, in vitro kinase assays were carried out by incubating recombinant CRK1as with crude trypanosome cell lysate in the presence of N6-(benzyl)ATPγS. To compete off nonspecific use of N6-(benzyl)ATPγS by metabolic enzymes and nucleotide exchange factors, GTP was added to the reaction. The results showed that numerous trypanosome proteins were thiophosphorylated by CRK1as (Fig. 1D). With increased concentration of GTP, nonspecific background signal was significantly decreased, and specific labeling of proteins by CRK1as was increased (Fig. 1D). These results validated the chemical genetic approach for CRK1 substrate labeling and identification.
Purification of thiophosphopeptides for mass spectrometric analysis was carried out essentially as described (30, 31). The total trypanosome proteins that had been incubated with N6-(benzyl)ATPγS in the presence (experimental sample) or absence (control sample) of the CRK1as-CYC2 complex were digested to peptides with trypsin, and the products were incubated with iodoacetyl-agarose, which allows the thio-containing groups to react to form covalent bonds. Thiophosphopeptides were liberated by oxidation-based hydrolysis of the sulfur-phosphorus bond, resulting in the replacement of the thiophosphoryl sulfur atom by oxygen (30). Purified phosphopeptides were then analyzed by LC-MS/MS and searched against the T. brucei proteome database. The proteins thus identified were compared between the experimental sample and the control sample, and 17 proteins were only present in the experimental sample but not the control sample. Ten out of the 17 proteins contain the consensus CDK phosphorylation sequence Ser*/Thr*-Pro (the asterisks indicate the site of phosphorylation), suggesting that they are potential CRK1 substrates. Of the 10 proteins, seven are hypothetical proteins of unknown function, two are protein translation initiation factors (eIF4E4 and Polyadenylate-binding protein 2), and one is Sec31, a subunit of the COPII coat protein complex (Table 1). Sec31 was of particular interest and thus was further characterized in this study.
TABLE 1.
CRK1 substrates identified by chemical genetics
| Gene ID | Phospho-type | Phosphopeptide (underlined, phosphosite) | Protein description |
|---|---|---|---|
| Tb927.11.6170 | CDK (Ser/Thr-Pro) | KNDRTPILSY | Coat protein II complex subunit Sec31 |
| Tb927.6.1870 | CDK (Ser/Thr-Pro) | IPTRMSPVAHP | Eukaryotic translation initiation factor 4E4 |
| Tb927.9.9290 | CDK (Ser/Thr-Pro) | VPRTPQASPAIAPDTPP | Polyadenylate-binding protein1 (PABP1) |
| Tb927.8.1780 | CDK (Ser/Thr-Pro) | TSGFTTPRPTF | Putative protein kinase |
| Tb927.9.12360 | CDK (Ser/Thr-Pro) | SSPTSSPSLRS | Putative RNA-binding protein |
| Tb927.3.4270 | CDK (Ser/Thr-Pro) | NAGQGSPSFSPKSPS | Hypothetical protein |
| Tb927.3.3300 | CDK (Ser/Thr-Pro) | PAPALSPRL | Hypothetical protein |
| Tb927.10.13800 | CDK (Ser/Thr-Pro) | HGEPSTPHTFS | Hypothetical protein |
| Tb927.4.2750 | CDK (Ser/Thr-Pro) | VTGDASPILSS | Hypothetical protein |
| Tb927.7.6820 | CDK (Ser/Thr-Pro) | WALETPPMFAQE | Hypothetical protein |
Sec31 Is an in Vivo Substrate of CRK1
Mass spectrometry identified Thr-442 in Sec31 as an in vitro CRK1 phosphosite (Table 1), but it is unclear whether this site is phosphorylated in vivo and whether Sec31 possesses additional phosphosites. To identify the in vivo phosphorylation sites in Sec31, we tagged Sec31 with a triple HA epitope at one of its endogenous loci and then immunoprecipitated Sec31–3HA for mass spectrometric analysis. This identified eight phosphosites, Thr-21, Thr-442, Thr-695, Thr-960, Thr-970, Ser-1013, Ser-1014, and Ser-1016. Except for Ser-1013, the other seven phosphosites are each followed by a proline residue, which are characteristic CDK phosphorylation sites. To test whether Sec31 is phosphorylated by CRK1 on the seven CDK consensus phosphosites, we carried out in vitro kinase assays with purified wild-type Sec31 and the Sec31-7A mutant, in which all of the seven putative CRK1 phosphosites were mutated to alanine (Fig. 2A), and we found that immunoprecipitated CRK1 was capable of phosphorylating Sec31 but not Sec31-7A (Fig. 2B), suggesting that Sec31 is an in vitro substrate of CRK1. To further confirm the in vivo phosphorylation of these seven CDK phosphosites, we overexpressed wild-type Sec31 and the Sec31-7A mutant in trypanosomes. 3HA-tagged Sec31 and Sec31-7A were immunoprecipitated with anti-HA antibody and then immunoblotted with anti-phosphothreonine-proline (α-Thr(P)-Pro) antibody, which detects the phosphorylated threonine and serine residues followed by a proline. The results showed that Sec31, but not Sec31-7A, was detected by anti-Thr(P)-Pro antibody (Fig. 2C), further confirming that these sites are phosphorylated in vivo. Finally, to examine whether CRK1 is responsible for Sec31 phosphorylation in trypanosomes, 3HA-tagged Sec31 was expressed from its endogenous locus in the CRK1 RNAi cell line, immunoprecipitated, and then immunoblotted with anti-Thr(P)-Pro antibody. Depletion of CRK1 caused reduced phosphorylation of Sec31 in trypanosome cells (Fig. 2D). The lack of complete elimination of Sec31 phosphorylation by CRK1 depletion could be due to the incomplete knockdown of CRK1 by RNAi. Nevertheless, this result confirms that Sec31 is an in vivo substrate of CRK1.
FIGURE 2.

Sec31 is phosphorylated by CRK1 at multiple serine and threonine residues. A, schematic drawing of the Sec31 protein, showing the seven serine and threonine residues that are phosphorylated in vivo and are putative CRK1 phosphosites. B, phosphorylation of Sec31 by CRK1 in vitro. Sec31 and Sec31-7A were purified as GST fusion protein from E. coli. CRK1-PTP was immunoprecipitated from trypanosome cell lysate. Phosphorylated GST-Sec31 (p-GST-Sec31) was detected by Western blotting with anti-phosphothreonine-proline (α-pT-P) antibody. GST-Sec31 was detected with anti-GST antibody, whereas CRK1-PTP was detected with anti-protein A antibody (α-ProtA). C, confirmation of in vivo Sec31 phosphorylation by Western blotting with anti-phosphothreonine-proline antibody. Sec31 and Sec31-7A were overexpressed in T. brucei, immunoprecipitated (IP) with anti-HA antibody, and then immunoblotted (IB) with anti-phosphothreonine-proline (α-pT-P) antibody to detect the phosphorylated serine and threonine residues followed by a proline in Sec31–3HA and with anti-HA antibody to detect 3HA-tagged Sec31 and Sec31-7A. D, depletion of CRK1 caused dephosphorylation of Sec31 in T. brucei. Sec31 was endogenously tagged with a C-terminal triple HA epitope in the T. brucei cell line harboring the CRK1 RNAi construct. RNAi was induced for 2 days. Sec31–3HA was immunoprecipitated from non-induced control and CRK1 RNAi cells, and immunoblotted with anti-phosphothreonine-proline (α-pT-P) antibody to detect phosphorylated Sec31–3HA and with anti-HA antibody to detect Sec31–3HA. The wild-type 29-13 cell line served as a mock control.
Subcellular Localization of Sec31 and Its Co-localization with CRK1
To determine the subcellular localization of Sec31, we tagged the endogenous Sec31 with EYFP at its C terminus in trypanosome cells expressing mCherry-tagged Sec13, which has previously been shown to localize to the ERES (32). Fluorescence microscopy showed that Sec31-EYFP co-localizes with Sec13-mCherry (Fig. 3A). We also examined the localization of Sec31 relative to the Golgi, and the results showed that Sec31-EYFP was juxtaposed to the Golgi, which was labeled by the Golgi stack marker TbGRASP (Golgi reassembly stacking protein) (Fig. 3B). These results confirmed that Sec31 localizes to the ERES.
FIGURE 3.
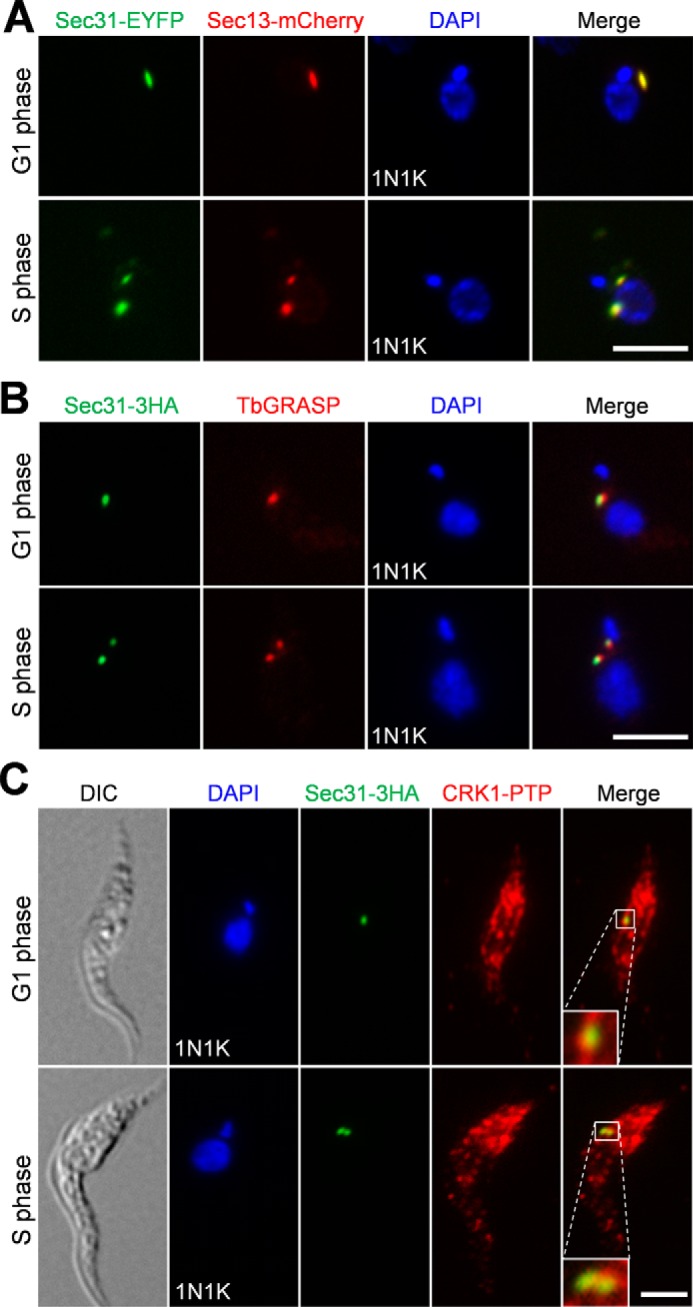
Subcellular localization of Sec31 and its co-localization with CRK1. A, Sec31 co-localizes with Sec13 in the ERES. Cells co-expressing endogenously EYFP-tagged Sec31 and mCherry-tagged Sec13 were fixed, stained with DAPI, and examined under a fluorescence microscope. B, Sec31 was juxtaposed to the Golgi. Cells expressing EYFP-tagged Sec31 were immunostained with the anti-TbGRASP antibody, which stains the Golgi stack protein TbGRASP, and counterstained with DAPI for DNA. C, co-localization of Sec31 and CRK1. Cells co-expressing 3HA-tagged Sec31 and PTP-tagged CRK1 were co-immunostained with FITC-conjugated anti-HA mAb and anti-protein A polyclonal antibody to label Sec31–3HA and CRK1-PTP, respectively. Scale bars, 5 μm. DIC, differential interference contrast.
We next investigated the co-localization of Sec31 with CRK1. To this end, CRK1 was endogenously tagged with a PTP epitope, and Sec31 was endogenously tagged with a triple HA epitope in the same cell line. Immunofluorescence microscopy showed that CRK1-PTP was detected in many punctate dots in the cytosol (Fig. 3C), as reported previously (27), and it co-localized with Sec31-HA at the ERES (Fig. 3C).
Sec31 Is Required for Cell Division, Cell Morphogenesis, and Golgi Protein Localization
To understand the function of Sec31 in T. brucei, we carried out RNAi in the procyclic form of T. brucei. Knockdown of Sec31 by RNAi was confirmed by quantitative RT-PCR, which showed that the mRNA level of Sec31 was reduced ∼75% after RNAi induction for 24 h (Fig. 4A), and by Western blotting and immunofluorescence microscopy, which showed that Sec31 protein was decreased to an undetectable level after RNAi induction for 48 h (Fig. 4, B and C). This depletion of Sec31 caused severe growth inhibition and eventual cell death after RNAi induction for 3 days (Fig. 4D), suggesting that Sec31 is essential for cell proliferation and cell viability in the procyclic form.
FIGURE 4.

Sec31 is required for cell viability and for localization of Golgi proteins TbGRASP and TbGntB. A, levels of Sec31 mRNA in non-induced control and Sec31 RNAi cells as measured by quantitative RT-PCR. RNAi was induced for 24 h. The error bar represents S.D. from three independent experiments. B, levels of Sec31–3HA in non-induced control and Sec31 RNAi cells were detected by Western blotting. Sec31 was endogenously tagged with a triple HA epitope in cells harboring the Sec31 RNAi construct. Levels of TbPSA6, the α-6 subunit of the 26S proteasome, served as the loading control. C, immunofluorescence microscopy to detect Sec31 in non-induced control and Sec31 RNAi cells. Cells were immunostained with FITC-conjugated anti-HA antibody and counterstained with DAPI to stain DNA. Arrows indicate Sec31–3HA signal. Scale bar, 5 μm. D, depletion of Sec31 resulted in growth inhibition and cell death. E, depletion of Sec31 caused G2/M arrest. Shown are flow cytometry histograms of the control and Sec31 RNAi cells induced for 24 and 48 h. Tet, tetracycline. F, quantification of the flow cytometry data. The error bars represent S.D. calculated from three independent experiments. G, tabulation of cells with different numbers of nuclei (N) and kinetoplasts (K). A total of 300 cells for each time point were counted, and error bars indicate S.D. calculated from three independent experiments. H, percentage of round-shaped cells in control and Sec31 RNAi cells. A total of 200 cells from each time point were counted, and error bars represent S.D. calculated from three independent experiments. I, depletion of Sec31 disrupted the localization of two Golgi proteins TbGRASP and TbGntB. TbGntB was endogenously tagged with a triple HA epitope in Sec31 RNAi cell line. Control and Sec31 RNAi (48 h) cells were co-immunostained with anti-TbGRASP antibody and FITC-conjugated anti-HA antibody and counterstained with DAPI for DNA. Arrows and arrowheads indicate the TbGRASP and TbGntB signal in the Golgi, respectively. Scale bar, 5 μm. J, quantification of cells with discrete TbGRASP and TbGntB spots in control and Sec31 RNAi (48 h) cells. A total of 300 cells from each time point were counted, and the error bar indicates S.D. calculated from three independent experiments. ***, p < 0.001. K, effect of Sec31 RNAi on the protein level of TbGRASP and TbGntB. Control and Sec31 RNAi (48 h) cells were immunoblotted with anti-TbGRASP and anti-HA to detect TbGRASP and TbGntB-3HA, respectively. TbPSA6 served as the loading control. DIC, differential interference contrast.
To further characterize the cell proliferation defect, flow cytometry was performed, which detected a significant increase of cells with 4C DNA content after Sec31 RNAi for 48 h (Fig. 4, E and F), indicating that the Sec31 RNAi cells were arrested at the G2/M phase. To identify the specific cell cycle stage that was blocked by Sec31 RNAi, cells were stained with DAPI and counted for the numbers of nuclei and kinetoplasts. The results showed that binucleate (2N2K and 2N1K) cells were increased from ∼3 to ∼65% of the total cell population after Sec31 RNAi for 48 h (Fig. 4G), suggesting a cytokinesis defect. Moreover, the accumulation of two nuclei and one kinetoplast (2N1K) cell (Fig. 4G) indicated that kinetoplast division was also inhibited. In addition to the cell cycle defect, Sec31 RNAi also disrupted cell morphology, as ∼84% of the Sec31 RNAi cells lost typical slender morphology after RNAi induction for 48 h, becoming somewhat round in shape (Fig. 4, C, H, and I).
Previous work reported that depletion of TbSec24.1, another COPII subunit in T. brucei, disrupted the localization of several Golgi proteins, such as the Golgi stack proteins TbGRASP and TbGolgin63, likely due to the general defect in Golgi structure (33). Depletion of TbSec24.1 also caused an accumulation of the Golgi enzyme TbGntB, an N-acetyl-d-glucosamine (GlcNAc) transferase, in the ER and a slight decrease of TbGntB in the Golgi (33). Given that Sec31 is another key component of the COPII complex, we reasoned that Sec31 RNAi might cause a similar defect in the localization of these Golgi proteins. To test this possibility, we tagged TbGntB with a triple HA epitope in the Sec31 RNAi cell line. Immunofluorescence microscopy showed that TbGRASP and TbGntB co-localize to the Golgi in control cells, and depletion of Sec31 impaired the localization of both proteins in most of the cells (Fig. 4, I and J). The protein levels of TbGRASP and TbGntB were not altered in Sec31 RNAi cells (Fig. 4K). Together, these results suggest that Sec31 is also required for TbGRASP and TbGntB localization. Unfortunately, despite numerous attempts, we were unable to tag TbGolgin63 in Sec31 RNAi cells due to unknown reasons, and thus the effect of Sec31 depletion on TbGolgin63 localization was not assessed. Given that TbGRASP is a Golgi stack protein required for Golgi stacking in T. brucei (32), the mis-localization of TbGRASP in Sec31 RNAi cells suggests that Golgi structure may be disturbed, as is the case of TbSec24.1 depletion (33).
CRK1 Is Also Required for Golgi Protein Localization
Given that CRK1 phosphorylates Sec31 (Fig. 2), we investigated whether depletion of CRK1 also impairs the localization of TbGRASP to the Golgi. Indeed, depletion of CRK1 by RNAi caused the mis-localization of TbGRASP in more than 80% of the cells after CRK1 RNAi induction for 3 days (Fig. 5, A and B), suggesting that CRK1 is required for TbGRASP localization. CRK1 is known to associate with four PHO80-like cyclins, CYC2, CYC4, CYC5, and CYC7 (27). However, only the depletion of CYC2 or CYC7 generated cells with an elongated posterior, as the depletion of CRK1 (27), suggesting that CYC2 and CYC7 play specialized functions with CRK1. Therefore, we examined whether depletion of CYC2 and CYC7 also affected TbGRASP localization, and the results showed that knockdown of CYC2 or CYC7 alone did not affect TbGRASP localization, but depletion of CYC2 and CYC7 simultaneously disrupted TbGRASP localization in ∼65% of the cells after RNAi induction for 3 days (Fig. 5, A and B). Depletion of CRK1 or CYC2 and CYC7 did not alter TbGRASP protein levels (Fig. 5C). These results further confirmed that CYC2 and CYC7 play redundant roles and suggest that the two cyclins function together with CRK1 to regulate COPII-mediated protein transport.
FIGURE 5.

Depletion of CRK1 disrupted the localization of Golgi proteins TbGRASP, TbGntB, and TbGolgin63. A, effect of CRK1, CYC2, and CYC7 depletion on TbGRASP localization. Non-induced control cells and cells induced for CRK1 RNAi (3 days), CYC2 RNAi (3 days), CYC7 RNAi (3 days), and CYC2-CYC7 double RNAi (3 days) were immunostained with anti-TBGRASP antibody and counterstained with DAPI for DNA. Arrows indicate the TbGRASP signal in the Golgi. Scale bar, 5 μm. B, quantification of cells with discrete TbGRASP spots in control and RNAi cells. A total of 300 cells were counted for each cell line, and results were presented as mean percentage ± S.D. (n = 3). ***, p < 0.001. C, effect of depletion of CRK1 and its associated cyclins on TbGRASP protein level. TbGRASP was detected by anti-TbGRASP antibody. TbPSA6 served as the loading control. D and E, effect of CRK1 depletion on TbGntB and TbGolgin63 localization. TbGntB and TbGolgin63 (TbG63) were each endogenously tagged with a triple HA epitope in CRK1 RNAi cell line. RNAi was induced for 3 days and collected for immunofluorescence microscopy with FITC-conjugated anti-HA antibody. Arrows indicate TbGntB and TbGolgin63 signal in the Golgi. Scale bar, 5 μm. F and G, effect of CRK1 RNAi on the protein level of TbGntB and TbGolgin63. Western blotting was performed with anti-HA antibody to detect TbGntB-3HA and TbGolgin63–3HA (TbG63–3HA). TbPSA6 served as the loading control. DIC, differential interference contrast.
We next investigated the effect of CRK1 depletion on the localization of TbGntB and TbGolgin63. TbGntB and TbGolgin63 were each tagged at their respective endogenous locus in CRK1 RNAi cell line. Immunofluorescence microscopy showed that the localizations of both TbGntB and TbGolgin63 were disrupted in CRK1 RNAi-induced cells (Fig. 5, D and E). The levels of both proteins were not changed upon CRK1 RNAi (Fig. 5, F and G). Given that CRK1 RNAi impaired the localization of the Golgi stack proteins TbGRASP and TbGolgin63 and the Golgi enzyme TbGntB, these results suggest that CRK1 depletion may cause a general defect in the Golgi structure.
CRK1 Is Not Required for Sec31 Localization and Sec31-Sec13 Complex Formation
To investigate whether CRK1-mediated phosphorylation of Sec31 is required for COPII complex formation, we carried out co-immunofluorescence microscopy and co-immunoprecipitation experiments to examine the co-localization and the in vivo interaction of Sec31 and Sec13, which were endogenously tagged with a PTP epitope and a triple HA epitope, respectively, in the CRK1 RNAi cell line. The results showed that Sec31-PTP and Sec13–3HA still co-localized in CRK1 RNAi cells (Fig. 6A) and were co-precipitated by immunoprecipitation from the CRK1 RNAi cell lysate (Fig. 6B). These results suggest that phosphorylation of Sec31 by CRK1 is not involved in Sec31 localization and Sec31-Sec13 complex formation.
FIGURE 6.
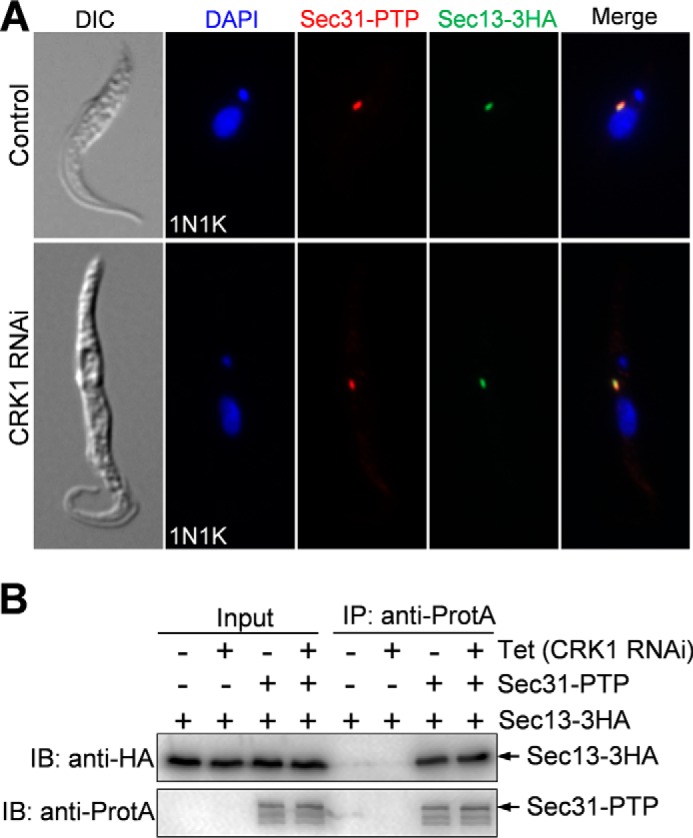
Depletion of CRK1 does not affect the localization of Sec31 to the ERES and the interaction of Sec31 with Sec13. A, effect of CRK1 depletion on Sec31 localization. Sec31 and Sec13 were endogenously tagged with PTP and a triple HA epitope, respectively, in CRK1 RNAi cell line. CRK1 RNAi was then induced for 3 days, and co-immunofluorescence microscopy was then performed with anti-protein A polyclonal antibody and FITC-conjugated anti-HA monoclonal antibody. Cells were counterstained with DAPI for DNA. B, effect of CRK1 depletion on Sec31-Sec13 complex formation, as measured by co-immunoprecipitation. Sec31 and Sec13 were tagged with a PTP epitope and a triple HA epitope, respectively, in cells containing the CRK1 RNAi construct. CRK1 RNAi was induced for 3 days, and co-immunoprecipitation was carried out in non-induced control (−Tet) and CRK1 RNAi (+Tet) cells. Tet, tetracycline. Immunoprecipitation (IP) was performed by incubating the cell lysate with IgG-Sepharose beads, and the immunoprecipitated proteins were separated on SDS-PAGE and immunoblotted (IB) with anti-HA antibody and anti-protein A antibody (anti-ProtA) to detect Sec13–3HA and Sec31-PTP, respectively. As negative controls, non-induced control and CRK1 RNAi cells expressing the 3HA-tagged Sec13 were used. DIC, differential interference contrast.
Phosphorylation of Sec31 by CRK1 Is Essential for Sec31 Function
To investigate whether CRK1-mediated phosphorylation on Sec31 is required for the cellular function of Sec31, we mutated all seven CRK1 phosphosites on Sec31 to alanine (Sec31-7A) or aspartate (Sec31-7D) for expression of phosphodeficient and phosphomimic mutants in trypanosomes. Wild-type Sec31 and the two Sec31 mutants were each tagged with a C-terminal triple HA epitope and overexpressed in the procyclic form of T. brucei under a tetracycline-inducible promoter, which was confirmed by Western blotting with the anti-HA antibody (Fig. 7A). Overexpression of Sec31-7A, but not Sec31, caused severe growth inhibition and cell death after tetracycline induction for 3 days (Fig. 7B), suggesting a dominant-negative effect caused by Sec31-7A overexpression. Overexpression of Sec31-7D caused slight growth defects (Fig. 7B), suggesting that the phosphomimic mutation partially restored the function of Sec31.
FIGURE 7.

Requirement of Sec31 phosphorylation by CRK1 for Sec31 function. A, Western blotting to detect the overexpression (OE) of Sec31 and its phosphodeficient mutant (Sec31-7A) and phosphomimic mutant (Sec31-7D), which was induced in the presence (+) of 100 ng/ml tetracycline. Levels of TbPSA6 served as the loading control. B, effect of overexpression of Sec31 phosphodeficient and phosphomimic mutants on cell proliferation. Cells were incubated with 0.5 μg/ml tetracycline to induce the overexpression of wild-type and mutant Sec31 proteins. C, tabulation of cells with different numbers of nuclei (N) and kinetoplasts (K) of the overexpression cell lines. A total of 200 cells were counted for each time point, and error bars represent S.D. calculated from three independent experiments. D, quantification of abnormal cells in the overexpression cell lines. A total of 300 cells were counted for each time point, and error bars indicate S.D. calculated from three independent experiments. E, quantification of cells with discrete TbGRASP spots in control and overexpression cells. A total of 300 cells were counted for each cell line, and results were presented as mean percentage ± S.D. (n = 3). ***, p < 0.001. F, effect of overexpression of mutant and wild-type Sec31 on TbGRASP localization. Non-induced control and tetracycline-induced overexpression cells (Sec31 wild-type and mutants) were co-immunostained with anti-TbGRASP and FITC-conjugated anti-HA antibodies. Arrows indicate the TbGRASP signal in the Golgi. Scale bar, 5 μm. G, effect of overexpression of Sec31, Sec31-7A, and Sec31-7D on the protein level of TbGRASP, which was detected by anti-TbGRASP. TbPSA6 served as the loading control. H, effect of overexpression of mutant and wild-type Sec31 on TbGntB localization. TbGntB was endogenously tagged with a PTP epitope in cells containing the overexpression construct. Overexpression of wild-type and mutant Sec31 was induced with tetracycline, and control and overexpression cells were immunostained with anti-protein A antibody to detect TbGntB-PTP. Arrows indicate TbGntB-PTP signal in the Golgi. Scale bar, 5 μm. I, effect of overexpression of Sec31, Sec31-7A, and Sec31-7D on the protein stability of TbGntB, which was tagged with PTP and detected by anti-protein A antibody. TbPSA6 served as the loading control. DIC, differential interference contrast.
To further characterize the defects caused by Sec31-7A overexpression, we analyzed which cell cycle stage was affected by counting the cells with different numbers of nuclei and kinetoplasts. The three overexpression cell lines (Sec31, Sec31-7A, and Sec31-7D) were thus compared. Overexpression of Sec31 exerted little effect on the cell cycle progression, but overexpression of Sec31-7A resulted in an accumulation of 2N2K and 2N1K cells to ∼40 and ∼30% of the total cell population, respectively (Fig. 7C), and disrupted cell morphology, with ∼90% of the cells becoming rounded up after tetracycline induction for 48 h (Fig. 7D). In all of these round-shaped cells caused by Sec31-7A overexpression, TbGRASP localization was impaired (Fig. 7, E and F), although the protein level of TbGRASP was not affected (Fig. 7G). Similarly, overexpression of Sec31-7A also disrupted TbGntB localization (Fig. 7H) but did not alter the TbGntB protein level (Fig. 7I). The defects caused by Sec31-7A overexpression mimic that by Sec31 RNAi (Fig. 4). In contrast, overexpression of Sec31-7D only resulted in an increase of 2N1K cells to ∼6% of the total cell population (Fig. 7C) and an accumulation of round-shaped cells to ∼14% of the total population (Fig. 7D), which is consistent with the fact that overexpression of Sec31-7D caused a slight growth defect (Fig. 7B). Taken together, these results suggest that phosphorylation of Sec31 at the seven Ser/Thr residues by CRK1 is essential for Sec31 function.
Phosphorylation of Sec31 by CRK1 Is Required for Anterograde Protein Trafficking
We next investigated whether Sec31 is required for anterograde trafficking of proteins, which are transported from the ER, via the Golgi, to the cell surface. To this end, we monitored the secretion of the secretory reporter protein BiPNAVRG-HA9 in control and Sec31 RNAi cells. BiPNAVRG-HA9 is an artificial protein consisting of 415 amino acids of the TbBiP ATPase domain fused with the C-terminal hexapeptide QPAVRG and a 9×HA epitope. Because of the lack of the C-terminal ER retention sequence MDDL, BiPNAVRG-HA9 is not retained in the ER but is secreted into the culture medium (34). BiPNAVRG-HA9 was stably expressed in cells harboring Sec31 RNAi construct, and RNAi was induced for 24 h. The control and Sec31 RNAi cells were first washed with fresh medium and then treated with cycloheximide for up to 4 h. BiPNAVRG-HA9 was immunoprecipitated with anti-HA antibody from cell lysates and media that were collected at different time points of cycloheximide treatment and then detected by Western blotting with anti-HA antibody. In the non-induced control cells, BiPNAVRG-HA9 was gradually secreted from the cells into the medium, with the half-time (t½) estimated to be around 90–100 min (Fig. 8A). Strikingly, depletion of Sec31 by RNAi completely blocked BiPNAVRG-HA9 secretion into the medium (Fig. 8A), suggesting that Sec31 is required for anterograde protein trafficking in T. brucei.
FIGURE 8.
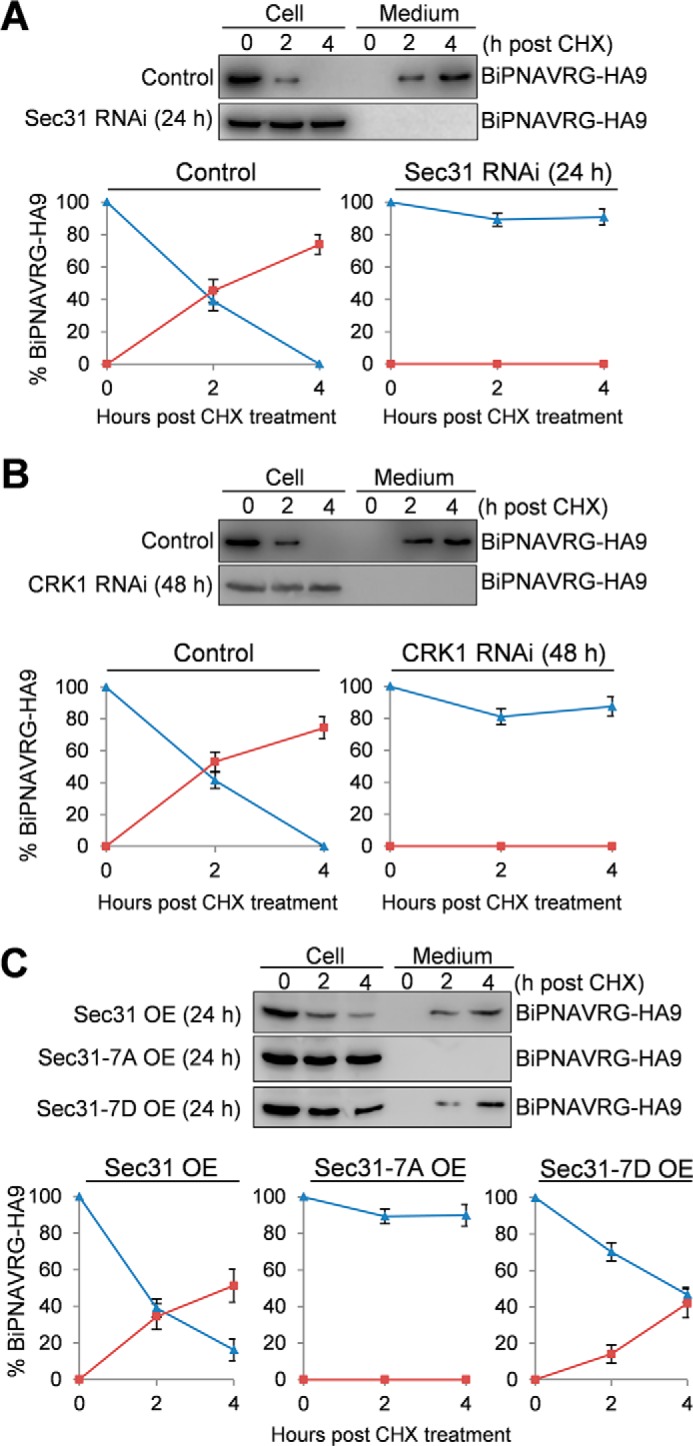
CRK1-mediated phosphorylation of Sec31 is required for anterograde transport of the secreted protein. Cells harboring the Sec31 RNAi construct (A), the CRK1 RNAi construct (B), or the overexpression construct containing either wild-type Sec31, Sec31-7A, or Sec31-7D (C) were stably transfected with a construct encoding the secretory protein BiPNAVRG-HA9. After induction with tetracycline for 24 h (Sec31 RNAi and overexpression of wild-type and mutant Sec31 proteins) or 48 h (CRK1 RNAi), cells were washed with fresh medium, resuspended in fresh medium, and treated with cycloheximide (CHX) for 4 h. Samples from cells and the medium were collected at the indicated times. BiPNAVRG-HA9 was immunoprecipitated from cell lysate and culture medium by anti-HA antibody, fractionated by SDS-PAGE, and detected by anti-HA antibody. Band intensity was determined using the ImageJ software, plotted against the time of cycloheximide treatment, and presented below the Western blots. Blue lines indicate the BiPNAVRG-HA9 protein detected in cells (non-secreted protein), and red lines indicate the BiPNAVRG-HA9 protein detected in the medium (secreted protein). Error bars indicate S.D. calculated from three independent experiments.
We next investigated whether CRK1 is also required for anterograde protein trafficking by monitoring the secretion of BiPNAVRG-HA9. Similar to Sec31 RNAi, knockdown of CRK1 by RNAi also completely blocked the secretion of BiPNAVRG-HA9 into the medium (Fig. 8B). Furthermore, to examine whether CRK1-mediated phosphorylation of Sec31 is required for its function in anterograde protein transport, BiPNAVRG-HA9 was stably expressed in cells overexpressing Sec31, Sec31-7A, or Sec31-7D. In Sec31-overexpressing cells and Sec31-7D-overexpressing cells, BiPNAVRG-HA9 was secreted into the medium, albeit the latter had a slower rate than the former, with the half-time (t½) of BiPNAVRG-HA9 export estimated to be about 220–230 min in Sec31-7D-overexpressing cells and about 90–100 min in Sec31-overexpressing cells (Fig. 8C). However, in Sec31-7A-overexpressing cells, secretion of BiPNAVRG-HA9 into the medium was completely blocked (Fig. 8C). These results suggest that phosphorylation of Sec31 at the seven Ser/Thr sites by CRK1 is essential for its function in anterograde protein trafficking.
Discussion
In this report, we applied the chemical genetic approach to screen for substrates of CRK1, a crucial cyclin-dependent kinase involved in the G1/S transition in T. brucei, and we identified Sec31 as a novel substrate of CRK1. Several lines of evidence suggest that Sec31 is a bona fide CRK1 substrate. First, it was specifically thiophosphorylated in the crude cell lysate by CRK1as in the presence of N6-(benzyl)ATPγS (Fig. 1 and Table 1). Second, purified recombinant Sec31 protein was phosphorylated in vitro by CRK1 (Fig. 2B). Third, depletion of CRK1 caused significant de-phosphorylation of Sec31 in trypanosome cells (Fig. 2D). Finally, Sec31 was phosphorylated at multiple CDK consensus sites in vivo in trypanosomes (Fig. 2, A and B), and mutation of these phosphosites made it unable to be phosphorylated by CRK1 in vitro (Fig. 2B). Sec31 is the first CRK1 substrate identified and functionally characterized so far.
The ER-to-Golgi secretory pathway in T. brucei shares similarities with that in other model organisms (35). T. brucei expresses the orthologs of all the COPII components, including one Sec13 (32), one Sec31, and two each for Sec23 and Sec24 (33, 36). All of these proteins localize to the ERES (32, 33, 36), which is in close proximity to the Golgi apparatus (Fig. 3B). The two Sec23 homologs and two Sec24 homologs form exclusive heterodimers, TbSec23.1-TbSec24.2 and TbSec23.2-TbSec24.1, and are all essential in the bloodstream form of T. brucei (36). The two Sec23-Sec24 heterodimers appear to be involved in transport of distinct secretory cargos (33, 36). Our RNAi data demonstrated that Sec31 is essential in the procyclic form of T. brucei (Fig. 4D) and is required for localization of Golgi proteins TbGRASP and TbGntB and for anterograde transport of the secretion marker BiPNAVRG-HA9 (Figs. 4, I and J, and 8A), similar to the defects caused by knockdown of TbSec24.1 in the procyclic form (33). These results provided compelling evidence that the T. brucei Sec31 homolog functions in the same secretory pathway as TbSec24.1.
RNAi of Sec31 also arrested cytokinesis and distorted cell morphology (Fig. 4, E–H), suggesting that Sec31 plays additional roles in cell division and cell morphogenesis. The requirement of Sec31 in cell cycle progression was also observed in the fission yeast, in which Sec31 null mutation caused accumulation of bi-nucleate cells (37). Importantly, this cell cycle defect was not attributed to a general defect in protein secretion (37). This notion is also supported by the finding that TbSec24.1 and TbSec24.2 RNAi did not cause cell cycle arrest, despite the protein secretion defect (33). It should be noted that depletion of Sec31 also caused defective kinetoplast duplication/segregation, producing 2N1K cells (Fig. 4G), which was due to inhibition of basal body segregation (data not shown). Although inhibited basal body segregation can lead to cytokinesis arrest and disrupts cell morphology in T. brucei (38), it is unlikely to contribute directly to the defects in cytokinesis and cell morphology in Sec31 RNAi cells, as the Sec31-deficient 2N2K cells were similarly arrested in cytokinesis and lost cell morphology (Fig. 4, C and G). The precise mechanisms underlying the role(s) of Sec31 in cell division and cell morphogenesis remain to be explored. Given that Sec31 forms the outer layer of the COPII complex with Sec13, it would be interesting to investigate whether depletion of Sec13 causes similar defects in cell division and cell morphology.
CDKs are known to also play important roles in a vast array of cellular processes that, in the traditional viewpoint, are not under cell cycle control. These include transcription, mRNA processing, translation, etc. (39). Our work uncovered a new function for CRK1, a G1 CDK in T. brucei, in the regulation of COPII-mediated anterograde protein trafficking, which is supported by the findings that CRK1 phosphorylates Sec31 in vivo in trypanosomes (Fig. 2) and mutation of CRK1 phosphorylation sites on Sec31 disrupted the function of Sec31 in protein secretion (Fig. 8C) and that depletion of CRK1 abolished protein secretion (Fig. 8B). This new function for CRK1 appears to be independent of its role in the G1/S cell cycle control, as the Sec31 phospho-deficient (Sec31-7A) mutant was arrested at cytokinesis but not at the G1/S boundary of the cell cycle (Fig. 7C).
The mechanistic role of CRK1-mediated Sec31 phosphorylation is still unclear. Our data showed that CRK1 is neither required for Sec31 localization to ERES nor for Sec31-Sec13 complex formation (Fig. 6). Human Sec31 is phosphorylated by casein kinase II, and this phosphorylation is also not required for Sec31 localization, but the non-phosphorylatable form of Sec31 becomes less dynamic at ERES in HeLa cells (21). Surprisingly, phosphorylation of Sec31 by casein kinase II appears to decrease its binding to Sec13, despite that the depletion of casein kinase II caused ER-to-Golgi trafficking defects (21). It is not clear whether CRK1 phosphorylation of Sec31 alters the dynamics of Sec31 at the ERES, but clearly it does not have any impact on Sec31-Sec13 interaction. Given that the Sec31-Sec13 complex is involved in vesicle fission from the ER membrane (40) and in accommodating large cargo molecules (41), CRK1 phosphorylation might affect Sec31 function in assembly of the outer layer Sec13-Sec31 coat to incorporate larger cargo molecules or in deformation of ER membrane into vesicles, which needs to be further explored.
Sec31 from Saccharomyces cerevisiae was also identified as a substrate of Cdk1 (42), but whether Cdk1-mediated phosphorylation of Sec31 is required for protein trafficking in the budding yeast was not investigated in that study. Cdk1 phosphorylates yeast Sec31 at Ser-836 and Ser-980 (42), both of which do not have equivalent sites in T. brucei Sec31. Conversely, the seven CRK1 phosphosites in T. brucei Sec31 (Fig. 2) also do not have equivalent sites in yeast and human Sec31 proteins. It should be noted that Sec31 homologs from T. brucei, budding yeast, and humans only share an overall sequence identity of ∼20–25%. Therefore, the lack of equivalent Cdk1 phosphorylation sites among these Sec31 homologs does not necessarily reflect any functional distinction. It is not known whether human Sec31 is a substrate of CDK1, but a CDK-related PCTAIRE protein kinase interacts with Sec23 and regulates ER-to-Golgi trafficking (19). Nevertheless, the identification of Sec31 from S. cerevisiae and T. brucei, two diverged eukaryotes, as a CDK substrate suggests a likely conserved function for CDK in regulating ER-to-Golgi protein trafficking by modulating the function of Sec31 via phosphorylation.
Experimental Procedures
Expression and Purification of Recombinant CRK1, Analog-sensitive CRK1, and CYC2 in Escherichia coli
The full-length coding sequence of CYC2 was PCR-amplified and cloned into the pGEX-4T-3 vector for expression of GST-tagged CYC2. Full-length coding sequences of CRK1 and the budding yeast Cak1p, the CDK activating kinase, were PCR-amplified and cloned into pCDF-Duet1 vector (Novagen) for co-expression of His6-tagged CRK1 and Cak1p. To generate the analog-sensitive CRK1 mutant, site-directed mutagenesis was performed to mutate Phe-81 to alanine. The pGEX-CYC2 plasmid and the pCDF-CRK1-Cak1p plasmid were transformed into E. coli BL21 Rosetta strain (Novagen). The strain with the transformed plasmids was cultured in LB medium supplemented with ampicillin, spectinomycin, and chloramphenicol.
Expression of recombinant proteins was induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 6 h at 27 °C. Cells were lysed in BugBuster reagent (Novagen) supplemented with 1 mm ATP and 10 mm MgCl2, and the cleared cell lysate was incubated with glutathione-Sepharose 4B beads for 30 min at 4 °C. Beads were washed with 60 column volumes of lysis buffer (50 mm Tris-Cl, pH 7.5, 0.3 m NaCl, 0.1% Triton X-100, and 0.01% glycerol) supplemented with 1 mm ATP and 10 mm MgCl2. Proteins bound to the beads were eluted with 2 column volumes of elution buffer (50 mm Tris-Cl, pH 7.5, 10 mm reduced glutathione) and concentrated and dialyzed against the storage buffer (50 mm HEPES, pH 7.5, 0.1 m NaCl, 1 mm DTT, 0.1 mm EDTA, 0.01% Tween 20, and 30% glycerol). Purified proteins were analyzed by SDS-PAGE followed by Coomassie Blue staining and Western blotting with anti-GST and anti-His antibodies. Western blotting showed that recombinant CRK1-His6 (and CRK1as-His6) was co-purified with GST-CYC2, due to binding to the latter.
In Vitro Kinase Assay
For in vitro kinase assay to test whether CRK1as can use N6-(benzyl)ATPγS, the CRK1-CYC2 complex and the CRK1as-CYC2 complex purified above were used, and recombinant histone H1 was used as the substrate (Upstate). 400 ng of purified kinase-cyclin complex was incubated with 1 μg of histone H1 in the kinase buffer (50 mm HEPES, pH 7.5, 0.1 m NaCl, 10 mm MgCl2) containing 1 mm ATPγS or N6-(benzyl)ATPγS for 1 h at room temperature. Kinase reaction was stopped by adding 2 mm EDTA. Subsequently, 2.5 mm PNBM (Epitomics) was added to the kinase reaction and incubated for 1 h at room temperature. Samples were run on an SDS-polyacrylamide gel and transferred onto a PVDF membrane. Thiophosphorylation was detected by Western blotting with a thiophosphate ester rabbit monoclonal antibody (Clone 51-8, Epitomics).
For the in vitro kinase assay to confirm that CRK1 can phosphorylate Sec31, Sec31 was cloned into the pGEX-4T-3 vector. Site-directed mutagenesis was carried out with pGEX-Sec31 vector to mutate all seven CRK1 phosphosites in Sec31 to alanine, thus generating pGEX-Sec31-7A vector. The two plasmids were transformed into E. coli BL21 strain, and recombinant GST-Sec31 and GST-Sec31-7A were purified through binding to glutathione-Sepharose beads. CRK1-PTP, which was immunoprecipitated from T. brucei cell lysate, was mixed with purified GST-Sec31 and GST-Sec31-7A in the kinase buffer (see above) in the presence of 1 mm ATP. Phosphorylated GST-Sec31 was detected by Western blotting with anti-Thr(P)-Pro antibody (Cell Signaling Technologies), which detects phosphorylated serine and threonine followed by a proline.
Identification of CRK1 Substrates Using Analog-sensitive CRK1 and Bulky ATP Analog
Kinase assay and purification of thiophosphopeptides were carried out according to the published protocol (31). T. brucei cells (5 × 108) were lysed by sonication in RIPA buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40), and cell lysate was cleared by centrifugation. The cleared cell lysate was split into four fractions, and one fraction was incubated with 1 μg of purified CRK1as-CYC2 complex or CRK1-CYC2 complex in the presence of N6-(benzyl)ATPγS. To compete off nonspecific labeling, 0.5 or 3 mm GTP was added to the kinase reaction. After incubating for 1 h at room temperature, proteins were digested with trypsin. The peptide mix was incubated with iodoacetyl-agarose beads for 16 h at room temperature. Beads were then washed successively with 80% acetonitrile, 50 mm Tris-Cl, pH 7.5, 50% acetonitrile, 50 mm Tris-Cl, pH 7.5, 50 mm Tris-Cl, pH 7.5, 5 m NaCl, 5% formic acid. Thiophosphopeptides bound to the beads were eluted with 1 mg/ml oxone and then cleaned with a C-18 ZipTips. Peptides were analyzed on an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) interfaced with an Eksigent nano-LC 2D Plus chipLC system (Eksigent Technologies) at the Proteomics Core Facility of the University of California at San Francisco.
Data analysis was performed according to our previous publications (43, 44). Raw data files were searched against the T. brucei genome database (version 4) using the Mascot search engine. The search conditions used peptide tolerance of 10 ppm and MS/MS tolerance of 0.8 Da with the enzyme trypsin and two missed cleavages.
Trypanosome Cell Lines
The procyclic 427 cell line was cultured in SDM-79 medium containing 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Inc.) at 27 °C. The procyclic 29-13 cell line (45) was grown in SDM-79 medium supplemented with 10% heat-inactivated fetal bovine serum, 15 μg/ml G418, and 50 μg/ml hygromycin at 27 °C. The CRK1 RNAi cell line, CYC2 RNAi cell line, CYC7 RNAi cell line, and CYC2-CYC7 double RNAi cell line, which have been described previously (27), were cultured in SDM-79 medium containing 10% heat-inactivated fetal bovine serum, 15 μg/ml G418, 50 μg/ml hygromycin, and 2.5 μg/ml phleomycin at 27 °C.
RNA Interference
To knock down Sec31 by RNAi, a 500-bp DNA fragment corresponding to the N-terminal coding region of Sec31 was PCR-amplified from the genomic DNA and cloned into the Stem-Loop vector pSL (43). The resulting plasmid, pSL-Sec31, was linearized with NotI and then transfected into the 29-13 cell line by electroporation according to our published procedures. The successful transfectants were selected with 2.5 μg/ml phleomycin in addition to 15 μg/ml G418 and 50 μg/ml hygromycin and then cloned by limiting dilution in a 96-well plate containing SDM-79 medium supplemented with 20% fetal bovine serum and all three antibiotics. RNAi was induced with 1.0 μg/ml tetracycline.
In Situ Epitope Tagging of Proteins
For C-terminal epitope tagging of Sec31 at one of its endogenous loci, a 400-bp fragment corresponding to the C-terminal coding region of Sec31 was PCR-amplified from the genomic DNA and cloned into pC-3HA-PAC and pC-EYFP-PAC vectors. The resulting plasmids were linearized and electroporated into the wild-type 427 cell line and the cell line harboring the Sec31 RNAi construct pSL-Sec31. Transfectants were selected with 1 μg/ml puromycin and cloned by limiting dilution in a 96-well plate.
For co-localization of Sec31-EYFP and Sec31-mCherry, the cell line harboring the pC-Sec31-PAC vector was transfected with pC-Sec13-mCherry-NEO. For co-localization of Sec31–3HA with CRK1-PTP, the cell line harboring the pC-Sec31–3HA-PAC vector was transfected with pC-CRK1-PTP-NEO. Both transfectants were selected with 40 μg/ml G418 in addition to 1 μg/ml puromycin and then cloned by limiting dilution as described above. For endogenous tagging of TbGntB and TbGolgin63, the PCR-based epitope tagging method was used according to previous publication (46).
For endogenous tagging of Sec31 and Sec13 with PTP and the triple HA epitope in the CRK1 RNAi cell line, a 500-bp fragment corresponding to the C-terminal coding region of Sec31 was cloned into the pC-PTP-PAC vector and electroporated into the CRK1 RNAi cell line. Transfectants were selected by 1 μg/ml puromycin and cloned by limiting dilution. Subsequently, a 500-bp DNA fragment corresponding to the C-terminal coding region of Sec13 was cloned into the pC-3HA-BSD vector, and the resulting construct was electroporated into the CRK1 RNAi cell line harboring the pC-Sec31-PTP-PAC vector. Transfectants were further selected with 10 μg/ml blasticidin and cloned by limiting dilution.
Co-immunoprecipitation
Control and CRK1 RNAi-induced cells (3 days) co-expressing endogenously PTP-tagged Sec31 and 3HA-tagged Sec13 or expressing Sec13–3HA alone were harvested by centrifugation, lysed in 1 ml of cell lysis buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm DTT, 1% Nonidet P-40, and protease inhibitor mixture), and cleared by centrifugation at the highest speed in a microcentrifuge. The supernatant was then incubated with 50 μl of IgG-Sepharose 6 fast flow beads (GE Healthcare) at 4 °C for 1 h. Beads were then washed six times with the cell lysis buffer, and bound proteins were eluted with 10% SDS. Eluted proteins were then separated on SDS-PAGE, transferred onto a PVDF membrane, and immunoblotted with anti-HA antibody and anti-protein A antibody to detect Sec13–3HA and Sec31-PTP, respectively.
Overexpression of Phosphodeficient and Phosphomimic Mutants of Sec31
Full-length coding sequence of Sec31 was PCR-amplified and cloned into the pLew100–3HA vector. Subsequently, site-directed mutagenesis was then performed to mutate all of the seven CRK1 phosphorylation sites on Sec31 to alanine (Sec31-7A) and aspartate (Sec31-7D). The three plasmids were linearized with NotI digestion and electroporated into the 29-13 cell line. Successful transfectants were selected with 2.5 μg/ml phleomycin in addition to 15 μg/ml G418 and 50 μg/ml hygromycin and then cloned by limiting dilution. Overexpression of Sec31 and its mutants were induced by adding 0.1 μg/ml tetracycline into the culture medium, and cell growth was monitored by counting the cells every day for 3 days.
Immunofluorescence Microscopy
Cells were washed once with PBS, adhered to the coverslips for 30 min, and fixed and permeabilized with cold methanol (−20 °C) for 30 min. Cells were then rehydrated with PBS for 10 min. Cells were blocked with 3% BSA in PBS for 45 min at room temperature and then incubated with the primary antibody for 1 h at room temperature. The following primary antibodies were used: FITC-conjugated anti-HA antibody (1:400 dilution, Sigma); anti-protein A antibody (1:400 dilution, Sigma); and anti-TbGRASP antibody (1:400 dilution) (32). Cells were washed three times with PBS and then incubated with FITC-conjugated anti-mouse IgG (1:400 dilution, Sigma) or Cy3-conjugated anti-rabbit IgG (1:400 dilution, Sigma) for 1 h at room temperature. Cells on the coverslips were washed three times with PBS, mounted with DAPI-containing VectaShield mounting medium (Vector Laboratories), and imaged using an inverted fluorescence microscope (Olympus IX71) equipped with a cooled CCD camera (model Orca-ER, Hamamatsu) and a PlanApo N 60 × 1.42-NA lens. Images were acquired using the Slidebook 5 software.
Secretion of the BiPNAVRG-HA9 Reporter
The pXS5-BiPNAVRG-HA9 plasmid (34) was linearized and electroporated into cell lines harboring the Sec31 RNAi construct (pSL-Sec31), the CRK1 RNAi construct (pZJM-CRK1), and each of the three overexpression constructs (pLew100-Sec31–3HA, pLew100-Sec31-7A-3HA, and pLew100-Sec31-7D-3HA) for constitutive expression of BiPNAVRG-HA9 fusion protein. For BiPNAVRG-HA9 secretion assays, non-induced control cells and tetracycline-induced cells were washed three times with fresh medium and then incubated with 100 μg/ml cycloheximide to inhibit protein synthesis. Time course samples of equal volume (5 ml of cell culture) immediately before (0 h) and after cycloheximide treatment (2 and 4 h) were collected, and cells were centrifuged to separate cell pellet and culture medium. Cell pellets were then lysed in immunoprecipitation buffer (25 mm Tris-Cl, pH 7.6, 100 mm NaCl, 1 mm DTT, 1% Nonidet P-40, and protease inhibitor mixture) of the equal volume (5 ml) of the culture medium. Protease inhibitor mixture was also added to the culture medium to prevent the degradation of secreted proteins. Both the crude cell lysate and the culture medium were incubated with anti-HA antibody for 4 h at 4 °C with gentle rotation, and then incubated with protein G-Sepharose beads for 1 h at 4 °C. Immunoprecipitates were eluted, separated on SDS-PAGE, transferred onto a PVDF membrane, and immunoblotted with anti-HA antibody to detect BiPNAVRG-HA9 in both the cells and the medium. Protein band intensity was determined using ImageJ software.
Statistical Analysis
Statistical analysis was performed using the t test provided in the Microsoft Excel software. For immunofluorescence microscopy experiments, images were randomly taken, and all of the cells in each image were counted.
Author Contributions
H. H., S. G., C. C. W., and Z. L. conceived and designed the experiments; H. H. and S. G. performed the experiments; H. H., S. G., C. C. W., and Z. L. analyzed the data; Z. L. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. George Cross of Rockefeller University for providing the 29-13 cell line and the pLew100 vector; Dr. Cynthia He of National University of Singapore for providing the anti-TbGRASP antibody; Dr. Arthur Günzl of University of Connecticut Health Center for epitope-tagging vectors; and Dr. James Bangs of University at Buffalo for providing the pXS5-BiPNAVRG-HA9 plasmid. We also thank Drs. Justin D. Blethrow, Jasmina J. Allen, and Kevan M. Shokat from University of California at San Francisco for assistance with CRK1 substrate identification using the chemical genetic approach.
This work was supported by National Institutes of Health Grants R01 AI118736 and AI101437 (to Z. L.) and Grant R01 AI21786 (to C. C. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ER
- endoplasmic reticulum
- COPII
- coat protein II
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- ERES
- ER exit site
- CDK
- cyclin-dependent kinase
- PNBM
- p-nitrobenzyl mesylate
- α-thioP
- anti-thiophosphate
- EYFP
- enhanced YFP
- PTP
- protein A-tobacco etch virus cleavage site-protein C.
References
- 1. Palade G. (1975) Intracellular aspects of the process of protein synthesis. Science 189, 867. [DOI] [PubMed] [Google Scholar]
- 2. Bonifacino J. S., and Glick B. S. (2004) The mechanisms of vesicle budding and fusion. Cell 116, 153–166 [DOI] [PubMed] [Google Scholar]
- 3. Farhan H., and Rabouille C. (2011) Signalling to and from the secretory pathway. J. Cell Sci. 124, 171–180 [DOI] [PubMed] [Google Scholar]
- 4. Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., and Schekman R. (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 [DOI] [PubMed] [Google Scholar]
- 5. Barlowe C. (2002) COPII-dependent transport from the endoplasmic reticulum. Curr. Opin. Cell Biol. 14, 417–422 [DOI] [PubMed] [Google Scholar]
- 6. Nakańo A., and Muramatsu M. (1989) A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J. Cell Biol. 109, 2677–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bielli A., Haney C. J., Gabreski G., Watkins S. C., Bannykh S. I., and Aridor M. (2005) Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J. Cell Biol. 171, 919–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee M. C., Orci L., Hamamoto S., Futai E., Ravazzola M., and Schekman R. (2005) Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122, 605–617 [DOI] [PubMed] [Google Scholar]
- 9. Bi X., Corpina R. A., and Goldberg J. (2002) Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature 419, 271–277 [DOI] [PubMed] [Google Scholar]
- 10. Fath S., Mancias J. D., Bi X., and Goldberg J. (2007) Structure and organization of coat proteins in the COPII cage. Cell 129, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 11. Aridor M., Weissman J., Bannykh S., Nuoffer C., and Balch W. E. (1998) Cargo selection by the COPII budding machinery during export from the ER. J. Cell Biol. 141, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuehn M. J., Herrmann J. M., and Schekman R. (1998) COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature 391, 187–190 [DOI] [PubMed] [Google Scholar]
- 13. Miller E. A., Beilharz T. H., Malkus P. N., Lee M. C., Hamamoto S., Orci L., and Schekman R. (2003) Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114, 497–509 [DOI] [PubMed] [Google Scholar]
- 14. Lederkremer G. Z., Cheng Y., Petre B. M., Vogan E., Springer S., Schekman R., Walz T., and Kirchhausen T. (2001) Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc. Natl. Acad. Sci. U.S.A. 98, 10704–10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuoka K., Schekman R., Orci L., and Heuser J. E. (2001) Surface structure of the COPII-coated vesicle. Proc. Natl. Acad. Sci. U.S.A. 98, 13705–13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stagg S. M., Gürkan C., Fowler D. M., LaPointe P., Foss T. R., Potter C. S., Carragher B., and Balch W. E. (2006) Structure of the Sec13/31 COPII coat cage. Nature 439, 234–238 [DOI] [PubMed] [Google Scholar]
- 17. Lowe M., Rabouille C., Nakamura N., Watson R., Jackman M., Jämsä E., Rahman D., Pappin D. J., and Warren G. (1998) Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94, 783–793 [DOI] [PubMed] [Google Scholar]
- 18. Kano F., Tanaka A. R., Yamauchi S., Kondo H., and Murata M. (2004) Cdc2 kinase-dependent disassembly of endoplasmic reticulum (ER) exit sites inhibits ER-to-Golgi vesicular transport during mitosis. Mol. Biol. Cell 15, 4289–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palmer K. J., Konkel J. E., and Stephens D. J. (2005) PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. J. Cell Sci. 118, 3839–3847 [DOI] [PubMed] [Google Scholar]
- 20. Sharpe L. J., Luu W., and Brown A. J. (2011) Akt phosphorylates Sec24: new clues into the regulation of ER-to-Golgi trafficking. Traffic 12, 19–27 [DOI] [PubMed] [Google Scholar]
- 21. Koreishi M., Yu S., Oda M., Honjo Y., and Satoh A. (2013) CK2 phosphorylates Sec31 and regulates ER-to-Golgi trafficking. PLoS ONE 8, e54382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Q., Hu H., and Li Z. (2014) New insights into the molecular mechanisms of mitosis and cytokinesis in trypanosomes. Int. Rev. Cell Mol. Biol. 308, 127–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hammarton T. C. (2007) Cell cycle regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 153, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Z., and Wang C. C. (2003) A PHO80-like cyclin and a B-type cyclin control the cell cycle of the procyclic form of Trypanosoma brucei. J. Biol. Chem. 278, 20652–20658 [DOI] [PubMed] [Google Scholar]
- 25. Hammarton T. C., Clark J., Douglas F., Boshart M., and Mottram J. C. (2003) Stage-specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J. Biol. Chem. 278, 22877–22886 [DOI] [PubMed] [Google Scholar]
- 26. Tu X., and Wang C. C. (2004) The involvement of two cdc2-related kinases (CRKs) in Trypanosoma brucei cell cycle regulation and the distinctive stage-specific phenotypes caused by CRK3 depletion. J. Biol. Chem. 279, 20519–20528 [DOI] [PubMed] [Google Scholar]
- 27. Liu Y., Hu H., and Li Z. (2013) The cooperative roles of PHO80-like cyclins in regulating the G1/S transition and posterior cytoskeletal morphogenesis in Trypanosoma brucei. Mol. Microbiol. 90, 130–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., and Shokat K. M. (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 [DOI] [PubMed] [Google Scholar]
- 29. Allen J. J., Li M., Brinkworth C. S., Paulson J. L., Wang D., Hübner A., Chou W. H., Davis R. J., Burlingame A. L., Messing R. O., Katayama C. D., Hedrick S. M., and Shokat K. M. (2007) A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blethrow J. D., Glavy J. S., Morgan D. O., and Shokat K. M. (2008) Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 105, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hertz N. T., Wang B. T., Allen J. J., Zhang C., Dar A. C., Burlingame A. L., and Shokat K. M. (2010) Chemical genetic approach for kinase-substrate mapping by covalent capture of thiophosphopeptides and analysis by mass spectrometry. Curr. Protoc. Chem. Biol. 2, 15–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He C. Y., Ho H. H., Malsam J., Chalouni C., West C. M., Ullu E., Toomre D., and Warren G. (2004) Golgi duplication in Trypanosoma brucei. J. Cell Biol. 165, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Demmel L., Melak M., Kotisch H., Fendos J., Reipert S., and Warren G. (2011) Differential selection of Golgi proteins by COPII Sec24 isoforms in procyclic Trypanosoma brucei. Traffic 12, 1575–1591 [DOI] [PubMed] [Google Scholar]
- 34. Bangs J. D., Brouch E. M., Ransom D. M., and Roggy J. L. (1996) A soluble secretory reporter system in Trypanosoma brucei. Studies on endoplasmic reticulum targeting. J. Biol. Chem. 271, 18387–18393 [DOI] [PubMed] [Google Scholar]
- 35. Silverman J. S., and Bangs J. D. (2012) Form and function in the trypanosomal secretory pathway. Curr. Opin. Microbiol. 15, 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sevova E. S., and Bangs J. D. (2009) Streamlined architecture and glycosylphosphatidylinositol-dependent trafficking in the early secretory pathway of African trypanosomes. Mol. Biol. Cell 20, 4739–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matynia A., Salus S. S., and Sazer S. (2002) Three proteins required for early steps in the protein secretory pathway also affect nuclear envelope structure and cell cycle progression in fission yeast. J. Cell Sci. 115, 421–431 [DOI] [PubMed] [Google Scholar]
- 38. Robinson D. R., Sherwin T., Ploubidou A., Byard E. H., and Gull K. (1995) Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J. Cell Biol. 128, 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lim S., and Kaldis P. (2013) Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093 [DOI] [PubMed] [Google Scholar]
- 40. Zanetti G., Pahuja K. B., Studer S., Shim S., and Schekman R. (2012) COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 14, 20–28 [DOI] [PubMed] [Google Scholar]
- 41. Jin L., Pahuja K. B., Wickliffe K. E., Gorur A., Baumgärtel C., Schekman R., and Rape M. (2012) Ubiquitin-dependent regulation of COPII coat size and function. Nature 482, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., and Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wei Y., Hu H., Lun Z. R., and Li Z. (2014) Centrin3 in trypanosomes maintains the stability of a flagellar inner-arm dynein for cell motility. Nat. Commun. 5, 4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou Q., Hu H., He C. Y., and Li Z. (2015) Assembly and maintenance of the flagellum attachment zone filament in Trypanosoma brucei. J. Cell Sci. 128, 2361–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wirtz E., Leal S., Ochatt C., and Cross G. A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 [DOI] [PubMed] [Google Scholar]
- 46. Shen H., Arhin G. K., Ullu E., and Tschudi C. (2001) In vivo epitope tagging of Trypanosoma brucei genes using a one-step PCR-based strategy. Mol. Biochem. Parasitol. 113, 171–173 [DOI] [PubMed] [Google Scholar]