

Abstract
One mode of γ-globin gene silencing involves a GATA-1·FOG-1·Mi2β repressor complex that binds to the −566 GATA site relative to the Aγ-globin gene cap site. However, the mechanism of how this repressor complex is assembled at the −566 GATA site is unknown. In this study, we demonstrate that the O-linked N-acetylglucosamine (O-GlcNAc) processing enzymes, O-GlcNAc-transferase (OGT) and O-GlcNAcase (OGA), interact with the Aγ-globin promoter at the −566 GATA repressor site; however, mutation of the GATA site to GAGA significantly reduces OGT and OGA promoter interactions in β-globin locus yeast artificial chromosome (β-YAC) bone marrow cells. When WT β-YAC bone marrow cells are treated with the OGA inhibitor Thiamet-G, the occupancy of OGT, OGA, and Mi2β at the Aγ-globin promoter is increased. In addition, OGT and Mi2β recruitment is increased at the Aγ-globin promoter when γ-globin becomes repressed in postconception day E18 human β-YAC transgenic mouse fetal liver. Furthermore, we show that Mi2β is modified with O-GlcNAc, and both OGT and OGA interact with Mi2β, GATA-1, and FOG-1. Taken together, our data suggest that O-GlcNAcylation is a novel mechanism of γ-globin gene regulation mediated by modulating the assembly of the GATA-1·FOG-1·Mi2β repressor complex at the −566 GATA motif within the promoter.
Keywords: GATA transcription factor, O-GlcNAcylation, O-linked N-acetylglucosamine (O-GlcNAc), post-translational modification (PTM), transcription, CHD4, FOG, Globin, O-GlcNAc-transferase, O-GlcNAcase
Introduction
The human β-globin gene cluster comprises five functional β-like globin genes, an embryonic gene (ϵ), duplicated fetal γ genes (Gγ and Aγ), a minor adult gene (δ), and the adult β gene, which are expressed during development in the same order as they are arrayed, 5′-ϵ-Gγ-Aγ-δ-β-3′. The expression of these genes is not only controlled by an upstream locus control region but also regulated by epigenetic signals as well as transcription factors and their DNA binding motifs (1–4). There are two major switches of human β-like globin gene expression during development. The first switch is from embryonic to fetal globin, characterized by silencing of the embryonic ϵ-globin gene in the yolk sac and activation of the fetal γ-globin genes in the liver. The second switch is from the fetal to adult globins, characterized by the progressive silencing of γ-globin gene expression in the liver and activation of adult globins (δ and β) in bone marrow (5).
Patients with sickle cell disease, caused by a point mutation in the β-globin gene, suffer chronic damage of multiple organs, increased risk of stroke, and cardiovascular abnormalities and dysfunction (6, 7). However, sickle cell disease pathophysiology is ameliorated if the patients carry compensatory mutations that result in continued expression of the fetal γ-globin genes (fetal hemoglobin), a condition called hereditary persistence of fetal hemoglobin (8). Thus, a logical clinical goal for treatment of this β-hemoglobinopathy is to up-regulate γ-globin synthesis (8). There are several mechanisms of γ-globin silencing. One of the well known γ-globin silencers is B-cell lymphoma-leukemia A (BCL11A),3 which forms a protein complex with nucleosome remodeling and deacetylase complex (NuRD), GATA-1, and SOX6, to repress γ-globin expression (5). Krüppel-like factor 1 (KLF1), required for β-globin gene activation, also stimulates BCL11A expression, which in turn represses γ-globin expression (4, 5, 8). Both BCL11A and KLF1 are positively regulated by binding of Mi2β (chromodomain helicase DNA-binding protein 4), a component of the NuRD complex (9). We previously demonstrated another modality of fetal globin repression using human β-globin locus yeast artificial chromosome (β-YAC) transgenic mice. The Aγ-globin gene is silenced during development following sequential binding of GATA-1, friend of GATA-1 (FOG-1), and Mi2β proteins at the −566 GATA site relative to the Aγ-globin gene cap site in the fetal liver between postconception days 16 and 18 (E16 and E18) (10, 11). GATA-1 is the DNA binding moiety in this complex, but GATA-1 can recruit both coactivators and corepressors in both FOG-1-dependent and -independent pathways (12–15). The determinants of repressor versus activator complex assembly during development are not well understood. We postulate that one potential mechanism is through post-translational modifications such as O-GlcNAcylation.
O-GlcNAcylation is the attachment of a single N-acetylglucosamine moiety to serine or threonine residues in mitochondrial, nuclear, and cytoplasmic proteins. The enzyme O-GlcNAc-transferase (OGT) adds the modification to proteins, whereas O-GlcNAcase (OGA) removes the modification (16). O-GlcNAcylation integrates signals from a variety of nutrients to regulate cell signaling pathways, transcription, and cellular metabolism (17). Furthermore, the rate of O-GlcNAc cycling (the addition and then removal of O-GlcNAc) affects cell cycle progression, mitotic signaling and spindle formation, and cellular respiration (16, 18–22). Both OGT and OGA are essential for cellular function. Knock-out mutations of OGT or OGA are embryonic lethal (23, 24). Disruption of O-GlcNAcylation contributes to the development of diseases including cancer, diabetes, and Alzheimer disease (18, 19, 25–27).
Growing evidence demonstrates that O-GlcNAcylation plays an important role in regulating transcription. O-GlcNAcylation is part of the histone code, and histones are O-GlcNAcylated under diverse cellular conditions (28–30). A recent study suggests that O-GlcNAc cycling also plays an important role in maintaining the epigenetic machinery in Drosophila (31). The RNA Pol II C-terminal domain (CTD) is modified by O-GlcNAc (32), facilitating preinitiation complex formation (33). Furthermore, numerous transcription factors are modified by O-GlcNAc and interact with OGT (16, 34–39). Thus, O-GlcNAcylation could potentially regulate γ-globin gene transcription.
In this study, we asked the question whether O-GlcNAcylation plays a role in organizing the Aγ-globin promoter. Herein, we demonstrate that both OGT and OGA interact with the Aγ-globin promoter in both immortalized β-YAC bone marrow cells (BMCs) and in β-YAC transgenic mouse fetal liver. The occupancy of OGT, OGA, and Mi2β at the Aγ-globin promoter is increased after Thiamet-G (TMG) (an OGA inhibitor) treatment. Furthermore, we show that Mi2β is modified by O-GlcNAc, and both OGT and OGA interact with GATA-1·FOG-1·Mi2β repressor complex. Taken together, our data suggest that O-GlcNAcylation is a novel mechanism that regulates Aγ-globin gene expression by modulating GATA-1·FOG-1·Mi2β repressor complex recruitment to the −566 GATA site in the promoter during development.
Results
OGT and OGA Interact with the Aγ-Globin Gene Promoter
To determine whether OGT and OGA interact with the Aγ-globin gene promoter, we utilized wild-type (WT) and −566 mutant murine chemical inducer of dimerization (CID)-dependent β-YAC BMCs (10, 11). At the −566 region of the Aγ-globin promoter relative to the cap site, WT β-YAC BMCs have a GATA motif that is bound by a GATA-1·FOG-1·Mi2β repressor complex when γ-globin is silenced during the adult stage of hematopoiesis. In the −566 mutant β-YAC BMCs, a T→G point mutation at nucleotide −566 (Fig. 1a) results in a hereditary persistence of fetal hemoglobin phenotype. This mutation changes the GATA site to a GAGA site, preventing binding of the GATA-1·FOG-1·Mi2β repressor complex, and results in continued γ-globin gene transcription in the adult stage of erythropoiesis (Fig. 1b). Both WT and −566 mutant β-YAC BMCs express OGT and OGA and have similar overall O-GlcNAc levels (Fig. 1c). Next, we performed chromatin immunoprecipitation (ChIP) assays in both of these BMC populations to test for OGT and OGA occupancy at the Aγ-globin promoter. In WT β-YAC BMCs, both OGT and OGA associated with the −566 region of the Aγ-globin promoter; however, in −566 mutant β-YAC BMCs, this association was dramatically decreased (Fig. 1d). These data suggest that OGT and OGA interact with a component(s) of the GATA-1·FOG-1·Mi2β repressor complex at the −566 GATA site of Aγ-globin promoter.
FIGURE 1.

OGT and OGA interact with the Aγ-globin promoter in CID-dependent β-YAC BMCs. a, WT CID-dependent β-YAC BMCs have a normal GATA motif that is bound by a GATA-1·FOG-1·Mi2β repressor complex, mediating γ-globin repression; whereas −566 mutant CID-dependent β-YAC BMCs have a T→G point mutation at nucleotide −566, which alleviates γ-globin repression. b, γ-globin mRNA level in WT and −566 mutant β-YAC BMCs was analyzed by qPCR. Mouse HPRT was used as an internal control. c, overall O-GlcNAc levels and OGA and OGT protein expression in total cell lysates from WT and −566 mutant CID-dependent β-YAC BMCs were analyzed by immunoblotting. Actin was used as a loading control. d, OGT and OGA ChIP assays were performed on WT and −566 mutant β-YAC BMCs, respectively. ChIP DNA was analyzed by qPCR using a set of primers targeting the −566 GATA site of the Aγ-globin promoter. Normal rabbit (Rb) IgG served as a negative control. All experiments were performed with at least three biological replicates (* indicates p < 0.05, Student's t test). Error bars represent S.E. WB, Western blotting.
OGT, OGA, and Mi2β Increase at the Aγ-Globin Gene Promoter after TMG Treatment
Our data suggest that OGT and OGA play a role in regulating γ-globin transcription by modulating GATA-1·FOG-1·Mi2β repressor complex activity at the Aγ-globin promoter. To test this hypothesis, we treated the WT β-YAC BMCs for 4 days with TMG (S. D. Specialty Chemicals), a competitive inhibitor of OGA, to increase overall O-GlcNAc levels. After TMG treatment for 4 days, the overall O-GlcNAc levels were increased with increased OGA and decreased OGT protein levels (Fig. 2a), an effect similar to that we observed previously (40). Interestingly, the γ-globin transcription level was decreased (Fig. 2b). To seek the mechanism of decreased γ-globin transcription after TMG treatment, we performed ChIP assays on WT β-YAC BMCs. After TMG treatment for 4 days, the occupancy of both OGT and OGA was increased at the Aγ-globin promoter compared with the cells without TMG treatment (Fig. 2c). We also observed an increase of Mi2β occupancy at Aγ-globin promoter after TMG treatment (Fig. 2d). These data demonstrate that OGT, OGA, and Mi2β increase at the Aγ-globin promoter after TMG treatment.
FIGURE 2.

OGT, OGA, and Mi2β increase at the Aγ-globin gene promoter after TMG treatment in WT β-YAC BMCs. a, overall O-GlcNAc levels and OGA and OGT protein expression in total cell lysates from control and 4-day TMG-treated WT β-YAC BMCs cells were analyzed by immunoblotting. GAPDH was used as a loading control. b, γ-globin mRNA level in control and 4-day TMG-treated WT β-YAC BMCs cells was analyzed by qPCR. Mouse HPRT was used as an internal control. OGT/OGA (c) and Mi2β (d) ChIP assays were performed on control and 4-day TMG-treated WT β-YAC BMCs cells, respectively. ChIP DNA was analyzed by qPCR using a set of primers targeting the −566 GATA site of the Aγ-globin promoter. Normal rabbit IgG served as a negative control. All experiments were performed with four biological replicates (* indicates p < 0.05, Student's t test). Error bars represent S.E. WB, Western blotting.
OGT and OGA Interact with the Aγ-Globin Promoter during Fetal Liver Development
In β-YAC transgenic mice, the γ-globin gene is expressed during early fetal development; however, prior to birth, γ-globin expression is silenced in hematopoietic cells of the fetal liver, partly through the gradual recruitment of the GATA-1·FOG-1·Mi2β repressor complex (10, 11). To determine whether OGT and OGA associate with the Aγ-globin promoter during different fetal stages of hematopoietic development as gene silencing progresses, we performed ChIP assays in β-YAC transgenic mouse fetal liver single cell suspensions to test for recruitment of Mi2β, OGT, and OGA. At day E12, γ-globin is expressed in the murine fetal liver. However, γ-globin is repressed at day E18 when the GATA-1·FOG-1·Mi2β repressor complex is recruited to the −566 Aγ-globin GATA silencer site (10). As a positive control for repressor complex binding, we confirmed our previous data (10) that Mi2β binds to the Aγ-globin promoter at day E18 when γ-globin is repressed but not in day E12 fetal liver when γ-globin is expressed (Fig. 3a). Interestingly, OGT occupancy was slightly increased at the Aγ-globin promoter at day E18 compared with day E12 (Fig. 3b). We did not observe an increase of OGA occupancy at the Aγ-globin promoter at day E18 compared with day E12, whereas OGA interacts with the promoter on both days (Fig. 3c). These data suggest that recruitment of OGT and OGA to the Aγ-globin promoter may contribute to Aγ-globin gene silencing.
FIGURE 3.

OGT and OGA interact with the Aγ-globin promoter during fetal liver development in β-YAC transgenic mice. Mi2β (a), OGT (b), and OGA (c) ChIP assays were performed using postconception day E12 (γ-globin is expressed) and E18 (γ-globin is repressed) fetal liver single cell suspension prepared from β-YAC transgenic mouse conceptuses. ChIP DNA was analyzed by qPCR using a set of primers targeting the −566 GATA site of the Aγ-globin promoter. Normal rabbit IgG served as a negative control. All experiments were performed with at least three biological replicates. Error bars represent S.E.
Mi2β Is Modified by O-GlcNAc
Because OGT and OGA interact with the Aγ-globin promoter where the GATA-1·FOG-1·Mi2β repressor complex binds, we determined whether any component(s) of the repressor complex is modified by O-GlcNAc. For these experiments, we used human erythroleukemia K562 cells, a well established model system for globin studies (41). The addition of sodium butyrate (NaB) to K562 cells increases γ-globin expression (41). Our experiments confirmed this phenotype; the γ-globin mRNA level was increased by NaB as expected (Fig. 4a). After 4 days of treatment, overall O-GlcNAc levels were decreased with an increase of OGA protein level and a decrease of OGT protein level (Fig. 4b), which corresponded to an increased OGA mRNA level (Fig. 4c) and a decreased OGT mRNA level (Fig. 4d). We also used hemin and N,N′-hexamethylenebisacetamide (HMBA) (H-H), another well established inducer of differentiation in K562 cells (10), that also relieve γ-globin repression (data not shown). We used these two different methods to alleviate Aγ-globin promoter repression to eliminate any artifacts from these chemical inducers that could skew the data.
FIGURE 4.

K562 γ-globin expression is increased after NaB induction. K562 cells were treated with NaB for 4 days. Cells were collected daily. a, γ-globin mRNA levels were measured before and after NaB induction by qPCR. b, overall O-GlcNAc levels and OGA and OGT protein expression from total cell lysates were analyzed by immunoblotting. Actin was used as a loading control. OGA (c) and OGT (d) mRNA levels were measured before and after NaB induction by qPCR. Human HPRT was used as an internal control in all qPCR analyses. All experiments were performed with at least three biological replicates (* indicates p < 0.05, Student's t test). Error bars represent S.E. WB, Western blotting.
Next, we assessed whether any proteins of the GATA-1·FOG-1·Mi2β complex were modified by O-GlcNAc pre- or post-γ-globin promoter activation. We used a specific antibody to O-GlcNAc to immunoprecipitate potential O-GlcNAc-modified proteins from K562 lysates before and after NaB treatment. TMG was used to increase the overall O-GlcNAc levels by competitively inhibiting OGA. Blots were probed individually with Mi2β, FOG-1, and GATA-1 antibodies, respectively. We found that CTD 110.6 immunoprecipitated Mi2β before NaB treatment (day 0), and TMG treatment increased the amount of Mi2β immunoprecipitated by CTD 110.6. However, the CTD 110.6-immunoprecipitated Mi2β protein level was decreased after 4 days of NaB treatment (Fig. 5a, top panel). Similar results were observed when using H-H as the inducer instead of NaB (Fig. 5a, bottom panel).
FIGURE 5.

Mi2β is modified by O-GlcNAc. a, O-GlcNAcylated protein was immunoprecipitated by an O-GlcNAc-specific antibody (CTD 110.6) in K562 cells before and after NaB induction (top panel) or H-H induction (bottom panel); the blot was probed with Mi2β antibody. b, Mi2β protein was immunoprecipitated from K562 cells before and after NaB induction (top panel) or H-H induction (bottom panel); the blot was probed with CTD 110.6. c and d, Mi2β protein was immunoprecipitated from K562 cells, and the blot was probed with CTD 110.6 (c) or CTD 110.6 preincubated with 500 mm GlcNAc (d). e, Mi2β protein was immunoprecipitated from K562 cells and treated with hexosaminidase CpNagJ at 37 °C for 3 h; the blot was probed with CTD 110.6. The OGA inhibitor TMG was used to increase overall O-GlcNAc levels. Isotype immunoprecipitation (normal mouse IgM or rabbit IgG) and antibody-alone immunoprecipitation (1°) were used as negative controls. All experiments were performed with at least three biological replicates. WB, Western blotting.
We also performed the converse immunoprecipitation using Mi2β antibody and probed the blot for O-GlcNAc. Mi2β was O-GlcNAc-modified prior to NaB induction, and this modification dramatically decreased after 4 days of NaB treatment (Fig. 5b, top panel). The same results were obtained when H-H were used as the inducer (Fig. 5b, bottom panel). These data suggest that O-GlcNAcylation of Mi2β changes during γ-globin induction.
We also performed two control experiments to further validate O-GlcNAcylation of Mi2β. Mi2β was immunoprecipitated from duplicate sets of lysates. Both sets were transferred to a PVDF membrane, which was cut in half. One set was probed with CTD 110.6 antibody detecting O-GlcNAcylated Mi2β (Fig. 5c). However, detection of O-GlcNAcylated Mi2β on the other set was abolished when CTD 110.6 antibody was preincubated with 500 mm free GlcNAc (Fig. 5d). In another experiment, Mi2β immunoprecipitates were treated with CpNagJ hexosaminidase (a gift from Daan van Aaltan, University of Dundee), which removes oxygen-linked hexoses from proteins (42). In the absence of CpNagJ treatment, Mi2β immunoprecipitates were O-GlcNAcylated, whereas detection of the Mi2β O-GlcNAcylation was completely lost after CpNagJ treatment (Fig. 5e).
Mi2β Interacts with OGT
Because Mi2β is modified by O-GlcNAc, we next explored whether Mi2β interacts with OGT. First, we performed IP experiments using OGT antibody on lysates prepared from K562 cells that had been treated with or without NaB. OGT interacts with Mi2β prior to NaB induction but not after 4 days of NaB induction (Fig. 6a). The lost interaction of OGT and Mi2β after NaB treatment corresponds to the decreased O-GlcNAcylation of Mi2β after NaB induction (Fig. 5, a and b, top panel). Similar IP results were observed when cells were differentiated by H-H for 3 days (Fig. 6b).
FIGURE 6.

Mi2β interacts with OGT. a and b, OGT was immunoprecipitated with an OGT-specific antibody (AL-34) in K562 cells before and after NaB induction (a) or hemin and HMBA induction (b), and the blot was probed with Mi2β antibody. c and d, Mi2β protein was immunoprecipitated from K562 cells before and after NaB induction (c) or hemin and HMBA induction (d), and the blot was probed with OGT antibody. The OGA inhibitor TMG was used to increase the overall O-GlcNAc levels. Isotype immunoprecipitation (normal rabbit or mouse IgG) and antibody-alone precipitation (1°) were used as negative controls. All experiments were performed with at least three biological replicates. WB, Western blotting.
We also performed IPs against Mi2β in K562 cell lysates prior to and following NaB induction. Mi2β antibody was able to co-immunoprecipitate OGT prior to NaB treatment but unable to immunoprecipitate OGT after 4 days of NaB treatment (Fig. 6c). The same results were seen when we used H-H to differentiate cells (Fig. 6d). These data demonstrate that Mi2β interacts with OGT before induction of γ-globin in K562 cells or prior to their terminal differentiation but not after treatments that induce fetal hemoglobin or terminal erythroid differentiation.
Mi2β Interacts with OGA
Because our data demonstrated that OGA also interacts at the −566 region of the Aγ-globin promoter, we next tested whether Mi2β interacts with OGA as well. We performed co-IP experiments as described in the previous section. OGA antibody was able to co-immunoprecipitate Mi2β prior to and after NaB induction (Fig. 7a). Similar co-IP results were observed when cells were differentiated by H-H (Fig. 7b). As described above for the Mi2β-OGT interaction experiments, we performed the converse co-IP using Mi2β antibody and looked for interactions with OGA. Mi2β antibody was able to co-immunoprecipitate OGA prior to and after NaB treatment (Fig. 7c). The same results were obtained when we used H-H to differentiate cells (Fig. 7d). These data indicate that Mi2β interacts with OGA before and after induction of γ-globin in K562 cells.
FIGURE 7.

Mi2β interacts with OGA. a and b, OGA was immunoprecipitated using an OGA-specific antibody in K562 cells before and after NaB induction (a) or hemin and HMBA induction (b), and the blot was probed with Mi2β antibody. c and d, Mi2β protein was immunoprecipitated from K562 cells before and after NaB induction (c) or hemin and HMBA induction (d), and the blot was probed with OGA antibody. The OGA inhibitor TMG was used to increase overall O-GlcNAc levels. Isotype immunoprecipitation (normal chicken IgY or rabbit IgG) and antibody-alone immunoprecipitation (1°) were used as negative controls. All experiments were performed with at least three biological replicates. WB, Western blotting.
OGT and OGA Interact with GATA-1 and FOG-1
Our data demonstrated that both OGT and OGA interact with Mi2β, a component of the GATA-1·FOG-1·Mi2β repressor complex. Thus, OGT and OGA might also interact with GATA-1 and FOG. To confirm the interaction of OGT/OGA with GATA-1/FOG-1, we performed IPs in murine erythroleukemia (MEL) birA cells (43) because they express relatively higher GATA-1 and FOG-1 protein compared with K562 and β-YAC BMCs (data not shown). GATA-1 antibody was able to pull down both OGT and OGA (Fig. 8a); FOG-1 antibody can also pull down OGT and Mi2β (Fig. 8b), and OGA antibody was able to immunoprecipitate FOG-1 (Fig. 8c). These data suggest that OGT and OGA can also interact with GATA-1 and FOG-1 besides Mi2β.
FIGURE 8.
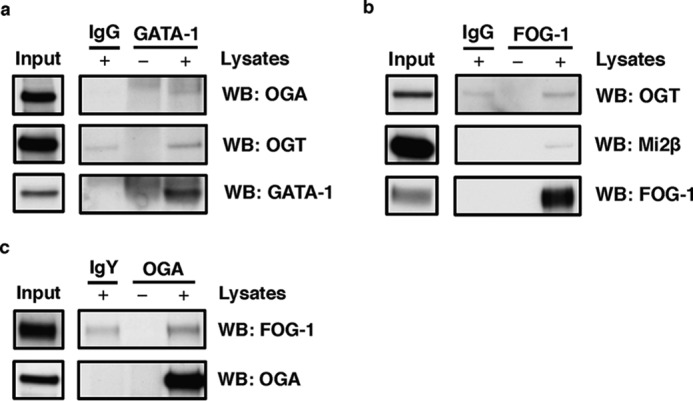
OGT and OGA interact with GATA-1 and FOG-1 in MEL birA cells. a, GATA-1 was immunoprecipitated using a GATA-1-specific antibody in MEL birA cells, and the blot was probed with OGA and OGT antibodies. b, FOG-1 was immunoprecipitated from MEL birA cells, and the blot was probed with OGT and Mi2β antibodies. c, OGA was immunoprecipitated from MEL birA cells, and the blot was probed with FOG-1 antibody. Isotype (IgY or IgG) immunoprecipitation and antibody-alone immunoprecipitation (without lysates) were used as negative controls. All experiments were performed with three biological replicates. WB, Western blotting.
Discussion
Using WT and −566 mutant CID-dependent β-YAC BMCs, we demonstrated that both OGT and OGA interact with the Aγ-globin promoter (Fig. 1) at the −566 GATA site occupied by the GATA-1·FOG-1·Mi2β repressor complex when γ-globin is silenced. In addition, the occupancy of OGT, OGA, and Mi2β at the Aγ-globin promoter is increased after TMG treatment (Fig. 2). In β-YAC transgenic mouse fetal liver, OGT and Mi2β recruitment was increased at the −566 GATA site of the Aγ-globin promoter at postconception day E18 when γ-globin is repressed compared with day E12 when γ-globin is expressed (Fig. 3). Furthermore, we determined that Mi2β is modified by O-GlcNAc (Fig. 5), and OGT/OGA interacts with GATA-1·FOG-1·Mi2β repressor complex (Figs. 6–8). Taken together, our data suggest that O-GlcNAcylation plays a role in regulating Aγ-globin gene expression via recruitment of the GATA-1·FOG-1·Mi2β repressor complex at the −566 GATA site of the Aγ-globin promoter.
O-GlcNAcylation plays an important role in controlling transcription. O-GlcNAc regulates gene transcription by directly modifying transcription factors/cofactors or RNA Pol II CTD (33) or by altering how the activity of these factors is modulated by other post-translational modifications (39). A previous study by Ranuncolo et al. (33) suggested that an O-GlcNAcylation cycle on RNA Pol II CTD occurs at gene promoters. In this cycle, unphosphorylated RNA Pol II (Pol IIA) CTD is first modified with O-GlcNAc (Pol IIγ) by OGT, promoting the association of the general transcription factors with Pol IIγ. Next, O-GlcNAc is removed from Pol IIγ by OGA converting Pol IIγ back to Pol IIA. Subsequently, Pol IIA is phosphorylated (Pol IIO) by transcription factor IIH and positive transcription elongation factor b, and transcription elongation occurs (33). Correspondingly, we demonstrated that both OGT and OGA interact with the Aγ-globin promoter in β-YAC BMCs (Fig. 1d) and β-YAC transgenic mouse fetal liver (Fig. 3, b and c). The addition of TMG to the β-YAC BMCs decreased OGA activity and reduced O-GlcNAc cycling of Mi2β at the Aγ-globin promoter, leading to the recruitment of more OGA to the Aγ-globin promoter to maintain the normal O-GlcNAc cycling (Fig. 2c). In addition, TMG can also increase the Mi2β O-GlcNAc levels, resulting in the extended occupancy of Mi2β (Fig. 2d), other components of the GATA-1·FOG-1·Mi2β repressor complex, and OGT (Fig. 2c) at the Aγ-globin promoter. All the above effects caused by TMG treatment could stabilize GATA-1·FOG-1·Mi2β repressor complex and reduce the γ-globin transcription level (Fig. 2b). Furthermore, these data suggest that both OGT and OGA interactions at the Aγ-globin promoter are important for the recruitment and organization of the GATA-1·FOG-1·Mi2β repressor complex at the −566 GATA site. Similar to the assembly of the preinitiation complex at gene promoters described by Ranuncolo et al. (33), the assembly of the GATA-1·FOG-1·Mi2β repressor complex at Aγ-globin promoter might also require the O-GlcNAcylation of Mi2β first to recruit other factors followed by OGA removal of the O-GlcNAc.
Although the GATA-1·FOG-1·Mi2β repressor complex is assembled sequentially during development in a murine model (11), the mechanisms controlling this assembly are unknown. Our data show that Mi2β is modified by O-GlcNAc (Fig. 5). O-GlcNAcylation of Mi2β may facilitate the sequential recruitment of Mi2β to the GATA-1/FOG-1 proteins already bound at the Aγ-globin promoter. Mi2β/NuRD also mediates Aγ-globin gene silencing independently of the GATA-1·FOG-1·Mi2β repressor complex at the −566 GATA site of the Aγ-globin promoter. Mi2β binds directly to BCL11A and KLF1 and positively regulates BLC11A and KLF1 expression, which in turn leads to γ-globin silencing (9). Thus, O-GlcNAcylation of Mi2β likely plays a role in mediating γ-globin silencing by affecting the action of several previously identified γ-globin repressor complexes that interact with different cis-regulatory motifs in the promoters of the γ-globin genes.
The GATA-1·FOG-1·Mi2β repressor complex mediates one mode of Aγ-globin gene silencing, the components of which are sequentially recruited to the −566 GATA silencer of the Aγ-globin promoter (10, 11). How this repressor complex is assembled and organized during development is unclear. Our data show that OGT and OGA interact with Mi2β (a component of NuRD), GATA-1, and FOG-1 (Figs. 6–8). These data suggested that OGT and OGA are part of the GATA-1·FOG-1·Mi2β repressor complex. Mi2β or other components of the GATA-1·FOG-1·Mi2β repressor complex may mediate the recruitment of OGT and OGA to the Aγ-globin promoter. Interestingly, we show that, besides OGT, OGA also interacts with Mi2β, suggesting that O-GlcNAc cycling on Mi2β may be a potentially important mechanism for repressor complex assembly and transcriptional repression. Of note, OGT interacts with numerous transcription factors and mediates gene activation and silencing. For example, OGT interacts with mSin3A, recruiting OGT to promoters to repress transcription. Furthermore, inactivation of transcription factors and RNA polymerase II by O-GlcNAc modifications contributes to gene repression (34). Drosophila polycomb group gene (sxc) encodes for OGT, and null mutations in OGT lead to polycomb defects, suggesting that OGT is critical for polycomb group-mediated gene silencing (44). Potentially, OGT is mediating Mi2β repression of the Aγ-globin promoter.
The transcription factor GATA-1 is essential for erythroid development. This DNA-binding protein functions in either transcriptional activator or repressor protein complexes as the DNA docking moiety in either a FOG-1-dependent or -independent fashion (12–15). A fundamental question remains as to what are the mechanisms that determine whether GATA-1 functions in an activation complex or a repressor complex. In some instances, binding by partner protein TAL1 at a neighboring recognition site assures the recruitment of an activator complex (45), and to a limited degree the WGATAR (in which W indicates A/T and R indicates A/G) binding site context plays a role (46). However, these mechanisms do not explain the outcome for the majority of protein complex binding events. The O-GlcNAcylation status of Mi2β during erythroid development may provide a mechanism by which different co-repressors or co-activators are recruited to interact with GATA-1. Moreover, GATA-1 itself may be a target of O-GlcNAcylation, influencing how it interacts with co-activators or co-repressors. In addition, FOG-1 also may be a target for O-GlcNAcylation, and the differential post-translational modification of FOG-1 might determine the recruitment of co-activators or co-repressors different from those recruited by GATA-1 alone.
O-GlcNAcylation and phosphorylation can compete for specific Ser/Thr sites on a protein or influence the post-translational modification state of nearby Ser/Thr sites. For example, threonine 58 on c-Myc is reciprocally modified; phosphorylation promotes its degradation, but O-GlcNAc promotes its stability (47). O-GlcNAcylation of CCAAT enhancer-binding protein β influences adjacent phosphorylation sites. O-GlcNAcylation of CCAAT enhancer-binding protein β Ser-180 and Ser-181 prevents the phosphorylation of Thr-188, Ser-184, and Thr-179 that is required for CCAAT enhancer-binding protein β DNA binding activity (48). Many phosphorylation sites have been mapped on Mi2β by mass spectrometry (49–54), and some of these sites could potentially be modified by O-GlcNAc. The reciprocal occupancy of GlcNAc or phosphate at these sites could fine-tune Mi2β activity or interactions. Moreover, O-GlcNAcylation of Mi2β may also influence the phosphorylation state of specific sites on Mi2β and therefore its function in the formation of and/or within the GATA-1·FOG-1·Mi2β repressor complex. In future studies, mapping Mi2β O-GlcNAc sites and exploring their biological function will be critical to understanding how O-GlcNAc cycling regulates the NuRD complex.
In summary, we demonstrated that OGT and OGA associate with the Aγ-globin promoter (Figs. 1 and 3). Inhibition of OGA by TMG leads to increased occupancy of OGT, OGA, and Mi2β at the Aγ-globin promoter and a decreased γ-globin transcription level (Fig. 2). Both OGT and OGA can interact with Mi2β, GATA-1, and FOG-1 (Figs. 6–8), and Mi2β is O-GlcNAcylated (Fig. 5). Our data indicate that Mi2β O-GlcNAcylation, OGT, and OGA facilitate assembly of the GATA-1·FOG-1·Mi2β (NuRD) repressor complex.
Based on our findings, we propose that OGT/OGA contributes to the regulation of GATA-1·FOG-1·Mi2β-mediated Aγ-globin gene repression. O-GlcNAc cycling on Mi2β is mediated by interactions with OGT and OGA (Fig. 9a). Potentially, O-GlcNAcylated Mi2β is recruited to −566 of the Aγ-globin promoter with OGT and OGA by GATA-1 and FOG-1. Subsequently, Mi2β recruits other potential cofactors (Fig. 9, a and b) to form a stable repressor complex. At this point, the Aγ-globin transcription is repressed (Fig. 9b). When Aγ-globin transcription is activated, Mi2β interaction with OGT or other cofactors is reduced, OGA removes O-GlcNAc from Mi2β, and the repressor complex is dissembled (Fig. 9c). However, several questions still need to be addressed. How is GATA-1·FOG-1·Mi2β assembly affected when the OGT is altered? Although Mi2β is modified by O-GlcNAc, are GATA-1, FOG-1, or other components of the NuRD complex modified by O-GlcNAc? Once the mechanistic role of O-GlcNAcylation in the assembly of the GATA-1·FOG-1·Mi2β repressor complex is understood, we may be able to target this complex to reverse γ-globin gene silencing and effectively treat sickle cell disease patients by increasing their fetal hemoglobin to beneficial therapeutic levels.
FIGURE 9.
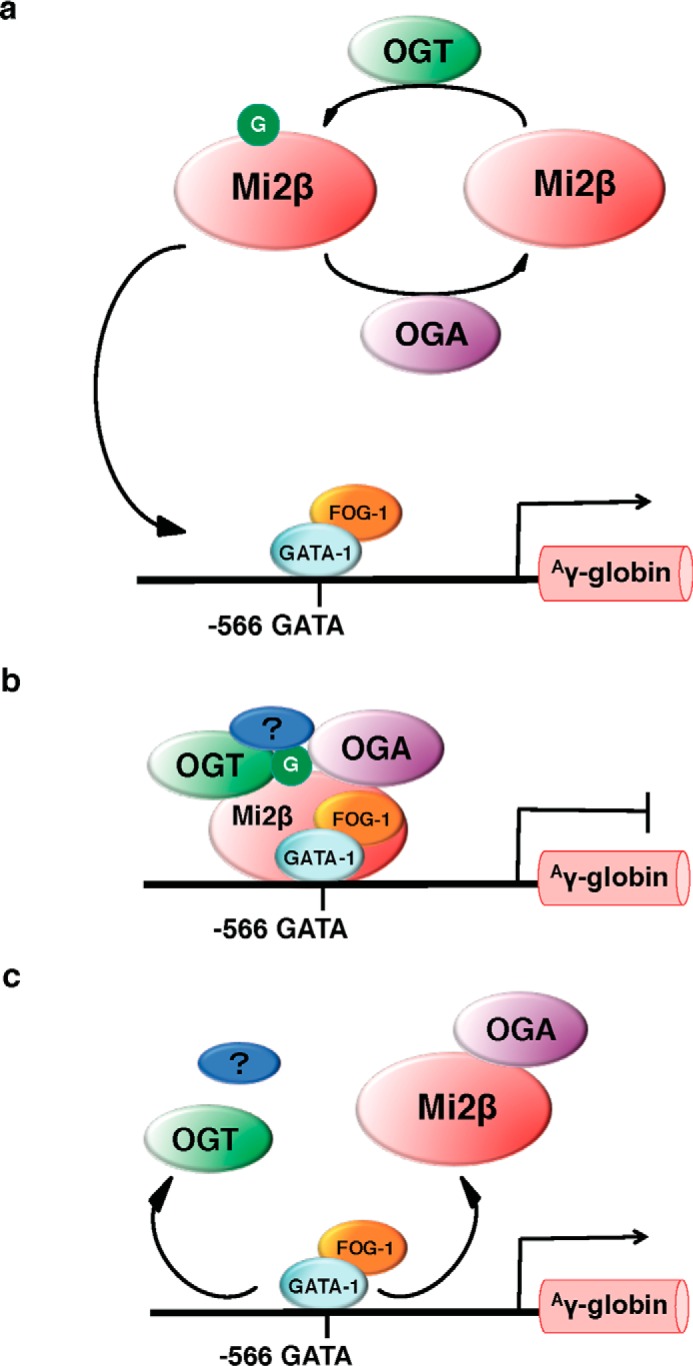
Proposed mechanism of OGT/OGA regulation of GATA-1·FOG-1·Mi2β mediated Aγ-globin repression. a, O-GlcNAc (G) cycling on Mi2β is processed by OGT and OGA. Potentially, O-GlcNAcylated Mi2β is recruited to −566 of Aγ-globin promoter with OGT and OGA by GATA-1 and FOG-1. b, once O-GlcNAcylated Mi2β is recruited to the promoter, other potential cofactors (blue oval with “?”) are recruited to the promoter as well to form a stable repressor complex, repressing Aγ-globin transcription. c, when Aγ-globin transcription is activated, Mi2β does not interact with OGT or other cofactors, and OGA removes O-GlcNAc from Mi2β, dissembling the repressor complex.
Materials and Methods
Antibodies
Primary antibodies and secondary antibodies for immunoblotting were used at a 1:1,000 and 1:10,000 dilutions, respectively. Antibodies for ChIP assay were used at 2 μg/reaction.
Antibodies for Immunoblotting
O-GlcNAc (CTD 110.6), OGT (AL-34), and OGA (345) were gracious gifts from the laboratory of Gerald Hart in the Department of Biological Chemistry at The Johns Hopkins University School of Medicine. Anti-O-linked N-acetylglucosamine antibody (RL2) (ab2739), anti-GAPDH antibody (ab9484), and anti-chromodomain helicase DNA-binding protein 4 antibody (3F2/4) (ab70469) were purchased from Abcam. Actin antibody (A2066), anti-chicken IgY-HRP (A9046), and anti-mouse IgM-HRP (A8786) were purchased from Sigma. Goat anti-rabbit IgG-HRP (170-6515) and goat anti-mouse IgG-HRP (170-6516) were purchased from Bio-Rad. Rabbit anti-goat IgG-HRP (31402) was purchased from Thermo Scientific. Goat anti-rat IgG-HRP (NA935V) was purchased from GE Healthcare. Mi2β antibody (H-242) (sc-11378 X; also used for ChIP assay), GATA-1 antibody (sc-265 X), FOG antibody (sc-9361 X), normal rabbit IgG (sc-2027), normal chicken IgY (sc-2718), normal mouse IgG (sc-2025) and IgM (sc-3881), rat IgG (sc-2026), and normal goat IgG (sc-2028) were purchased form Santa Cruz Biotechnology.
Antibodies for ChIP
Rabbit control IgG (ab46540) was purchased from Abcam. OGT antibody (61355) was purchased from Active Motif. Anti-OGA antibody (SAB4200267) was purchased from Sigma.
Cell Culture
K562 cells were cultured in RPMI 1640 medium (R8758, Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS; 100-106, Gemini), 1× minimum Eagle's medium non-essential amino acids solution (Sigma), 1 mm sodium pyruvate (Sigma), 2.5 mm HEPES (Sigma), and 1× penicillin/streptomycin (Sigma). K562 cells were treated with 10 μm TMG (S.D. Specialty Chemicals) for 6 h followed by 0.75 mm NaB (B5887, Sigma) for 4 days or 10 μm hemin (51280, Sigma) and 3 mm HMBA (208320500, Acros Organics) for 3 days (10).
WT and −566 Aγ-globin mutant murine CID-dependent human β-YAC BMCs were generated as described previously (10, 11). WT CID-dependent β-YAC BMCs have the normal GATA sequence at −566 of the Aγ-globin promoter, whereas −566 mutant CID-dependent β-YAC BMCs have a T→G point mutation at nucleotide −566. Cells were cultured in HyClone Iscove's modified Dulbecco's medium (SH30005.02, Thermo Scientific) supplemented with 10% heat-inactivated FBS, 1× penicillin/streptomycin, and the CID, 5 μm CL-COB-II-293 (synthesized by the University of Kansas Centers of Biomedical Research Excellence Center for Cancer Experimental Therapeutics Core C Synthesis Lab; commonly called AP20187). The cells were passaged at 2 × 105 cells/ml every 3 days in the presence of CL-COB-II-293 (55). WT β-YAC BMCs were treated with 10 μm TMG for 4 days with fresh TMG and culture medium supplied daily.
The MEL cell line stably expressing bacteria biotin ligase birA was a kind gift from the laboratory of Alan B. Cantor at Harvard Medical School and cultured as described previously (43). All cells were incubated at 37 °C in 5% CO2 in a 95% humidified incubator.
Immunoprecipitation
Cells were lysed on ice for 30 min in Nonidet P-40 lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm DTT, 40 mm GlcNAc, and 1% Nonidet P-40) supplemented with 1 mm PMSF, 1 mm sodium fluoride, 1 mm β-glycerol phosphate, and 1× protease inhibitor mixture I (1 μg/ml leupeptin, 1 μg/ml antipain, 10 μg/ml benzamidine, and 0.1% aprotinin). Two milligrams of cell lysates were incubated with 2 μg of antibody in a 1-ml reaction volume overnight with end-to-end rotation at 4 °C. The next day, 20 μl of anti-mouse IgM (μ-chain-specific)-agarose (A4540, Sigma), Protein G-Sepharose 4 Fast Flow (17-0618-01, GE Healthcare), or anti-chicken IgY-agarose (DAIgY-AGA-2, Gallus Immunotech) slurry were added into the mixture followed by end-to-end rotation at 4 °C for 2 h. Agarose beads were washed three times with 1 ml of Nonidet P-40 lysis buffer and mixed with 20 μl of 2× protein solubility mixture (50 mm Tris-HCl, pH 6.8, 5 mm EDTA, 4% SDS, 25% sucrose, 2.5% β-mercaptoethanol, and 0.04% Pyronin-Y). Forty micrograms of cell lysates were mixed with 4× protein solubility mixture as input. The beads and input were heated at 95 °C for 2 min and subjected to immunoblotting.
Immunoblotting
All electrophoresis was performed with 4–15% gradient Criterion TGX precast gels (567-1084, Bio-Rad) at 140 V followed by transfer of protein to PVDF membrane (IPVH00010, Millipore) at 0.4 A. Blots were blocked by 3% BSA in TBST (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, and 0.05% Tween 20) at room temperature for 20 min and then incubated with primary antibody at 4 °C overnight. After washing with TBST five times for 5 min each time, blots were incubated with HRP-conjugated secondary antibody for 1 h at room temperature followed by TBST washes again and developed using chemiluminescent substrate (E2400, Denville Scientific). Blots were stripped in 200 mm glycine, pH 2.5, at room temperature for 1 h and probed with other primary antibodies. All immunoblotting results were repeated in three independent experiments, and representative images are shown.
ChIP Assay
WT and −566 Aγ-globin mutant CID-dependent β-YAC BMCs were used, or WT β-YAC transgenic mouse fetal liver single cell suspensions were prepared as described previously (10, 11). Briefly, fetal liver from WT β-YAC transgenic mice at postconception days E12 or E18 were isolated and kept on ice in 1× PBS. The liver tissue was cut into small pieces with a scissor, and a single cell suspension was prepared by repeatedly passing the liver pieces through a 1-ml syringe with sequentially smaller needle sizes (16, 18, and 20 gauge; BD Biosciences) and then subjected to ChIP assay.
The ChIP assay was performed using a method described previously (40) with slight modifications. Briefly, cells were cross-linked by 2 mm ethylene glycol bis(succinimidylsuccinate) (21565, Pierce) in PBS for 30 min followed by 1% formaldehyde (BP531-25, Fisher) for 10 min at room temperature. The cross-linking reaction was terminated by the addition of 2.5 m glycine to a final concentration of 125 mm. Cells were lysed, and chromatin was collected by centrifugation at 1,000 × g. Three hundred microliters of nuclear lysis buffer (50 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS, 40 mm GlcNAc, and 25% glycerol) were used to resuspend chromatin pellets from 5 × 106 cells. Chromatin DNA was sheared to the size of 100–300 bp and immunoprecipitated with 2 μg of control IgG or specific antibody, respectively, at 4 °C overnight. The next day, 30 μl of PBS-washed M-280 sheep anti-rabbit IgG Dynabeads (11204D, Invitrogen) were added to the chromatin followed by rotation at 4 °C for 4 h. Dynabeads were separated using a DynaMag-2 Magnet (12321D, Invitrogen) and subsequently washed and eluted as described previously (40). The eluate was subjected to reverse cross-linking by sequential treatments with RNase A and proteinase K (40). ChIP DNA was extracted as described previously (40) and dissolved in 50 μl of nuclease-free water (AM9906, Ambion) followed by quantitative PCR (qPCR).
Total RNA Isolation and Reverse Transcription (RT)-PCR
Total RNA was isolated as described previously (40). Briefly, RNA was isolated from 2 × 106 cells using TRI Reagent solution (T9424, Sigma) according to the manufacturer's instructions. For RT, 0.5 μg of total RNA was used in iScript Reverse Transcription Supermix (170-8841, Bio-Rad) following the manufacturer's instructions. Ten microliters of each completed reaction mixture was incubated in a thermal cycler (Model 2720, Applied Biosystems) using the following protocol: priming, 5 min at 25 °C; RT, 30 min at 42 °C; and RT inactivation, 5 min at 85 °C. cDNA products were diluted with 90 μl of nuclease-free water and analyzed by qPCR.
qPCR Assay
cDNA and ChIP DNA were analyzed by qPCR as described previously (40) according to the manufacturer's instructions. Two microliters of cDNA or 5 μl of ChIP DNA sample, 10 μl of SsoAdvanced Universal SYBR Green Supermix (172-5271, Bio-Rad), nuclease-free water, and primers (Table 1) for the target gene were mixed together in a total reaction volume of 20 μl. The reactions were run in a CFX96 Touch Real-Time PCR Detection System (185-5195, Bio-Rad) using the following conditions: polymerase activation and DNA denaturation, 30 s at 95 °C; amplification denaturation, 5 s at 95 °C; and amplification annealing and extension, 30 s at 60 °C or 62 °C for 40 cycles.
TABLE 1.
Primer sequences used for qPCR
| Target gene | Primer sequence (5′ → 3′) |
|---|---|
| Human γ-globin | Forward, GTATTGCTTGCAGAATAAAGCC |
| Reverse, GACCGTTTTGGCAATCCATTTC | |
| Mouse HPRT | Forward, GGCCAGACTTTGTTGGATTTG |
| Reverse, CGCTCATCTTAGGCTTTGTATTTG | |
| Aγ-Globin promoter | Forward, CTAATCTATTACTGCGCTGA |
| Reverse, GTTTCTAAGGAAAAAGTGCT | |
| Human OGT | Forward, CATCGAGAATATCAGGCAGGAG |
| Reverse, CCTTCGACACTGGAAGTGTATAG | |
| Human OGA | Forward, TTCACTGAAGGCTAATGGCTCCCG |
| Reverse, ATGTCACAGGCTCCGACCAAGT | |
| Human HPRT | Forward, ATTGGTGGAGATGATCTCTCAACTTT |
| Reverse, GCCAGTGTCAATTATATCTTCCACAA |
qPCR Data Analysis
Quantification cycle (Cq) values were calculated by CFX ManagerTM software. For cDNA qPCR data, the dynamic range of RT and amplification efficiency was evaluated before applying the ΔΔCq method to calculate relative gene expression change. The transcription level of the target gene was normalized to the internal control as -fold change. For ChIP DNA qPCR data, the Cq value was normalized as percentage of input. Data generated in three independent experiments are presented as means ± S.E.; the two-tailed Student's t test statistic was applied with p < 0.05 considered to be a significant difference.
Author Contributions
Z. Z. conducted most of the experiments, analyzed the results, and wrote the paper. F. C. C. designed and performed the experiments and analyzed the data shown in Fig. 2. E. P. T., N. B., and L. D. provided technical assistance. C. E. C., M. E. M., and S. A. W. contributed to the manuscript preparation. K. R. P. and C. S. conceived the idea for the project and contributed to manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Zhuan Li from the University of Kansas Medical Center Liver Center for assistance with the ChIP assay.
This work was supported by an institutional development award from the NIGMS under Grant P20GM104936 and NIDDK Grant R01DK100595 (to C. S. and K. R. P.), NHLBI Grant R01HL111264 (to K. R. P.), and NHLBI Contract HHSN268201000031C and NIGMS Grant P41 GM104603 (to C. E. C.) all from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BCL11A
- B-cell lymphoma-leukemia A
- NuRD
- nucleosome remodeling and deacetylase complex
- KLF1
- Krüppel-like factor 1
- FOG
- friend of GATA
- O-GlcNAc
- O-linked N-acetylglucosamine
- OGT
- O-GlcNAc-transferase
- OGA
- O-GlcNAcase
- β-YAC
- β-globin locus yeast artificial chromosome
- BMC
- bone marrow cell
- TMG
- Thiamet-G
- Pol
- polymerase
- CTD
- C-terminal domain
- CID
- chemical inducer of dimerization
- NaB
- sodium butyrate
- HMBA
- N,N′-hexamethylenebisacetamide
- H-H
- hemin and HMBA
- Cp
- Clostridium perfringens
- IP
- immunoprecipitation
- MEL
- murine erythroleukemia
- qPCR
- quantitative PCR
- Cq
- quantification cycle
- HPRT
- hypoxanthine phosphoribosyltransferase.
References
- 1. Orkin S. H. (1990) Globin gene regulation and switching: circa 1990. Cell 63, 665–672 [DOI] [PubMed] [Google Scholar]
- 2. Fraser P., and Grosveld F. (1998) Locus control regions, chromatin activation and transcription. Curr. Opin. Cell Biol. 10, 361–365 [DOI] [PubMed] [Google Scholar]
- 3. Li Q., Peterson K. R., Fang X., and Stamatoyannopoulos G. (2002) Locus control regions. Blood 100, 3077–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginder G. D. (2015) Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl. Res. 165, 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bauer D. E., and Orkin S. H. (2011) Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr. Opin. Pediatr. 23, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verduzco L. A., and Nathan D. G. (2009) Sickle cell disease and stroke. Blood 114, 5117–5125 [DOI] [PubMed] [Google Scholar]
- 7. Gladwin M. T., and Sachdev V. (2012) Cardiovascular abnormalities in sickle cell disease. J. Am. Coll. Cardiol. 59, 1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sankaran V. G. (2011) Targeted therapeutic strategies for fetal hemoglobin induction. Hematology Am. Soc. Hematol. Educ. Program 2011, 459–465 [DOI] [PubMed] [Google Scholar]
- 9. Amaya M., Desai M., Gnanapragasam M. N., Wang S. Z., Zu Zhu S., Williams D. C. Jr., and Ginder G. D. (2013) Mi2β-mediated silencing of the fetal γ-globin gene in adult erythroid cells. Blood 121, 3493–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harju-Baker S., Costa F. C., Fedosyuk H., Neades R., and Peterson K. R. (2008) Silencing of Aγ-globin gene expression during adult definitive erythropoiesis mediated by GATA-1-FOG-1-Mi2 complex binding at the −566 GATA site. Mol. Cell. Biol. 28, 3101–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costa F. C., Fedosyuk H., Chazelle A. M., Neades R. Y., and Peterson K. R. (2012) Mi2β is required for γ-globin gene silencing: temporal assembly of a GATA-1-FOG-1-Mi2 repressor complex in β-YAC transgenic mice. PLoS Genet. 8, e1003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Letting D. L., Chen Y. Y., Rakowski C., Reedy S., and Blobel G. A. (2004) Context-dependent regulation of GATA-1 by friend of GATA-1. Proc. Natl. Acad. Sci. U.S.A. 101, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miccio A., and Blobel G. A. (2010) Role of the GATA-1/FOG-1/NuRD pathway in the expression of human β-like globin genes. Mol. Cell. Biol. 30, 3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miccio A., Wang Y., Hong W., Gregory G. D., Wang H., Yu X., Choi J. K., Shelat S., Tong W., Poncz M., and Blobel G. A. (2010) NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J. 29, 442–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez P., Bonte E., Krijgsveld J., Kolodziej K. E., Guyot B., Heck A. J., Vyas P., de Boer E., Grosveld F., and Strouboulis J. (2005) GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J. 24, 2354–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hart G. W., Slawson C., Ramirez-Correa G., and Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hart G. W. (2014) Three decades of research on O-GlcNAcylation—a major nutrient sensor that regulates signaling, transcription and cellular metabolism. Front. Endocrinol. 5, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slawson C., and Hart G. W. (2011) O-GlcNAc signalling: implications for cancer cell biology. Nat. Rev. Cancer 11, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bond M. R., and Hanover J. A. (2013) O-GlcNAc cycling: a link between metabolism and chronic disease. Annu. Rev. Nutr. 33, 205–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan E. P., Caro S., Potnis A., Lanza C., and Slawson C. (2013) O-Linked N-acetylglucosamine cycling regulates mitotic spindle organization. J. Biol. Chem. 288, 27085–27099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan E. P., Villar M. T., E. L., Lu J., Selfridge J. E., Artigues A., Swerdlow R. H., and Slawson C. (2014) Altering O-linked β-N-acetylglucosamine cycling disrupts mitochondrial function. J. Biol. Chem. 289, 14719–14730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lanza C., Tan E. P., Zhang Z., Machacek M., Brinker A. E., Azuma M., and Slawson C. (2016) Reduced O-GlcNAcase expression promotes mitotic errors and spindle defects. Cell Cycle 15, 1363–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shafi R., Iyer S. P., Ellies L. G., O'Donnell N., Marek K. W., Chui D., Hart G. W., and Marth J. D. (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. U.S.A. 97, 5735–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Y. R., Song M., Lee H., Jeon Y., Choi E. J., Jang H. J., Moon H. Y., Byun H. Y., Kim E. K., Kim D. H., Lee M. N., Koh A., Ghim J., Choi J. H., Lee-Kwon W., Kim K. T., Ryu S. H., and Suh P. G. (2012) O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 11, 439–448 [DOI] [PubMed] [Google Scholar]
- 25. Dias W. B., and Hart G. W. (2007) O-GlcNAc modification in diabetes and Alzheimer's disease. Mol. Biosyst. 3, 766–772 [DOI] [PubMed] [Google Scholar]
- 26. Fardini Y., Dehennaut V., Lefebvre T., and Issad T. (2013) O-GlcNAcylation: a new cancer hallmark? Front. Endocrinol. 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu Y., Shan X., Yuzwa S. A., and Vocadlo D. J. (2014) The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 289, 34472–34481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakabe K., Wang Z., and Hart G. W. (2010) β-N-Acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl. Acad. Sci. U.S.A. 107, 19915–19920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujiki R., Hashiba W., Sekine H., Yokoyama A., Chikanishi T., Ito S., Imai Y., Kim J., He H. H., Igarashi K., Kanno J., Ohtake F., Kitagawa H., Roeder R. G., Brown M., and Kato S. (2011) GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480, 557–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fong J. J., Nguyen B. L., Bridger R., Medrano E. E., Wells L., Pan S., and Sifers R. N. (2012) β-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J. Biol. Chem. 287, 12195–12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akan I., Love D. C., Harwood K. R., Bond M. R., and Hanover J. A. (2016) Drosophila O-GlcNAcase deletion globally perturbs chromatin O-GlcNAcylation. J. Biol. Chem. 291, 9906–9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelly W. G., Dahmus M. E., and Hart G. W. (1993) RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J. Biol. Chem. 268, 10416–10424 [PubMed] [Google Scholar]
- 33. Ranuncolo S. M., Ghosh S., Hanover J. A., Hart G. W., and Lewis B. A. (2012) Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J. Biol. Chem. 287, 23549–23561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang X., Zhang F., and Kudlow J. E. (2002) Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110, 69–80 [DOI] [PubMed] [Google Scholar]
- 35. Chen Q., Chen Y., Bian C., Fujiki R., and Yu X. (2013) TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li M. D., Ruan H. B., Hughes M. E., Lee J. S., Singh J. P., Jones S. P., Nitabach M. N., and Yang X. (2013) O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 17, 303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramakrishnan P., Clark P. M., Mason D. E., Peters E. C., Hsieh-Wilson L. C., and Baltimore D. (2013) Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 6, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dehennaut V., Leprince D., and Lefebvre T. (2014) O-GlcNAcylation, an epigenetic mark. Focus on the histone code, TET family proteins, and polycomb group proteins. Front. Endocrinol. 5, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lewis B. A., and Hanover J. A. (2014) O-GlcNAc and the epigenetic regulation of gene expression. J. Biol. Chem. 289, 34440–34448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Z., Tan E. P., VandenHull N. J., Peterson K. R., and Slawson C. (2014) O-GlcNAcase expression is sensitive to changes in O-GlcNAc homeostasis. Front. Endocrinol. 5, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cioe L., McNab A., Hubbell H. R., Meo P., Curtis P., and Rovera G. (1981) Differential expression of the globin genes in human leukemia K562(S) cells induced to differentiate by hemin or butyric acid. Cancer Res. 41, 237–243 [PubMed] [Google Scholar]
- 42. Rao F. V., Dorfmueller H. C., Villa F., Allwood M., Eggleston I. M., and van Aalten D. M. (2006) Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 25, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu M., Riva L., Xie H., Schindler Y., Moran T. B., Cheng Y., Yu D., Hardison R., Weiss M. J., Orkin S. H., Bernstein B. E., Fraenkel E., and Cantor A. B. (2009) Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sinclair D. A., Syrzycka M., Macauley M. S., Rastgardani T., Komljenovic I., Vocadlo D. J., Brock H. W., and Honda B. M. (2009) Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proc. Natl. Acad. Sci. U.S.A. 106, 13427–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lahlil R., Lécuyer E., Herblot S., and Hoang T. (2004) SCL assembles a multifactorial complex that determines glycophorin A expression. Mol. Cell. Biol. 24, 1439–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Merika M., and Orkin S. H. (1993) DNA-binding specificity of GATA family transcription factors. Mol. Cell. Biol. 13, 3999–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chou T. Y., Hart G. W., and Dang C. V. (1995) c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 270, 18961–18965 [DOI] [PubMed] [Google Scholar]
- 48. Li X., Molina H., Huang H., Zhang Y. Y., Liu M., Qian S. W., Slawson C., Dias W. B., Pandey A., Hart G. W., Lane M. D., and Tang Q. Q. (2009) O-Linked N-acetylglucosamine modification on CCAAT enhancer-binding protein β: role during adipocyte differentiation. J. Biol. Chem. 284, 19248–19254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., and Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rigbolt K. T., Prokhorova T. A., Akimov V., Henningsen J., Johansen P. T., Kratchmarova I., Kassem M., Mann M., Olsen J. V., and Blagoev B. (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3. [DOI] [PubMed] [Google Scholar]
- 52. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Winiewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 [DOI] [PubMed] [Google Scholar]
- 53. Yi T., Zhai B., Yu Y., Kiyotsugu Y., Raschle T., Etzkorn M., Seo H. C., Nagiec M., Luna R. E., Reinherz E. L., Blenis J., Gygi S. P., and Wagner G. (2014) Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E2182–E2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trinidad J. C., Barkan D. T., Gulledge B. F., Thalhammer A., Sali A., Schoepfer R., and Burlingame A. L. (2012) Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 11, 215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peterson K. R., Costa F. C., Fedosyuk H., Neades R. Y., Chazelle A. M., Zelenchuk L., Fonteles A. H., Dalal P., Roy A., Chaguturu R., Li B., and Pace B. S. (2014) A cell-based high-throughput screen for novel chemical inducers of fetal hemoglobin for treatment of hemoglobinopathies. PLoS One 9, e107006. [DOI] [PMC free article] [PubMed] [Google Scholar]