

Abstract
G‐protein‐coupled receptors (GPCRs) are involved in a wide range of physiological processes, and they have attracted considerable attention as important targets for developing new medicines. A central and largely unresolved question in drug discovery, which is especially relevant to GPCRs, concerns ligand selectivity: Why do certain molecules act as activators (agonists) whereas others, with nearly identical structures, act as blockers (antagonists) of GPCRs? To address this question, we employed all‐atom, long‐timescale molecular dynamics simulations to investigate how two diastereomers (epimers) of dihydrofuroaporphine bind to the serotonin 5‐HT1A receptor and exert opposite effects. By using molecular interaction fingerprints, we discovered that the agonist could mobilize nearby amino acid residues to act as molecular switches for the formation of a continuous water channel. In contrast, the antagonist epimer remained firmly stabilized in the binding pocket.
Keywords: GPCRs, molecular dynamics simulations, proteins, stereoselectivity, water channels
The growing number of crystal structures and related computer simulations of G‐protein‐coupled receptors (GPCRs) have resolved a number of structural key features in the activation process of GPCRs, including ligand‐binding specificity, side‐chain molecular switches, rearrangement of transmembrane helices, and formation of internal water channels.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 In spite of this progress, many important mechanistic principles of GPCR‐mediated signalling remain poorly understood at the molecular level. An example is ligand stereoselectivity, which is a central concern in drug discovery since it substantially influences the efficacy, efficiency, and metabolic properties of drug candidates.11, 12 Molecular dynamics could be of great help towards addressing such unresolved issues.9, 10 In this work, we used all‐atom, long‐timescale molecular dynamics (MD) simulations to investigate the ligand stereoselectivity of the serotonin 5‐HT1A receptor and determine how the stereochemical arrangement of a single methyl group at a chiral carbon atom determines whether the ligand acts as an agonist or an antagonist.
For a pair of dihydrofuroaporphine epimers, functional assays have identified one epimer to be a full agonist and the other to be a full antagonist of the serotonin 5‐HT1A receptor.13, 14 The configuration of a single methyl group is the only structural difference between this pair of diastereomers, and it results in different functional properties as ligands for the receptor (Scheme 1).
Scheme 1.
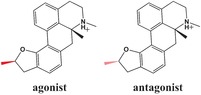
The dihydrofuroaporphine epimers used as 5‐HT1A ligands in the MD simulations. The configuration of the methyl group in the two epimers is highlighted in red.
To explain the structural basis of this stereoselectivity of a prototypical GPCR, we first built a homology model of the 5‐HT1A receptor by using the crystal structure of the 5‐HT1B receptor (PDB ID: 4IAQ)15 for an agonist‐bound receptor structure template and that of the M3 muscarinic receptor (PDB ID: 4U15)16 for an antagonist‐bound receptor structure template. Interestingly, the superimposed crystal structures of the two receptors are almost identical (Figure S1 in the Supporting Information), with an RMSD of less than 1.5 Å for the TM backbone. GPCRs are known to undergo large helix movements when activated by their G proteins. Since these signatures are not present in the homology model of the 5‐HT1A receptor, it represents the receptor in a non‐activated state (see the Supporting Information, including Figure S1). Since W6.48 adopts different rotamer states in the 5‐HT1B and M3 receptor template structures, we compared the side‐chain conformations of the highly conserved W6.48 across all available GPCR crystal structures (Figure S2 A). In almost all cases, the long axis of the indole ring orients preferentially parallel to the TM helices (Figure S2 B). The only exception is found in the M3 crystal structure, where the long axis of the indole ring of W6.48 is oriented perpendicular to the TM helix. In our homology model of the 5‐HT1A receptor, we adjusted the side‐chain conformation of W6.48 to that found in most GPCR structures (Figure S2 B). On this basis, we performed 3×1.2 μs all‐atom MD simulations for both the agonist‐bound and the antagonist‐bound 5‐HT1A receptor. This yielded a total MD simulation time of 7.2 μs.
Since ligand binding is a crucial step for GPCR activation, we first examined the binding modes for the agonist and the antagonist epimers. In the MD structure of the human 5‐HT1A receptor, both bound ligands form a salt bridge with D1163.38, which is similar to what is observed in the crystal structures of the related 5‐HT1B 15 and 5‐HT2B 17 receptors (Figure 1 A–D). Interaction fingerprints obtained from the initial 50 ns MD simulations (Figure 1 E, G) show that the bound agonist forms strong hydrophobic interactions with residues I189ECL2, W3586.48, F3616.51 and F3626.52, and additionally forms weak hydrophobic interactions with I1133.35, D1163.38, V1173.39, and Y3907.43. The bound antagonist formed hydrophobic interactions with residues V1173.39, I189ECL2, S1995.42 and F3616.51, however, hydrophobic contacts with W3586.48 and F3626.52 were not observed.
Figure 1.

Interactions of ligands with side chains in the binding pocket of the 5‐HT1A receptor. A) Antagonist; beginning of MD simulation. B) Antagonist; last frame of MD simulation. C) Agonist; beginning of MD simulation. D) Agonist; last frame of the MD simulation. Cyan: antagonist epimer, yellow: agonist epimer, green: highlighted side chains in the binding pocket of the 5‐HT1A receptor. Blue dashed lines: ionic interactions between D1163.38 and the ligand, red dashed lines: hydrogen bond between D1163.38 and Y3907.43. E) Interaction fingerprint between the 5‐HT1A receptor and the antagonist in the initial 50 ns. F) Interaction fingerprint between the 5‐HT1A receptor and antagonist in the final 50 ns. G) Interaction fingerprint between the 5‐HT1A receptor and agonist in the initial 50 ns. H) Interaction fingerprint between the 5‐HT1A receptor and agonist in the final 50 ns. Blue, green, and red areas represent the three different MD simulations presented in this work.
Next, we investigated how these interactions changed during the final 50 ns of the MD simulation. In the agonist/receptor complex, most of the ligand–receptor interactions were preserved except for hydrophobic interactions between the agonist and W3586.48 (Figure 1 H). The highly conserved W3586.48 plays a crucial role during the activation of the majority of rhodopsin‐like GPCRs, and it is an essential residue in the transmission switch.18 In contrast, all ligand–receptor interactions in the antagonist/receptor complex were preserved during the entire MD simulation (Figure 1 F). Activation of the transmission switch in the agonist‐bound 5‐HT1A receptor started with the rotation of F3626.52 during the 150–220 ns period (Figure 2 A). F3626.52 has been shown elsewhere to play a pivotal role in the activation of 5‐HT1A. 19, 20
Figure 2.

Molecular switches in the 5‐HT1A receptor. A) χ2 angles of F3626.52. B) χ2 angles of W3586.48. C) H‐bond lengths between D1163.38 and Y3907.43. Black, red, brown: trajectories of three independent MD simulations (ago‐1, ago‐2, ago‐3) for the agonist‐bound receptor. Blue, green, purple: trajectories of three independent MD simulations (anta‐1, anta‐2, anta‐3) for the antagonist‐bound receptor.
In the antagonist‐bound receptor, F3626.52 remained in the initial conformation throughout the MD simulation. Additionally, the hydrogen bond between D1163.38 and Y3907.43 was observed to break in the 300–600 ns period in the agonist/receptor complex (Figures 1 D and 2 C), while it remained stable in the antagonist‐bound form of the receptor complex. Interestingly, D1163.38 formed a salt bridge with both the agonist and antagonist, and such interactions were stable in both complexes throughout the MD simulations (Figure 1 E and Figure S3).
More importantly, W3586.52, a highly conserved residue in transmembrane helix 6 (TM6), was found to undergo an abrupt rotational switch between 350 ns and 650 ns in our MD simulations (Figure 2 B). Subtle fluctuations also were observed for W3586.48 in the complex with the antagonist at the beginning of the simulations, but these returned to the starting conformation after 700 ns (Figure 2 B). Our previous studies of the adenosine and opioid receptors6, 21 revealed W3586.48 to be a central molecular switch that enables the formation of an internal continuous water channel within the receptor, which was proposed to be a hallmark of GPCR activation. In the present case of the 5‐HT1A receptor, we again observed the side‐chain rotational switch associated with W3586.48, and therefore we also analyzed water movement inside the 5‐HT1A receptor. Similar to our previous findings, we detected a distinct difference in the relocation of water molecules inside the receptor after binding of the agonist compared to the antagonist (Figure 3 and Figure S4). The final structures obtained from MD simulations showed fewer water molecules at the ligand binding site of the 5‐HT1A receptor when complexed with the antagonist compared to the agonist (Figure 3 A,B and Figure S4). The movement of water molecules in the agonist‐bound receptor was initiated by the preceding conformational changes of the molecular switches. The first event was a rotational switch of F3626.52 located very close to the stereocenter of the agonist (Figure 1). This was followed by rotational switching of W3586.48 and breaking of the 3–7 lock (a hydrogen bond between helices TM3 and TM7; Figure 2). Only after some delay (50 ns in one simulation and 100 ns in the second simulation) did the number of water molecules increase in the allosteric site at D822.50 (Figure S2). Contrary to the case of bound agonist, two hydrophobic layers of amino acids prevent the formation of a continuous intrinsic water channel in the antagonist‐bound receptor at the end of the MD simulations (Figure 3 A). The first hydrophobic layer is located between the orthosteric and the allosteric sites, with a thickness of 5 Å. Notably, water molecules from the bulk solvent next to the bound antagonist only rarely diffused into the deep pocket of the receptor. The second, 8 Å hydrophobic layer is positioned close to the highly conserved D822.50 in TM2 and the cytoplasmic end of the receptor (close to the Y4007.53 residue). The existence of such hydrophobic layers of amino acids agrees with the disruption of water‐mediated hydrogen‐bond networks observed in GPCR crystal structures22, 23 and in MD simulations.6, 7, 21, 24
Figure 3.

The 5‐HT1A receptor at the end of the MD simulations: Internal water molecules and the side‐chain interaction network. A) Receptor with bound antagonist (cyan). Two layers of hydrophobic amino acids with thicknesses of 5 Å and 8 Å were observed. Red dots: water molecules. B) The 5‐HT1A receptor with bound agonist (yellow). C) The residue interaction network in the antagonist‐bound receptor. The large circle indicates multiple residue interactions. (D) The residue interaction network in the agonist‐bound receptor. Small circles and scattered dots indicate fewer residue interactions. In (C) and (D), residues in helices and in loops are shown as red and blue dots, respectively, and line connections indicate contacts between residues (for details, see Supporting Information).
The bending of transmembrane helices is another signature of GPCR activation.25, 26 Such events were observed in the present study of the 5‐HT1A receptor. As captured and quantified for the final 50 ns period of each of our MD simulations, the agonist‐bound receptor underwent bending of helices TM5 (Figures S3, S4), TM6, and TM7, which is not seen in the antagonist‐bound complexes. Changes in the D/ERY ionic lock have also been thought to play an important role during GPCR activation. We found such changes (Figure S5) in the salt bridge between D1333.49 and R1343.50 in two trajectories of the agonist‐bound receptor but not in the antagonist‐bound counterpart. This salt bridge was broken in the simulations at about 450 ns and 780 ns.
The sudden increase in water at the R822.50 residue at 350 ns and 780 ns, the dissolution of the salt bridge, and the appearance of water appear highly correlated, likely making the presence of water inside GPCRs an important event. In contrast, this salt bridge was quite stable during our MD simulations of the antagonist‐bound complex.
Analysis of the interaction network between residue side chains (Figure 3) indicates that in the antagonist‐bound receptor, most of the residues inside firmly contact multiple neighbors (depicted in a large circle, Figure 3 C). In the agonist‐bound receptor (Figure 3 D), however, the interactions between side chains inside the receptor were disrupted by helix bending (Figures S3, S4) accompanied by water influx (Figure 3 B). Such disruptions are characterized by fewer residue contacts (chain of dots at the bottom of the figure) and multiple local small‐group interactions (scattered dots).
In conclusion, our MD simulations reveal structural differences upon the binding of a pair of optical isomers, one acting as an agonist and the other as an antagonist of the 5‐HT1A receptor. The results reveal in molecular detail the central steps of agonist‐induced activation of the 5‐HT1A receptor (Figure 4). First, the methyl group at the chiral center of the agonist molecule contacts the F3626.52 of the receptor through hydrophobic interactions, thereby resulting in a rotamer switch of the phenyl group of this residue. This first movement induces structural changes in the transmission switch, including the central residue in this switch, the highly conserved W3586.48, which opens a gate, followed by opening of the 3–7 lock of the receptor, thereby eventually allowing diffusion of water molecules from the bulk extracellular phase towards the central cytoplasmic internal space of the receptor. Moreover, the successive movement of water molecules into the receptor induces structural changes in TM5, TM6, and TM7, first bending and then rotation, thereby finally enabling the binding and activation of a G protein at the intracellular site of the receptor. The agonist and antagonist share a similar binding mode, including residues from extracellular loop 2 (ECL2) stabilizing the methyl group of the ligand at the stereocenter. One of these residues, I189ECL2, has a dual role: it stabilizes the methyl group of the antagonist when it is far from F3626.52, but in the case of the agonist, it keeps contact with the methyl group but is moved closer to F3616.51 This can facilitate the interaction between switching residues F3626.52 and W3586.48 and result in a large distortion of the central part of TM6, followed by bending of TM7 and breaking of the 3–7 lock.
Figure 4.
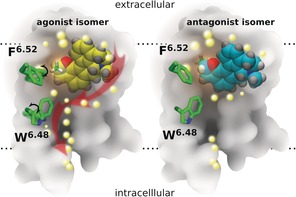
Schematic representation of ligand interactions and water influx in the binding site of the 5‐HT1A receptor (transmembrane cross‐section). Left: agonist isomer (yellow); right: antagonist isomer (cyan). The methyl group at the chiral carbon atom of the ligand is shown in green (partially hidden behind the rest of the molecule in the antagonist). The back part of the 5‐HT1A receptor is represented in gray, and the location of the lipid bilayer is indicated by horizontal dotted lines. The central amino acid side chains F6.52 and W6.48 (green), together with the 3–7 lock (residues D3.38 and Y7.43), act as a gate, which after binding of the agonist undergoes a rotational switch (black arrows, left) to form a continuous water channel inside the receptor. Water molecules are depicted as bright yellow spheres, and red arrows (left) indicate the movement of water molecules after opening of the water channel inside the receptor.
The structural details reported herein provide new insight into the unresolved issue of ligand stereoselectivity of GPCRs. As such, the findings could find application in innovative drug discovery.
Experimental Section
Membrane systems were built with the g_membed27 tool in Gromacs with each receptor structure pre‐aligned in the Orientations of Proteins in Membranes (OPM) database.28 Pre‐equilibrated 140 POPC lipids coupled with 10,200 TIP3P water molecules in a periodic box of 72 Å×72 Å×100 Å were used to build the protein/membrane system. Proteins, lipids, water molecules, and ions were modeled with the CHARMM36 force field29 parameter set; and the ligands were modeled with the CHARMM CGenFF small‐molecule force field.30 Ligands were submitted to the GAUSSIAN 09 program31 for structure optimization at the B3LYP/6‐31G* level prior to the generation of force‐field parameters. All bond lengths to hydrogen atoms in each protein/membrane system were constrained with M‐SHAKE.32 Van der Waals and short‐range electrostatic interactions were cut off at 10 Å.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Interdisciplinary Centre for Mathematical and Computational Modeling in Warsaw for support (grant G07‐13). Part of the work was done at the Pittsburgh Supercomputing Center using the Anton supercomputer (NIH grant P41GM103712‐S1; grant PSCA15060P) and at the Shanghai Supercomputer Center. S.F. was funded by the National Center of Science, Poland, grant 2013/08/M/ST6/00788. Research in H.V.′s group was supported by the European Community (project SynSignal, grant FP7‐KBBE‐2013‐613879), and internal funds of the EPFL. KP was funded by the Arnold and Mabel Beckman Foundation. H.V. and S.F. participate in the European COST Action CM1207 (GLISTEN), and K.P. is the John H. Hord Professor of Pharmacology.
S. Yuan, Q. Peng, K. Palczewski, H. Vogel, S. Filipek, Angew. Chem. Int. Ed. 2016, 55, 8661.
Contributor Information
Dr. Shuguang Yuan, Email: shuguang.yuan@gmail.com
Prof. Slawomir Filipek, Email: sfilipek@chem.uw.edu.pl
References
- 1. Kruse A. C., Kobilka B. K., Gautam D., Sexton P. M., Christopoulos A., Wess J., Nat. Rev. Drug Discovery 2014, 13, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K., Cell 2013, 152, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M., Nature 2013, 494, 185. [DOI] [PubMed] [Google Scholar]
- 4. Park J. H., Morizumi T., Li Y., Hong J. E., Pai E. F., Hofmann K. P., Choe H. W., Ernst O. P., Angew. Chem. Int. Ed. 2013, 52, 11021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11227. [Google Scholar]
- 5. Rose A. S., Elgeti M., Zachariae U., Grubmuller H., Hofmann K. P., Scheerer P., Hildebrand P. W., J. Am. Chem. Soc. 2014, 136, 11244. [DOI] [PubMed] [Google Scholar]
- 6. Yuan S., Palczewski K., Peng Q., Kolinski M., Vogel H., Filipek S., Angew. Chem. Int. Ed. 2015, 54, 7560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7670. [Google Scholar]
- 7. Yuan S., Filipek S., Palczewski K., Vogel H., Nat. Commun. 2014, 5, 4733. [DOI] [PubMed] [Google Scholar]
- 8. Lakkaraju S. K., Lemkul J. A., Huang J., A. D. MacKerell, Jr. , J. Comput. Chem. 2016, 37, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kappel K., Miao Y., McCammon J. A., Q. Rev. Biophys. 2015, 48, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miao Y., Nichols S. E., Gasper P. M., Metzger V. T., McCammon J. A., Proc. Natl. Acad. Sci. USA 2013, 110, 10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehvar R., Brocks D. R., Vakily M., Clin. Pharmacokinet. 2002, 41, 533. [DOI] [PubMed] [Google Scholar]
- 12. Lu H., Expert Opin. Drug Metab. Toxicol. 2007, 3, 149. [DOI] [PubMed] [Google Scholar]
- 13. Dosa P. I., Amin E. A., J. Med. Chem. 2016, 59, 810. [DOI] [PubMed] [Google Scholar]
- 14. Liu Z., Zhang H., Ye N., Zhang J., Wu Q., Sun P., Li L., Zhen X., Zhang A., J. Med. Chem. 2010, 53, 1319. [DOI] [PubMed] [Google Scholar]
- 15. Wang C., Jiang Y., Ma J. M., Wu H. X., Wacker D., Katritch V., Han G. W., Liu W., Huang X. P., Vardy E., McCorvy J. D., Gao X., Zhou X. E., Melcher K., Zhang C. H., Bai F., Yang H. Y., Yang L. L., Jiang H. L., Roth B. L., Cherezov V., Stevens R. C., Xu H. E., Science 2013, 340, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thorsen T. S., Matt R., Weis W. I., Kobilka B. K., Structure 2014, 22, 1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wacker D., Wang C., Katritch V., Han G. W., Huang X. P., Vardy E., McCorvy J. D., Jiang Y., Chu M. H., Siu F. Y., Liu W., Xu H. E., Cherezov V., Roth B. L., Stevens R. C., Science 2013, 340, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trzaskowski B., Latek D., Yuan S., Ghoshdastider U., Debinski A., Filipek S., Curr. Med. Chem. 2012, 19, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorinski N., Kowalsman N., Renner U., Wirth A., Reinartz M. T., Seifert R., Zeug A., Ponimaskin E., Niv M. Y., Mol. Pharmacol. 2012, 82, 448. [DOI] [PubMed] [Google Scholar]
- 20. Franchini S., Baraldi A., Sorbi C., Pellati F., Cichero E., Battisti U. M., Angeli P., Cilia A., Brasili L., MedChemComm 2015, 6, 677. [Google Scholar]
- 21. Yuan S., Hu Z., Filipek S., Vogel H., Angew. Chem. Int. Ed. 2015, 54, 556–559; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 566–569. [Google Scholar]
- 22. Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., Stevens R. C., Science 2012, 337, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fenalti G., Giguere P. M., Katritch V., Huang X. P., Thompson A. A., Cherezov V., Roth B. L., Stevens R. C., Nature 2014, 506, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan S., Vogel H., Filipek S., Angew. Chem. Int. Ed. 2013, 52, 10112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10299. [Google Scholar]
- 25. Yuan S., Wu R., Latek D., Trzaskowski B., Filipek S., PLoS Comput. Biol. 2013, 9, e1003261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan S., Ghoshdastider U., Latek D., Trzaskowski B., Debinski A., Filipek S., PLoS One 2012, 7, e47114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolf M. G., Hoefling M., Aponte-Santamaria C., Grubmuller H., Groenhof G., J. Comput. Chem. 2010, 31, 2169. [DOI] [PubMed] [Google Scholar]
- 28. Lomize A. L., Pogozheva I. D., Mosberg H. I., J. Chem. Inf. Model. 2011, 51, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Pastor R. W., J. Phys. Chem. B 2010, 114, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vanommeslaeghe K., Raman E. P., A. D. MacKerell, Jr. , J. Chem. Inf. Model. 2012, 52, 3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.M. J. Frisch, et al., Gaussian, Inc., Wallingford, CT, USA, 2009.
- 32. Kräutler V., Van Gunsteren W. F., Hunenberger P. H., J. Comput. Chem. 2001, 22, 501. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary