

Abstract
Gastrointestinal stromal tumors (GISTs) have been recognized as a biologically distinctive type of tumor, different from smooth muscle and neural tumors of the gastrointestinal tract. The identification of genetic aberrations in proto-oncogenes that drive the growth of GISTs is critical for improving the efficacy of cancer therapy by matching targeted drugs to specific mutations. Research into the oncogenic mechanisms of GISTs has found that these tumors frequently contain activating gene mutations in either platelet-derived growth factor receptor A (PDGFRA) or a receptor tyrosine protein associated with a mast cell growth factor receptor encoded by the KIT gene. Mutant cancer subpopulations have the potential to disrupt durable patient responses to molecularly targeted therapy for GISTs, yet the prevalence and size of subpopulations remain largely unexplored. Detection of the cancer subpopulations that harbor low-frequency mutant alleles of target proto-oncogenes through the use of molecular genetic methods, such as polymerase chain reaction (PCR) target amplification technology, is hampered by the high abundance of wild-type alleles, which limit the sensitivity of detection of these minor mutant alleles. This is especially true in the case of mutant tumor DNA derived “driver” and “drug-resistant” alleles that are present in the circulating cell-free tumor DNA (cfDNA) in the peripheral blood circulation of GIST patients. So-called “liquid biopsy” allows for the dynamic monitoring of the patients’ tumor status during treatment using minimally invasive sampling. New methodologies, such as a technology that employs a xenonucleic acid (XNA) clamping probe to block the PCR amplification of wild-type templates, have allowed improved molecular detection of these low-frequency alleles both in tissue biopsy samples and in cfDNA. These new methodologies could be widely applied for minimally invasive molecular testing in the therapeutic management of GISTs.
Keywords: Gastrointestinal stromal tumors, Liquid biopsy, Mutations, Targeted therapy
Background
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract, accounting for approximately 20% of all sarcomas [1, 2]. Sarcomas are a rare type of cancer that can occur in the bones, muscles, nerves, blood vessels, connective tissue, fat, and cartilage. The neoplastic GIST cell appears to arise from a common precursor cell, which gives rise to the interstitial cells of Cajal in the normal myenteric plexus [3]. GISTs are characterized by mutations in a receptor tyrosine protein (encoded by the KIT gene) associated with a mast cell growth factor receptor or in the gene encoding platelet-derived growth factor receptor A (PDGFRA). These genetic aberrations lead to constitutive activation of these growth factor receptors and concomitant abnormal cellular proliferation, which leads to the development of GIST.
Early detection of these genetic alterations is important for diagnosis and therapy and for monitoring the progression of GIST. This article provides an overview of the current status of targeted molecular therapies for GISTs and the current state-of-the-art of high-sensitivity molecular genetic tests that are able to detect low-frequency tumor-derived mutations in cancer patients employing minimally invasive sampling, i.e., “liquid biopsy”.
Kinase mutations in GISTs
The biology of GISTs has been widely investigated at the genomic level. Mutations in KIT or the receptor tyrosine kinase PDGFRA are the hallmarks of molecular diagnosis of GIST. KIT and PDGFRA are mutated in approximately 85% and 5%, respectively, of GISTs. Mutations are also rarely (<1%) found in the serine-threonine protein kinase BRAF (Fig. 1) [4].
Fig. 1.
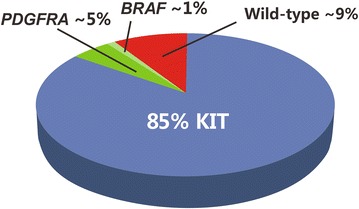
Molecular subsets of gastrointestinal stromal tumors (GISTs). Gene that encodes a receptor tyrosine protein associated with a mast cell growth factor receptor (KIT); platelet-derived growth factor receptor A (PDGFRA); gene that encodes an enzyme belonging to the RAF family of serine-threonine protein kinases participating in the RAS-RAF-ERK pathway (BRAF); wild-type (WT) normal gene sequence that does not contain any somatic mutations
Approximately 80% of GISTs have an oncogenic mutation in the tyrosine kinase KIT [5–7]. Most of these mutations affect the juxtamembrane domain encoded by KIT exon 11, allowing spontaneous (ligand-independent) receptor dimerization and kinase activation. However, mutations also occur in exons 9, 13, and 17, and these mutations may support constitutive KIT signaling through other mechanisms. A subset (5%–7%) of GISTs have an activating mutation in the KIT-homologous tyrosine kinase PDGFRA [8, 9]. Many of these PDGFRA-mutant GISTs have an epithelioid morphology and express little or no KIT; however, such features are not unique to these tumors, and mutation status can be determined only through molecular analysis. Approximately 10%–15% of GISTs are negative for KIT and PDGFRA gene mutations; these tumors are often referred to as wild-type GISTs (Fig. 2) [10, 11].
Fig. 2.
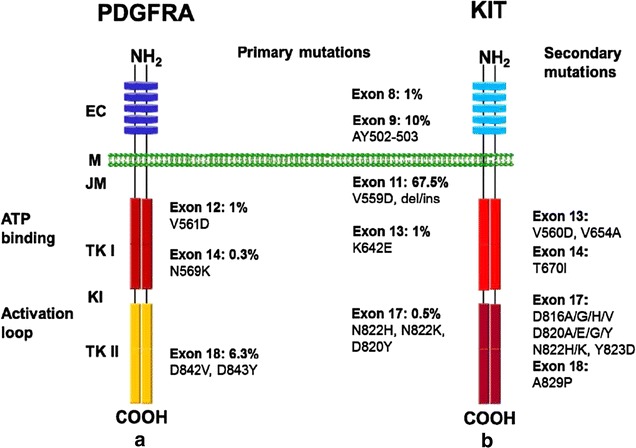
Schematic structures of PDGFRA and KIT. a Primary oncogenic mutation in PDGFRA. b Distribution of primary and secondary mutations in KIT. EC extracellular domain, M membrane, JM juxtamembrane domain, TK I tyrosine kinase domain I, KI kinase insert, TK II tyrosine kinase domain II
Mutant and wild-type GISTs show marked differences in imatinib response. The response rates were approximately 80% in GIST patients with KIT exon 11 mutants, 40% in those with KIT exon 9 mutants, and 14% in wild-type GIST patients [12]. GIST patients with PDGFRA mutations showed mild sensitivity to imatinib (66%), with the exception of those with the exon 18 point mutation D842V, who were totally resistant to imatinib [12]. In addition to the importance of GIST mutational status in predicting imatinib sensitivity, as described above, the acquisition of secondary mutations in either KIT or PDGFRA represents the most frequent mechanism of imatinib resistance in GISTs. Advances in whole genome analysis technology has shown that GISTs with wild-type KIT/PDGFRA should be considered more appropriately as a heterogeneous family of distinct disease entities, with different biological and clinical features [13].
BRAF belongs to the RAF family of serine-threonine protein kinases. These kinases participate in the RAS-RAF-ERK pathway, which regulates cell cycle through activation of the mitogen-activated protein kinase (MAPK) pathway. BRAF mutations have been detected in GIST patients with wild-type KIT/PDGFRA. In a recent study, BRAF exon 15 V600E mutation was detected in 13% (9 of 70) of patients with wild-type KIT/PDGFRA GISTs [14]. In naïve GIST patients carrying activating mutations in KIT or PDGFRA, a concomitant activating mutation in BRAF gene (in approximately 2% of the patients) was found to be consistent with resistance [15]. In this study, in vitro experiments showed that imatinib was able to switch off the mutated activated receptor KIT but not the downstream signaling triggered by the BRAF mutation [15].
The most common primary mutations in KIT affect the juxtamembrane domain encoded by exon 11. Two-thirds of GISTs harbor KIT mutations in exon 11, which disrupt the normal juxtamembrane secondary structure that prevents the kinase activation loop from swinging into active conformation [16]. These mutations include in-frame deletions, insertions, and substitutions, as well as combinations of these. The deletions are associated with shorter progression-free and overall survival in comparison to other exon 11 mutations. In particular, deletions involving codon 557 and/or codon 558 are associated with malignant behavior [17].
Aside from exon 11 mutations, 7%–10% of GISTs have a KIT mutation in an extracellular domain that is encoded by exon 9. These mutations are thought to mimic the conformational change that the KIT receptor undergoes when stem cell factor (SCF) is bound. Importantly, the kinase domain in exon 9-mutant KIT is essentially the same as in wild-type KIT and has an effect on inhibitor sensitivity. Also important is that these mutations occur in tumors that arise in the intestines but are rarely seen in the stomach. KIT mutations in the activation loop (which is encoded by exon 17) of the kinase are uncommon and stabilize the active conformation. KIT mutations in the ATP-binding region encoded by exon 13 (such as K642E) are also uncommon. Secondary mutations are concentrated in two regions of the KIT kinase domain. One target is the ATP-binding pocket, encoded by exons 13 and 14, the part of the protein that directly interferes with drug binding. The other target is the activation loop, where mutations can stabilize KIT in the active conformation and thereby hinder drug interaction. By contrast, the secondary ATP-binding pocket mutations do not cause intrinsic kinase activation.
Crystal structure analysis of KIT has shown that in the absence of the ligand, the juxtamembrane domain folds back into the active site of the kinase. Disruption of the juxtamembrane domain by mutation is believed to lead to activation of the kinase by removal of this auto-inhibition. The prognostic significance of mutations in the KIT and PDGFRA genes has been examined in GISTs in the pre-imatinib era, and tumors with a KIT exon 11 mutation are associated with a worse outcome than tumors with other KIT or PDGFRA mutant isoforms or with no detectable mutation [18–22]. Conversely, KIT exon 11 mutations have been found in mitotically inactive GISTs of 1 cm or smaller, suggesting that oncogenic KIT activity contributes to early tumor growth [23, 24]. GISTs with a KIT exon 9 mutation arise predominantly in the small intestine and colon and appear to be clinically more aggressive than tumors with KIT exon 11 mutations [25, 26]. In contrast, tumors with PDGFRA mutations are less aggressive than those with KIT mutations [27, 28].
Current targeted therapies for GISTs
The golden standard GIST therapy is the tyrosine kinase inhibitor (TKI) imatinib, which offers a good and stable response for approximately 18–36 months; however, development of resistance is very common. The detection of somatic mutations from cancer genome sequences is the key to understanding the genetic basis of disease progression, patient survival, therapy response, and toxicity. The DNA of the tumor serves as the primary source of information, which over the years has led to a more detailed characterization of the biological profile of GIST.
For GISTs, the main objective of treatment is complete resection. As discussed above, TKIs such as imatinib or sunitinib (Fig. 3) are used for neoadjuvant, adjuvant, or palliative treatment. Sunitinib malate is an oxindole molecule designed to interact selectively with the intracellular ATP-binding sites of tyrosine kinase vascular endothelial growth factor receptors 1–3 (VEGFR1–3), platelet-derived growth factor receptors (PDGFRs), stem cell growth factor receptor (KIT), fms-related tyrosine kinase 3 (FLT3), and colony-stimulating factor 1 receptor (CSF1R).
Fig. 3.
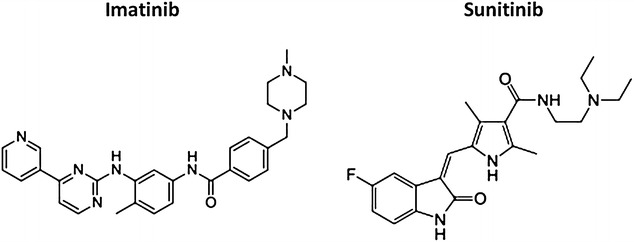
Chemical structures of the tyrosine kinase inhibitors (TKIs) imatinib [Gleevec, approved by the Food and Drug Administration (FDA) of the United States in 2002 for the treatment of advanced GIST patients and marketed by Novartis] and sunitinib (Sutent, approved by the FDA in 2006 for the treatment of patients with imatinib-resistant GIST and marketed by Pfizer)
Receptor inhibition has multiple effects on cellular processes, including tumor cell survival, endothelial cell growth and migration, vascular permeability, pericyte recruitment, and lymphangiogenesis. The final antitumor effects may be classified as follows: direct cytotoxic effects on tumor cells by induction of cell death, anti-angiogenic effects leading to growth delay and/or tumor regression by cytostatic inhibition of new blood-vessel formation, vascular disruption by inhibition of existing VEGF/VEGFR-dependent tumor blood vessels leading to central tumor cell necrosis, and cavitation that may or may not be associated with tumor regression. Responses to imatinib can be achieved; however, treatment is not curative unless complete resection is possible. In advanced GISTs, a partial remission can be attained in 50% of patients treated with imatinib, and there are single cases of complete responses. However, most patients receiving imatinib experience disease progression [29–34]. Patients receiving imatinib who progress only transiently respond to sunitinib [6, 35, 36].
For patients with GISTs, the response to therapy is evaluated by diagnostic imaging, which displays limited sensitivity and specificity. Computed tomography (CT) constitutes the golden standard of imaging for GIST [37, 38]. However, sensitivity of CT in detecting GISTs is insufficient. Currently, there is no biomarker available for detecting success or failure of therapy in GISTs. Activating mutations in KIT or PDGFRA can be found in at least 85% of all GISTs and constitute the transforming event in the pathogenesis of GIST tumors. In addition, mutated genomic DNA fragments are highly specific for the tumor. High sensitivity methods for the detection of these mutations are needed. Currently, there is limited clinical sensitivity of Sanger dideoxy sequencing; at least ~20% of the DNA derived from the tumors needs to be detectable mutants, meaning that GISTs harboring mutations below this threshold will go undetected.
Mutation detection of circulating cell-free tumor DNA (cfDNA) for diagnosis and cfDNA-targeted therapy for cancer
Circulating cfDNA can be detected in healthy individuals but is increased in patients with tumors [39–43]. Proposed models for tumor DNA release into the blood include DNA release by tumor cells that undergo apoptosis or necrosis and extravasation of tumor cells into the blood, where cells undergo lysis.
It has been shown that the levels of long interspersed nucleotide elements 1 (LINE1) DNA in plasma samples associate with tumor progression in different cancer entities [44–47]; however, LINE1 sequences are not tumor-specific. It has also been shown that growth factor mutations can be recovered in cfDNA in serum samples of patients with non-small cell lung cancer [48], and detection of circulating mutant tumor suppressors adenomatous polyposis coli (APC) and p53, or KRAS DNA was shown to associate with progression-free survival in patients with colorectal cancer [49].
cfDNA in GISTs
Understanding the genetic landscape of GISTs is important for selection of the most appropriate medical treatment. A sensitive, reliable biomarker for detection and quantitation of disease activity would be a highly valuable tool for the management of GIST. Traditionally, DNA extracted from formalin-fixed, paraffin-embedded specimens has been used for genetic analysis. However, a recent study showed that tumor-specific mutations in KIT or PDGFRA can be detected and quantified in circulating cfDNA in plasma samples from patients with GIST. This technique is known as “liquid biopsy” [50]. A target amplification methodology known as quantitative ligation polymerase chain reaction (PCR) was used, and the cfDNA levels were associated with the clinical course of the GIST lesions as measured by diagnostic imaging. Thus, tumor-specific cfDNA in plasma can be used as a highly specific biomarker in patients with GIST and can also be used to predict treatment responses and relapse, allowing for earlier changes in treatment.
A sponsored clinical trial is currently being conducted in Italy (ClinicalTrials.gov identifier: NCT0244398; https://clinicaltrials.gov/ct2/show/NCT02443948) to establish an association between the change in cfDNA and disease state of GISTs. Another recent study using next-generation sequencing (NGS) successfully detected secondary KIT mutations in circulating cfDNA from the peripheral blood of GIST patients undergoing imatinib therapy [51].
QClamp™ mutation assays using cfDNA
For the high-sensitivity detection of low-frequency mutations in tumor-derived cfDNA, a technology has been developed that uses a polymerase chain reaction xenonucleic acid clamping approach (XNA-PCR; QClamp™). This novel technology has been developed for the detection of low-frequency mutations in tumor-derived DNA [52] and the early detection of mutations in cfDNA from cancer patients [53] and from stool-derived DNA in colorectal cancer patients [54]. This technology allows for the selective primer-mediated DNA polymerase-based amplification of only mutant template in tumor-derived DNA because the wild-type templates are blocked by the highly specific XNA probes used during PCR (Fig. 4).
Fig. 4.
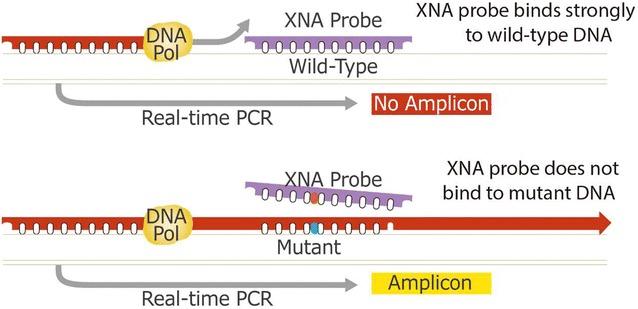
QClamp™ xenonucleic acid (XNA) probe hybridizes to wild-type DNA (WT-DNA) templates and prevents WT-DNA from being amplified, while melting off from the mutant DNA template so that only mutant DNA is amplified
Mutations in growth factor receptor pathways can guide a physician in choosing the appropriate precision therapy for a particular cancer. QClamp™ can detect mutations in tumor-derived cfDNA from plasma samples. The QClamp™ method is highly sensitive and can detect mutations at a level below 0.1% (1 mutant copy in 1000 wild-type copies). For example, KIT mutations in exon 17, particularly at codon 816, lead to activation of the kinase in GISTs. Figure 5 shows the real-time PCR profile for the high-sensitivity detection of this mutation at <0.1%.
Fig. 5.
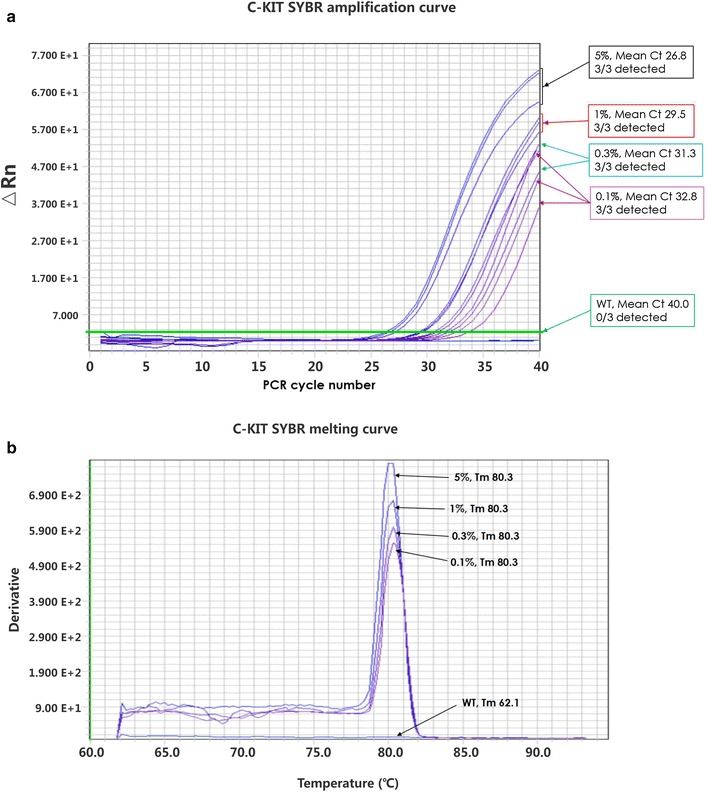
QClamp™ assay for KIT exon 17 D816V/Y/H mutation detection performed with template DNA containing D816V mutations at the allelic frequencies shown. a Real-time polymerase chain reaction (PCR) profiles KIT D816V amplification plots. b Melting curves for PCR amplicons clearly demonstrate detection sensitivity of >99.9% (i.e., 1 mutant allele in 1000 wild-type alleles)
The QClamp™ XNA-PCR technology has also been used to detect low-frequency mutations in breast cancer patient samples [55] and fine needle aspiration (FNA) biopsy samples [56], which further demonstrates the practical clinical use of the technology for detection of low-frequency mutant tumor alleles in tumor biopsy samples.
Conclusions
The prognosis for GIST patients has changed enormously over the past few decades. In particular, imatinib has radically changed life expectancy of patients with GIST, a group of patients previously considered to be largely untreatable. For those with disease refractory to imatinib, as well as the majority who develop resistance to imatinib, other TKIs, such as sunitinib and regorafenib, are available. As more drug options become available, mutation testing by pathologists will become more common to select the best drugs for treatment depending on the characteristics of the tumor, whether the treatment is for primary tumors or not, and whether there are metastases that are present at diagnosis or that develop during treatment.
The high sensitivity of QClamp™ technology and its ability to be performed using widely available real-time PCR instrumentation platforms such as ABI 7500, Roche LC480, and Rotor-Gene Q make it highly attractive for clinical applications where a rapid “sample-to-answer” result is needed for precise medical applications. It is expected that the QClamp™ technology will find wide application in the minimally invasive detection of tumor-specific mutations to aid in the early diagnosis of GISTs and the development of targeted therapies.
Authors’ contributions
MJP drafted the manuscript, genetic analysis was performed under supervision of MJP, AZ and WY revised the draft. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Weixin Yan, Email: xiaogu4524@sjtu.edu.cn.
Aiguo Zhang, Email: azhang@diacarta.com.
Michael J. Powell, Email: mpowell@diacarta.com
References
- 1.Zhang W. TCGA divides gastric cancers into four molecular subtypes: implications for individualized therapeutics. Chin J Cancer. 2014;33:469–470. doi: 10.5732/cjc.014.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Zeng H, Zhang S. The updated incidences and mortalities of major cancers in China, 2011. Chin J Cancer. 2015;34:53. doi: 10.1186/s40880-015-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol. 2000;15:1293–1301. doi: 10.14670/HH-15.1293. [DOI] [PubMed] [Google Scholar]
- 4.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain of function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 5.Maki R, Keedy V. Molecular profiling of gastrointestinal stromal tumor (GIST). My Cancer Genome. 2015. https://www.mycancergenome.org/content/disease/gist/. Accessed 16 Mar 2016.
- 6.Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- 7.Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130:1466–1478. doi: 10.5858/2006-130-1466-GSTROM. [DOI] [PubMed] [Google Scholar]
- 8.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 9.Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/S0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 10.Liegl B, Kepten I, Le C, Zhu M, Demetri G, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA. 2009;106:1542–1747. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joensuu H. Gastrointestinal stromal tumor (GIST) Ann Oncol. 2006;17(Supplement 10):x280–x286. doi: 10.1093/annonc/mdl274. [DOI] [PubMed] [Google Scholar]
- 13.Nannini M, Biasco G, Astolfi A, Pantaleo MA. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumors (GIST) J Med Genet. 2013;50:653–661. doi: 10.1136/jmedgenet-2013-101695. [DOI] [PubMed] [Google Scholar]
- 14.Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foester A, Hartmann A, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal tumors. J Clin Pathol. 2009;62:613–616. doi: 10.1136/jcp.2009.064550. [DOI] [PubMed] [Google Scholar]
- 15.Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:1769–1776. doi: 10.1158/1078-0432.CCR-11-2230. [DOI] [PubMed] [Google Scholar]
- 16.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 17.Wardelmann E, Losen I, Hans V, Neidt I, Speidel N, Bierhoff E, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer. 2003;106:887–895. doi: 10.1002/ijc.11323. [DOI] [PubMed] [Google Scholar]
- 18.Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008;21:476–484. doi: 10.1038/modpathol.2008.2. [DOI] [PubMed] [Google Scholar]
- 19.Singer S, Rubin BP, Lux ML, Chen CJ, Demetri GD, Fletcher CDM, et al. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol. 2002;20:3898–3905. doi: 10.1200/JCO.2002.03.095. [DOI] [PubMed] [Google Scholar]
- 20.Kim TW, Lee H, Kang YK, Choe MS, Ryn MH, Chang HM, et al. Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer Res. 2004;10:3076–3081. doi: 10.1158/1078-0432.CCR-03-0581. [DOI] [PubMed] [Google Scholar]
- 21.Andersson J, Bumming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology. 2006;130:1573–1581. doi: 10.1053/j.gastro.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 22.Martin J, Poveda A, Llombart-Bosch A, Ramos R, Lopez-Guerrero JA, Garcia del Muro J, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS) J Clin Oncol. 2005;23:6190–6198. doi: 10.1200/JCO.2005.19.554. [DOI] [PubMed] [Google Scholar]
- 23.Agaimy A, Wunsch PH, Hofstaedter F, Blaszyk H, Rummele P, Gaumann A, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113–120. doi: 10.1097/01.pas.0000213307.05811.f0. [DOI] [PubMed] [Google Scholar]
- 24.Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567–1572. doi: 10.1016/S0002-9440(10)61103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonescu CR, Sommer G, Sarran L, Tschernyavsky SJ, Riedel E, Woodruff JM, et al. Association of KIT exon 9 mutations with non-gastric primary site and aggressive behaviour: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–3337. [PubMed] [Google Scholar]
- 26.Lasota J, Kopczynski J, Sarlomo-Rikala M, Schneider-Stock R, Stachura T, Kordek R, et al. KIT 1530ins6 mutation defines a subset of predominantly malignant gastrointestinal stromal tumors of intestinal origin. Hum Pathol. 2003;34:1306–1312. doi: 10.1016/S0046-8177(03)00407-6. [DOI] [PubMed] [Google Scholar]
- 27.Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Investig. 2004;84:874–883. doi: 10.1038/labinvest.3700122. [DOI] [PubMed] [Google Scholar]
- 28.Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Investig. 2006;86:94–100. doi: 10.1038/labinvest.3700360. [DOI] [PubMed] [Google Scholar]
- 29.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 30.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 31.Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 32.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 33.Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–632. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 35.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 36.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 37.Hong X, Choi H, Loyer EM, Benjamin RS, Trent JC, Charnsangavej C. Gastrointestinal stromal tumor: role of CT in diagnosis and in response evaluation and surveillance after treatment with imatinib. Radiographics. 2006;26:481–495. doi: 10.1148/rg.262055097. [DOI] [PubMed] [Google Scholar]
- 38.Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–1759. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983;51:2116–2120. doi: 10.1002/1097-0142(19830601)51:11<2116::AID-CNCR2820511127>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 40.Anker P, Stroun M. Circulating DNA in plasma or serum. Medicina. 2000;60:699–702. [PubMed] [Google Scholar]
- 41.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210–219. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 42.Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 2003;21:3902–3908. doi: 10.1200/JCO.2003.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Gormally E, Caboux E, Vineis P, Hainaut P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat Res. 2007;635:105–117. doi: 10.1016/j.mrrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 44.Sunami E, Vu AT, Nguyen SL, Giuliano AE, Hoon DS. Quantification of LINE1 in circulating DNA as a molecular biomarker of breast cancer. Ann N Y Acad Sci. 2008;1137:171–174. doi: 10.1196/annals.1448.011. [DOI] [PubMed] [Google Scholar]
- 45.Wang BG, Huang HY, Chen YC, Bristow RE, Kassauei K, Cheng CC, et al. Increased plasma DNA integrity in cancer patients. Cancer Res. 2003;63:3966–3968. [PubMed] [Google Scholar]
- 46.Anker P, Mulcahy H, Chen XQ, Stroun M. Detection of circulating tumor DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev. 1999;18:65–73. doi: 10.1023/A:1006260319913. [DOI] [PubMed] [Google Scholar]
- 47.Tomita H, Ichikawa D, Ikoma D, Sai S, Tani N, Ikoma H, et al. Quantification of circulating plasma DNA fragments as tumor markers in patients with esophageal cancer. Anticancer Res. 2007;27:2737–2741. [PubMed] [Google Scholar]
- 48.Kimura H, Kasahara K, Kawaishi M, Kunitoh H, Tamura T, Holloway B, et al. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small cell lung cancer. Clin Cancer Res. 2006;12:3915–3921. doi: 10.1158/1078-0432.CCR-05-2324. [DOI] [PubMed] [Google Scholar]
- 49.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of mutant tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations in CKIT or PDGFRA. Clin Cancer Res. 2013;19(17):4854–4867. doi: 10.1158/1078-0432.CCR-13-0765. [DOI] [PubMed] [Google Scholar]
- 51.Wada N, Kurokawa Y, Takahashi T, Hamakawa T, Hirota S, Naka T, et al. Detecting secondary c-KIT mutations in the peripheral blood of patients with imatinib-resistant gastrointestinal stromal tumor. Oncology. 2016;90:112–117. doi: 10.1159/000442948. [DOI] [PubMed] [Google Scholar]
- 52.Powell MJ, Ganta M, Peletskaya E, Pastor L, Raymundo M, Wu G et al. High sensitivity detection of tumor gene mutations. BAOJ Cancer Res Ther. 2015;1:001. Weblink. http://bioaccent.org/cancer-sciences/cancer-sciences1.php.
- 53.Costa C, Molina MA, Drozdowsky A, Gimenez-Capitan A, Bertran-Alamillo J, Karachaliou N, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC Trial. Clin Cancer Res. 2014;20(7):1–10. doi: 10.1158/1078-0432.CCR-13-2233. [DOI] [PubMed] [Google Scholar]
- 54.Gerecke C, Mascher C, Gottschalk U, Kleuser B, Scholtka B. Ultrasensitive detection of unknown colon cancer-initiating mutations using the example of the adenomatous Polyposiscoli gene. Cancer Prev Res. 2013;6:898–907. doi: 10.1158/1940-6207.CAPR-13-0145. [DOI] [PubMed] [Google Scholar]
- 55.Myers MB, Banda M, McKim KL, Wang Y, Powell MJ, Parson BL. Breast Cancer Heterogeneity examined by high-sensitivity quantitation of PIK3CA, KRAS, HRAS, and BRAF mutations in normal breast and ductal carcinomas. Neoplasia. 2016;16(4):253–263. doi: 10.1016/j.neo.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oktay MH, Adler E, Hakima L, Grunblatt E, Pieri E, Seymour A, et al. The application of molecular diagnostics to stained cytology smears. J. Mol. Diag. 2016;18(3):407–415. doi: 10.1016/j.jmoldx.2016.01.007. [DOI] [PubMed] [Google Scholar]