

Cells must grow to reach a critical size before cell division. In 1977 Fantes and Nurse established that this critical size depends on the nutritional environment.1 This seminal work was carried out in the fission yeast Schizosaccharomyces pombe, but further work revealed that the environmental control of cell size at division was also conserved in mammalian cells.2 S. pombe is an ideal model organism for the analysis of cell size. Fission yeast cells are rod-shaped, grow by tip elongation, and their cell length is proportional to their volume. Nutrients in the media determine cell length: cells growing in rich medium are larger than those growing in poor medium, and a rapid re-adjustment of the cell size is produced when fission yeast cells are shifted from a nitrogen-rich to a nitrogen-poor medium or vice versa. However, the molecular mechanisms connecting the nutritional control of the cell size with the cell cycle machinery are poorly understood.
TORC1 (target of rapamycin 1), a central controller of cell growth, is highly conserved from yeast to mammals.3 In media with nutrients, TORC1 is active and promotes growth by inducing ribosome biogenesis and protein synthesis. In addition, TORC1 signalling inhibits catabolic processes such as autophagy. When nutrients are scarce, the TORC1 pathway is inhibited, and cells stop growing as a result of the inactivation of the anabolic processes induced by TORC1, while autophagy is active to allow survival under starvation conditions. Concomitant with the inactivation of TORC1, cells divide at a reduced size.2 Taking this observation into account, we wondered if there was a connection between the master regulator of cell growth, TORC1, and the basic cell cycle machinery to regulate cell size at division. Recent experiments in fission yeast suggest this may be the case.4
Phosphorylation of proteins by mitotic kinases such as Cdk1-CyclinB is essential for the G2/M transition and mitosis to proceed properly. But for the accurate progression of the cell cycle, the regulation of counteracting phosphatases is as essential as the regulation of kinases. As Cdk1 activity is reversed by PP2A·B55, this phosphatase must be inhibited at mitosis to achieve a complete and timely phosphorylation of Cdk1 substrates. Among these substrates there are two Cdk1 regulators, a positive one, the Cdc25 phosphatase, and a negative one, the Wee1 kinase. The phosphorylation of these regulators by Cdk1, that leads to the activation and inhibition of Cdc25 and Wee1, respectively, contributes to the amplification of the Cdk1 signal. In recent years, work from several laboratories using different model systems has shown that the inhibition of PP2A·B55 is carried out by the endosulfines, ENSA and Arpp19.5,6 These small proteins are phosphorylated by the Greatwall kinase, which is also activated by Cdk1, and become potent inhibitors of PP2A·B55 at mitotic entry and during mitosis. Accordingly, depletion of Greatwall induces defects of entry into and progression through mitosis in different model systems.
In S. pombe, the igo1+ gene encodes for the only endosulfine present in the fission yeast genome, and Ppk18 and Cek1 kinases are orthologous to Greatwall, although Ppk18 seems to carry out most of Igo1 phosphorylation.4 However, under standard laboratory conditions, that is, in media with plenty of nutrients, cells deleted for either of these genes do not show apparent problems to enter into and proceed through mitosis. Surprisingly, when cells are transferred into media with a poor-nitrogen source, such as phenylalanine, they are unable to accelerate mitosis and show a delay in G2, in contrast to wild-type cells, which reduce cell size to adapt to starvation conditions.4 This result suggests that in fission yeast the Greatwall-Endosulfine pathway is conserved and promotes the timely entry into mitosis, but its function is only required when cells are grown in media with low nutrients (Fig. 1). In this poor medium inhibition of the PP2A·B55 complex is required for the complete phosphorylation of mitotic substrates to occur at an earlier point in G2, thus allowing cells to enter mitosis with a smaller cell size.4 In fission yeast a shorter G2 phase makes the extension of the G1 phase necessary, which is cryptic in rich medium, for the cells to grow and reach the minimal size required to enter into a new cell cycle. This modification of the cell cycle phases is essential for survival under starvation conditions, because cells from G1 can exit the cell cycle to enter the G0 phase or, in the case of yeasts of opposite mating types, to conjugate and form spores, which will allow survival during starvation.
Figure 1.
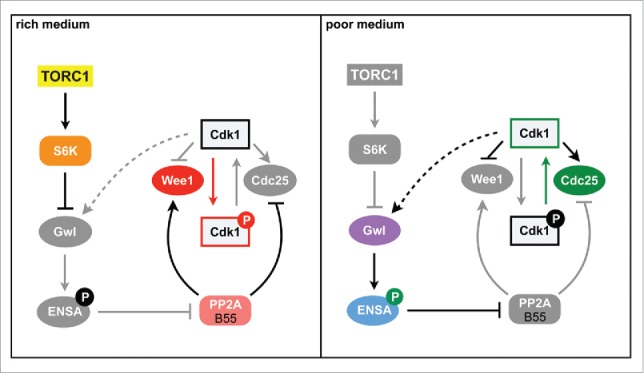
Nutritional control of cell size in fission yeast. In rich medium, TORC1/S6K phosphorylate and inhibit Greatwall/Ppk18. In these conditions, PP2A activity is high, and cells grow in G2 to a larger size before entering mitosis. In poor medium, PP2A activity is reduced because it is inhibited by the Greatwall-Endosulfine pathway, Cdk1 is activated prematurely, resulting in cells with a small size. Gwl: Greatwall.
These results suggest that in fission yeast the Greatwall-Endosulfine pathway is inhibited in rich medium. Interestingly, before the function of Greatwall-Endosulfines as inhibitors of PP2A·B55 was uncovered, the budding yeast Greatwall ortholog, Rim15, had been shown to play a central role in the entry into and in survival during stationary phase, and to be negatively regulated by TORC1, PKA and Pho80-Pho85 pathways.7 Similarly, our results suggest that fission yeast Ppk18 is negatively regulated in rich medium by phosphorylation by the S6-kinases acting downstream of TORC1, and by PKA, providing a mechanism to link the cell size to the nutritional environment through the regulation of the Greatwall-Endosulfine-PP2A pathway (Fig. 1). Future studies will determine if the regulation of Greatwall by TORC1 is conserved in other eukaryotes, and how it coexists with the regulation by Cdk1. In addition, studies in budding yeast open up the possibility that Greatwall and Endosulfines also play a role in the survival of G0 cells in other organisms.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Fantes P, Nurse P. Exp Cell Res 1977; 107:377-86; PMID:872891; http://dx.doi.org/ 10.1016/0014-4827(77)90359-7 [DOI] [PubMed] [Google Scholar]
- [2].Fingar DC, et al.. Genes Dev 2002; 12:1472-8; PMID:12080086; http://dx.doi.org/ 10.1101/gad.995802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Loewith R, Hall MN. Genetics 2011; 189:1177-1201; PMID:22174183; http://dx.doi.org/ 10.1534/genetics.111.133363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chica N, et al.. Curr Biol 2016; 26:319-30; PMID:26776736; http://dx.doi.org/ 10.1016/j.cub.2015.12.035 [DOI] [PubMed] [Google Scholar]
- [5].Mochida S, et al.. Science 2010; 330:1670-3; PMID:21164013; http://dx.doi.org/ 10.1126/science.1195689 [DOI] [PubMed] [Google Scholar]
- [6].Gharbi-Ayachi A, et al.. Science 2010; 330:1673-7; PMID:21164014; http://dx.doi.org/ 10.1126/science.1197048 [DOI] [PubMed] [Google Scholar]
- [7].Pedruzzi I, et al.. Mol Cell 2003; 12:1607-13; PMID:14690612; http://dx.doi.org/ 10.1016/S1097-2765(03)00485-4 [DOI] [PubMed] [Google Scholar]