

Abstract
The initiation and progression of human cancer is frequently linked to the uncontrolled activation of survival kinases. Two such pro-survival kinases that are commonly amplified in cancer are PIM and Akt. These oncogenic proteins are serine/threonine kinases that regulate tumorigenesis by phosphorylating substrates that control the cell cycle, cellular metabolism, proliferation, and survival. Growing evidence suggests that cross-talk exists between the PIM and Akt kinases, indicating that they control partially overlapping survival signaling pathways that are critical to the initiation, progression, and metastatic spread of many types of cancer. The PI3K/Akt signaling pathway is activated in many human tumors, and it is well established as a promising anticancer target. Likewise, based on the role of PIM kinases in normal and tumor tissues, it is clear that this family of kinases represents an interesting target for anticancer therapy. Pharmacological inhibition of PIM has the potential to significantly influence the efficacy of standard and targeted therapies. This review focuses on the regulation of PIM kinases, their role in tumorigenesis, and the biological impact of their interaction with the Akt signaling pathway on the efficacy of cancer therapy.
Keywords: PIM kinases, Akt, Oncogenes, Cancer, Therapeutic resistance
1. Introduction
Solid and hematopoietic cancers utilize intercellular signaling cascades mediated by oncogenic kinases to maintain tumor cell growth and survival. In normal cells, the activity of these kinases is tightly controlled, whereas their sustained activation promotes apoptotic resistance and uncontrolled proliferation. PIM and Akt are serine/threonine kinases that are frequently activated in human cancers, and they phosphorylate overlapping substrates to activate common pathways that control various physiological processes that ultimately dictate the balance between cell survival and apoptosis. As a result, these kinases represent promising targets for cancer therapy and they are the focus of intense drug development efforts. Understanding how these kinases control distinct and overlapping signal transduction pathways is important for designing rational drug combinations and improving the efficacy of PIM and Akt inhibitors in the clinic.
The Proviral Integration site for Moloney murine leukemia virus (PIM) kinases were initially discovered through a viral-insertion screen for genes that enhance the development of lymphoma in the Eu-myctumor model. The PIM family of serine/threonine kinases consists of three isoforms (PIM1, PIM2, and PIM3) that are highly conserved throughout evolution (Nawijn et al., 2011). Separate genes located on different human chromosomes encode the PIM isoforms, and they share high sequence homology at the amino acid level; PIM1 and PIM2 are 61% identical and PIM1 and PIM3 are 71% identical (Baytel et al., 1998). Multiple reports have demonstrated functional redundancy between PIM kinase isoforms both in vitro and in vivo (Mikkers et al., 2004; Bullock et al., 2005; Narlik-Grassow et al., 2012). Alternative translation initiation sites have been described for PIM1 and PIM2, giving rise to isoforms with different molecular weights. The PIM1 kinase encodes 33 and 44 kDa isoforms that display comparable kinase activity (Saris et al., 1991). However, the longer isoform of PIM1 was demonstrated to bind to the SH3 domain of the ETK/BMX tyrosine kinase, localizing it to the plasma membrane, whereas the shorter isoform is primarily localized in the cytosol and nucleus (Xie et al., 2006). PIM2 exists as three isoforms (34, 37, and 40 kDa), but no differences in protein function or interaction have been described. PIM3 has only been described as a single isoform (Nawijn et al., 2011). The PIM kinase isoforms are ubiquitously expressed, although some tissue specificity has been described: PIM1 is highly expressed in hematopoietic cells, gastric, head and neck, and prostate tumors (Bachmann et al., 2006; Cibull et al., 2006); PIM2 is highly expressed in lymphoid and brain tissues (Cohen et al., 2004; Mikkers et al., 2004); and PIM3 is highly expressed in breast, kidney and brain tissues (Feldman et al., 1998). In contrast to a majority of serine/threonine kinases, including Akt, the activity of PIM kinases is not regulated by cellular localization or post-translational modification. X-ray crystallography structural analyses of PIM1 revealed that this protein contains a conserved catalytic domain but lacks a regulatory domain (Qian et al., 2005). Therefore, it is thought that PIM kinases are constitutively active when expressed in cells, and their activity is directly correlated with their expression level.
The PKB/Akt family of serine/threonine protein kinases play a central role in signaling downstream of phosphatidylinositol 3-kinase (PI3K) (Cantley, 2002). The Akt gene was originally discovered as a transforming retrovirus isolated from the AKR mouse strain, which spontaneously developed many types of cancer. Subsequently, the oncogene encoding this virus was discovered and termed v-Akt. Thus, the later identified human analogues were named accordingly (Staal, 1987). Akt is a member of the AGC kinase family, which mediate a wide array of important cellular functions, and whose dysregulation is strongly associated with the pathogenesis of many human diseases, most notably cancer (Pearce et al., 2010). The activation of Akt is controlled by phosphorylation at two conserved residues known as the activation loop, which is controlled by the upstream kinase PDK-1 (phosphoinositide dependent kinase-1) (Toker & Newton, 2000), and the hydrophobic motif, which is regulated by the mTORC2 complex (Sarbassov et al., 2005a,b), as well as other kinases (Warfel et al., 2011; Xie et al., 2011); phosphorylation at both of these sites is necessary maximal activation of the enzyme. Once activated, Akt phosphorylates defined substrates throughout the cell, ultimately inducing pro-proliferation and anti-apoptotic signaling pathways (Cantley, 2002). The structure and function of Akt have been reviewed in great detail (see Vivanco & Sawyers, 2002; Hanada et al., 2004).
2. Regulation of Proviral integration site for Moloney murine leukemia virus kinase expression
2.1. Transcriptional regulation
The expression of PIM kinases is largely regulated at the transcriptional level. The PIM kinases are downstream of multiple oncogenic tyrosine kinase receptors, including Janus kinase (JAK) (Wernig et al., 2008) and FMS-like tyrosine kinase 3 (FLT3) (Kim et al., 2005). The JAK/STAT pathway plays a critical role in regulating the expression of PIM genes. The JAK/STAT pathway represents an alternative to the second messenger signaling system, which is required for the activation of Akt, PKC, and other oncogenic kinases. In response to cytokine binding to cell surface receptors, the JAK kinases phosphorylate receptor domains in the cytoplasm, creating intercellular binding sites for STATs. STAT proteins then bind to the phosphorylated receptors, are phosphorylated by JAKs, and subsequently form dimers and translocate to the nucleus. Once in the nucleus, they initiate the transcription of STAT-responsive genes (Shuai & Liu, 2003). STATs can also be directly phosphorylated and activated by receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR), as well as non-receptor tyrosine kinases, such as c-src (Quesnelle et al., 2007). STAT3 and STAT5 specifically bind to promoter sequences in PIM1, enhancing its transcription and ultimately protein expression in response to a variety of growth factors and cellular mitogens, such as interferons, interleukins, and other chemical messengers that activate STAT-mediated transcription (Fig. 1) (Yip-Schneider et al., 1995; Matikainen et al., 1999). Interestingly, PIM1 binds and activates suppressors of cytokine signaling (SOCS) proteins, which inhibit STAT phosphorylation by binding and inhibiting JAKs or competing with STATs for phospho-tyrosine binding sites on cytokine receptors (Peltola et al., 2004); thus, PIM1 functions as part of a negative feedback loop that serves to damper JAK/STAT signaling. In addition to STATs, several other transcription factors influence the expression of PIM isoforms, including nuclear factor kappa-B (NFκ-B) and krüppel-like factor 5 (KLF5) in response to CD40 and DNA damage, respectively (Zhu et al., 2002; Zhao et al., 2008). Notably, PIM3 transcription is enhanced in EWS/ETS-induced malignant transformation of NIH 3T3 cells, and the Ets-1 transcription factor constitutively binds to the promoter of PIM3 in pancreatic cancer cells, which may explain the high basal expression of this PIM isoform in pancreatic cancer (Deneen et al., 2003).
Fig. 1.

Regulation of PIM expression at the mRNA and protein levels. In the presence of cytokines and growth factors, cytokine receptors and RTKs induce the transcription of PIM isoforms through various transcription factors, and in response to hypoxia, PIM2 is transcriptionally activated by HIF-1. At the protein level, PIM kinases are rapidly degraded by the proteasome under normal conditions. Dephosphorylation of PIM by PP2A and binding to HSP70 promote PIM ubiquitination and degradation, whereas PIM is stabilized in the presence of HSP90 and under hypoxic conditions.
2.2. Regulation of messenger RNA and protein stability
At the translational level, PIM expression is regulated in large part by mRNA stability. PIM mRNA transcripts have a very short half-life due to the presence of several destabilizing AUUU(A) sequences located in the 3′ UTR. Initial studies on the PIM kinases revealed that the activation of pim genes in MuLV-induced lymphomas occurred due to selective proviral integration into their 3′ UTR sequences, which resulted in the removal of the destabilizing motifs and increased PIM expression (Domen et al., 1987). In addition, PIM mRNAs contain long GC-rich regions near the 5′ UTR, which suggests that they are “weak” transcripts that require cap-dependent translation (Wang et al., 2005). As a result, overexpression of eIF4E, which allows for assembly of the eIF4F complex, binding to the 5′-m7G cap structure and translation initiation, is sufficient to significantly enhance PIM1 protein expression (Hoover et al., 1997).
Because the PIM kinases are constitutively active and have very short protein half-life (<5 min), regulation of protein stability is critical for their cellular function and activity. PIM protein stability is largely controlled through ubiquitination and proteasomal degradation (Fig. 1). While the relevant E3-ligase complexes that mediate PIM kinase degradation remain unclear, other factors controlling their stability have been described. Binding to the cellular chaperone proteins HSP90 and HSP70 regulate PIM stability by altering its proteasomal degradation (Mizuno et al., 2001; Shay et al., 2005). Interestingly, the binding of PIM1 to Hsp70 occurs primarily when PIM1 is conjugated to ubiquitin, suggesting that PIM1 degradation may be regulated by E3 ubiquitin ligases associated with HSP70 (Shay et al., 2005). There is also evidence that PIM activity and stability are regulated by phosphorylation. One study reported by ETK tyrosine kinase phosphorylates PIM1 at Y218, which is located in the activation loop, increasing its activity (Kim et al., 2004). In addition, dephosphorylation of PIM kinases by the serine/threonine phosphatase, PP2A, promotes their ubiquitination and subsequent proteasomal degradation (Losman et al., 2003; Ma et al., 2007). Taken together, these reports suggest that while PIM may be constitutively active in cells, phosphorylation by itself or other kinases can influence PIM activity and function.
3. Proviral integration site for Moloney murine leukemia virus and protein kinase B kinase signaling in cancer
An investigation of the amino acid sequence specificity of PIM1 substrates revealed that PIM1 prefers to phosphorylate peptides with the following motif: K/R–X–X–X–S/T–X (X is neither a basic group nor a large hydrophobic residue) (Friedmann et al., 1992). Further screening of peptide libraries identified the consensus sequence that bound to PIM kinases, ARKRRRHPSGPPTA (termed “pimtide”) (Bullock et al., 2005). Notably, this consensus phosphorylation motif is strikingly similar to that of Akt (R–X–R–X–X–S/T) (Obata et al., 2000), indicative of the fact that these kinases recognize similar substrates and control overlapping pro-survival signaling pathways that are critical for tumorigenesis.
3.1. Regulation of cell cycle
PIM and Akt play critical roles in regulating tumor cell proliferation by influencing cell cycle progression. Most notably, PIM and Akt isoforms phosphorylate the cell cycle regulatory proteins, p21waf1 and p27kip. PIM1 and Akt directly phosphorylate p21waf1 at Thr145, stabilizing this cell cycle inhibitor, leading to its accumulation in the cytoplasm, which increases cell proliferation (Zhou et al., 2001a,b; Wang et al., 2002; Zhang et al., 2007). PIM and Akt regulate p27kip expression levels at both the transcriptional and protein levels: 1) repression of p27kip transcription via direct phosphorylation and inactivation of the FoxO1a and FoxO3a transcription factors, which drive p27kip expression and 2) direct phosphorylation of p27kip at Thr157, promoting its interaction with 14–3–3 proteins and proteasomal degradation in the cytoplasm (Fujita et al., 2002; Morishita et al., 2008). In addition, PIM kinases directly phosphorylate SKP2, the putative E3 ligase for p27, at Thr417, which stabilizes SKP2, enhancing the proteasomal degradation of p27 (Cen et al., 2010). Interestingly, SKP2 was reported to ubiquitinylate Akt and increase its activity in response to EGF, indicating that a positive feedback loop may exist between PIM and Akt in certain cellular contexts (Lin et al., 2009). PIM and Akt also phosphorylate Mdm2, the canonical E3 ubiquitin ligase that mediates p53 degradation, at Ser166/186. Thus, overexpression of these kinases in tumors prevents the degradation of both p53 and Mdm2, ultimately increasing p53 expression (Zhou et al., 2001a,b; Hogan et al., 2008). PIM kinases also promote cycle progression via direct phosphorylation and activation of the CDC25A (Mochizuki et al., 1999) and CDC25C (Bachmann et al., 2006) phosphatases, as well as inhibition of the CDC25A inhibitory kinase, c-Tak1 (Bachmann et al., 2004). Numerous studies have demonstrated that pharmacological inhibitors of Akt and PIM induced cell cycle arrest in multiple tumor types, indicative of the overlapping activity of these two kinase families.
3.2. Regulation of survival and apoptosis
One of the primary mechanisms by which Akt and PIM kinases elicit their pro-survival effect is via regulation of Bcl-2 family members. The Bcl-2 family is comprised of both pro-apoptotic proteins, such as BAD and BAX, and anti-apoptotic proteins, such as Bcl-2 and Bcl-XL. PIM and Akt phosphorylate BAD at Ser112, which disrupts its association with Bcl-2 and promotes binding to 14–3–3 and retention in the cytosol (del Peso et al., 1997; Yan et al., 2003); ultimately, the dissociation of BAD and Bcl-2 promotes anti-apoptotic activity (Aho et al., 2004). In addition, PIM and Akt influence the apoptotic-signaling pathway. In the context of hypoxia, increase PIM expression was sufficient to promote resistance to various chemotherapeutic drugs by preventing the loss of mitochondrial membrane potential and inhibiting the activation of caspases 3 and 9 (Chen et al., 2009a,b). Similarly, Akt inhibits caspase-induced cell death by directly phosphorylating caspase 9 at Ser196, which inhibits its proteolytic processing and promotes resistance to apoptosis (Cardone et al., 1998). PIM and Akt are also implicated in the regulation of apoptosis via the JNK signaling pathway. PIM1 and Akt directly phosphorylate Ask1 at Ser83, which decreases its ability to phosphorylate and activate its substrates, JNK and p38 (Zhang et al., 2005; Gu et al., 2009). As a result, PIM and Akt protect cells from stress-induced apoptosis by inhibiting caspase-3 activation.
Akt and PIM kinases also play a critical role in regulating the cell survival within the tumor microenvironment by regulating metabolism and the response to cellular stressors. Akt has been described as a “Warburg” kinase due to its marked influence on enzymes involved in the switch to aerobic glycolysis, which is a hallmark of solid tumors (Robey & Hay, 2009). Akt promotes glycolysis and oxidative phosphorylation through multiple, non-exclusive mechanisms, including the expression and membrane translocation of glucose transporters (Barthel et al., 1999) and effects on HK expression, activity, and mitochondrial interaction (Gottlob et al., 2001), respectively. PIM kinases also appear to play a critical role in regulating cellular metabolism. A recent report demonstrated that mouse embryo fibroblasts lacking all three PIM isoforms (TKO MEFs) undergo cell death in response to activated K-Ras (Song et al., 2014). Transduction of a constitutively active Ras, K-RasG12V, into TKO MEFs increased the level of cellular reactive oxygen species (ROS) to a toxic level. Importantly, the addition of N-acetyl cysteine, a ROS scavenger, reversed the cytotoxic effects of K-RasG12V in TKO MEFs. Expression profiling revealed that loss of PIM altered the cellular redox state by reducing metabolic intermediates in the glycolytic and pentose phosphate pathways, as well as abnormal mitochondrial oxidative phosphorylation. These findings indicate that PIM kinases play an important role in regulating cellular redox and metabolism, particularly in the context of K-Ras-stimulated cell growth. Moreover, recent reports indicate that both Akt and PIM are elevated in the stromal cells that surround epithelial tumors. Stromal Akt2 is an essential regulator of invasion and in oro-pharyngeal cancers (Cichon et al., 2013), whereas PIM1 is elevated in prostate fibroblasts, enhancing the ability of fibroblasts to differentiate into myofibroblasts and express known markers of cancer-associated fibroblasts (CAFs) (Zemskova et al., 2014), suggesting an important role for this kinase in epithelial/stromal crosstalk.
3.3. Regulation of translation and cell growth
The mammalian target of rapamycin (mTOR) is an evolutionarily conserved Ser/Thr protein kinase that serves as the catalytic component of two distinct signaling complexes in human cells, mTORC1 and mTORC2 (Sarbassov et al., 2005a,b). mTORC1, which is composed of mTOR, Raptor, PRAS40 and mLST8, is a regulator of the cellular translational machinery, cell growth and metabolism (Sengupta et al., 2010). Due to its critical role in growth, survival and proliferation, effectors that control the mTOR pathway represent promising cancer targets. Both Akt and PIM kinases signaling converge to control the output of the mTOR-signaling axis via the regulation of upstream and downstream effectors (Fig. 2). First, Akt and PIM kinases are important regulators of cellular energy levels and AMPK activity. Transgenic mice lacking PIM or Akt isoforms display increased cellular AMP:ATP ratio, inhibiting AMPK activation, which leads to the activation of TSC2, a negative regulator of mTOR (Beharry et al., 2011). The effect on AMPK activation is likely mediated via the upstream kinase, LKB1, as phosphorylation of AMPK (Thr172), the putative LKB1 site, was insensitive to PIM inhibitors in LKB-1 deficient cell lines (Hahn-Windgassen et al., 2005; Beharry et al., 2011). Second, both Akt and PIM directly phosphorylate an inhibitory subunit of the mTORC1 complex, PRAS40, at Thr246. Upon phosphorylation at this site, PRAS40 dissociates from the mTOR complex, increasing mTOR kinase activity, and ultimately increasing mTOR-mediated phosphorylation of 4EBP1 and p70S6 kinase. Treatment with small molecule inhibitors of Akt or PIM reduces both PRAS40 and 4EBP1 phosphorylation (Kovacina et al., 2003; Zhang et al., 2009). In addition, PIM2 and Akt directly phosphorylate TSC2 on distinct sites (Ser1798 and Ser939/Thr1462, respectively), relieving its suppression of TORC1 (Manning et al., 2002; Lu et al., 2013). Therefore, PIM and Akt inhibitors may elicit a synergistic or cooperative activating effect on TSC2, suppressing mTORC1 signaling. In addition, PIM controls mRNA translation at several different levels in an mTOR-independent manner. For example, in prostate cancer cells, transient overexpression of PIM kinases is sufficient to maintain 4EBP1 phosphorylation and translational activity in the presence of mTOR inhibitors or growth factor withdraw. In addition to phosphorylation of 4EBP1, PIM enhances the expression of eukaryotic initiation factor 4E (eIF-4E), the limiting component of the cap-dependent translation initiation complex, at the protein level (Hammerman et al., 2005). Thus, PIM expression can overcome 4EBP1-mediated inhibition of translation by generating an excess of eIF-4E. Moreover, PIM1 controls translation by regulating the phosphorylation of eIF-4B on Ser406 (Cen et al., 2014). Interestingly, Akt and PIM regulate eIF-4B through phosphorylation of separate sites that control its ability to bind to the eIF3 translation initiation complex: Akt1 preferentially phosphorylates eIF-4B at Ser422, whereas PIM1 directly phosphorylates Ser406.
Fig. 2.
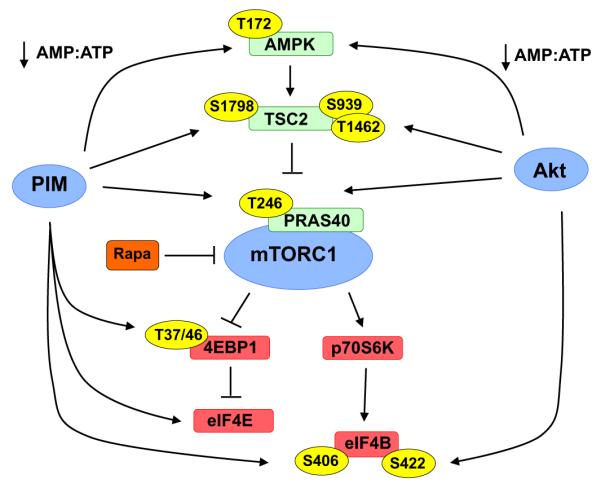
Akt and PIM kinases control translation and cell growth through distinct and overlapping mechanisms. PIM and Akt phosphorylate multiple substrates (phosphorylation sites noted in yellow ovals) upstream and downstream of mTORC1 to control the output of the mTORC1 signaling axis.
4. Proviral Integrations of Moloney virus kinases in cancer
4.1. Proviral Integrations of Moloney virus kinases promote carcinogenesis
Anton Berns and colleagues originally identified the pim1 gene as an integration site of the Moloney murine leukemia virus during a screen of viral carcinogenesis (Selten et al., 1984), and all three PIM isoforms were identified as genes co-activated with myc in murine lymphoid tumors (Nawijn et al., 2011). The Pim kinases are overexpressed in a wide variety of human tumors of both hematological and epithelial origin. PIM1 expression is correlated with tumor aggressiveness, and it is a marker of poor prognosis in several tumor types, including leukemia and prostate cancer (Dhanasekaran et al., 2001; Shah et al., 2008; Liu et al., 2010). Despite their frequent amplification in human tumors, the PIM kinases are considered weak oncogenes. Initial studies to validate the oncogenic activity of PIM revealed that transgenic mice overexpressing PIM1 in T- and B-cells developed spontaneous lymphomas with low incidence and high latency (van Lohuizen et al., 1989). Similarly, overexpression studies in prostate cancer cell lines revealed that PIM1 alone was not sufficient to transform benign cells (Kim et al., 2010). However, the overexpression of PIM enhanced the tumorigenic capabilities of prostate cancer cell lines that are representative of later stages of disease (i.e., PC3 and DU145 cells) both in vitro and in vivo (Chen et al., 2005).
The most substantial evidence supporting the cooperative oncogenic property of PIM kinases is illustrated by its synergism with c-MYC. Myc is a proto-oncogene whose overexpression triggers apoptosis in normal cells. Thus, for myc to act as an oncogene, anti-apoptotic signals are required to prevent myc-induced apoptosis. In addition to the loss of tumor suppressor genes, such as p53 and PTEN, Akt and PIM kinases have been described as potent suppressors of MYC-induced apoptosis. For example, whereas Eμ-myc mice develop lymphomas by three months of age (Adams et al., 1985), the formation of these tumors is greatly accelerated in Eμ-pim-1–Eμ-myc or Eμ-pim-2–Eμ-myc compound transgenic mice. In fact, lymphomas are prenatally lethal in these compound mice (Verbeek et al., 1991; Allen et al., 1997). Moreover, PIM levels are directly correlated with the onset of MYC-driven lymphoma; lymphomas occurred prenatally or at birth in PIM1/MYC bitransgenic mice, where lymphangiogenesis was dramatically delayed in MYC transgenic mice lacking all three PIM isoforms (Moroy et al., 1991). Importantly, evidence suggests that PIM1 cooperates with MYC to promote a malignant phenotype in human tumors as well, as PIM1 is the most frequently co-expressed gene in MYC-positive human prostate cancer. Mechanistic studies have shed light on several mechanisms to explain the synergism between MYC and PIM. PIM1 and PIM2 phosphorylate MYC on S62 and Ser329, respectively, inhibiting MYC protein degradation, increasing protein levels, and enhancing its transcriptional activity (Zhang et al., 2008; Kim et al., 2010). MYC has been shown to form a complex with PIM1, recruiting PIM to the E-boxes targeted by MYC, where PIM phosphorylates histone H3 at Ser10, stimulating the transcription of a subset of MYC-specific genes (Zippo et al., 2007). Thus, PIM kinases can contribute to tumorigenesis by enhancing MYC-regulated oncogenic signaling pathways.
4.2. Proviral Integrations of Moloney virus kinases as a therapeutic target
Investigations into PIM expression in human cancer revealed that PIM1 levels are elevated in lymphoid and myeloid leukemia and lymphomas (Cuypers et al., 1986; Nieborowska-Skorska et al., 2002; Adam et al., 2006), suggesting that these neoplasms may respond to PIM kinase inhibitors. In particular, PIM mRNA is increased in acute myeloid leukemia (AML), presumably due to constitutive activation of the FLT3 tyrosine-kinase receptor, a transcriptional activator of PIM that is constitutively activated in 15–30% of all AML cases (Nakao et al., 1996). In models of AML, forced expression of PIM1 increased resistance to FLT3 inhibition-mediated cytotoxicity and apoptosis. In contrast, expression of a dominant-negative PIM1 accelerated cytotoxicity in response to FLT3 inhibition and inhibited colony growth of FLT3/ITD-transformed BaF3 cells (Kim et al., 2005). Therefore, constitutively activated FLT3 signaling up-regulates Pim-1 expression in leukemia cells, and PIM is a key contributor to FLT3-induced proliferative and antiapoptotic pathways. In addition, expression of the CXCR-4 receptor is an independent prognostic marker of relapse in AML patients. A recent study revealed a correlation between PIM1 and CXCR-4 levels in the blast cells of AML patients, and ex vivo treatment of patient cells with a PIM inhibitor significantly reduced CXCR-4 expression (Grundler et al., 2009). Thus, PIM might regulate leukemia cell migration and potentially influence patient prognosis by modulating CXCR-4 expression. PIM is also reported to mediate the development of several B-cell tumors, including B cell non-Hodgkin's lymphoma (NHL), diffuse B-cell lymphoma, Burkitt's lymphoma, mantel-cell lymphoma, and multiple myeloma (Huttmann et al., 2006; Zhukova Iu et al., 2011). Specifically, PIM1 and PIM3 expressions are increased in lymphoproliferative diseases associated with Epstein–Barr virus (EBV) or Kaposi sarcoma-associated herpes virus (KSHV). PIM kinases play a critical role in the reactivation of these tumor-associated viruses by enhancing the activities of the viral transactivator, EBNA2, and the latency associated nuclear antigen (LANA), respectively (Rainio et al., 2005; Cheng et al., 2009).
Microarray analyses and immunohistochemical studies demonstrated that PIM isoforms are elevated in various solid tumors, including prostate, pancreatic, gastric, liver and colon cancers. Transcription analyses performed in prostate cancers showed no or weak PIM1 expression in benign lesions and moderate to strong PIM1 expression in over 50% of prostate cancer samples, correlating with a poor therapeutic outcome (Dhanasekaran et al., 2001). Indicative of a potential role in the early development of prostate cancer, PIM1 is highly expressed in prostate intraepithelial neoplasias (Dhanasekaran et al., 2001). In prostate cancer cell lines, PIM1 expression is also correlated with resistance to chemotherapy (Chen et al., 2009a,b; Mumenthaler et al., 2009). While all 3 PIM isoforms have been reported to be overexpressed in several types of cancer, including germ cell-derived tumors, isoform specific expression has been correlated with several tumor types: PIM1 expression represents a potential prognostic marker in prostate and pancreatic cancers (Dhanasekaran et al., 2001; Reiser-Erkan et al., 2008); PIM 2 expression is commonly associated with B-cell derived malignancies (Huttmann et al., 2006); and increased PIM 3 expression has been linked to tumors of the pancreas, liver, hepatocellular carcinoma and Ewing's sarcoma (Deneen et al., 2003; Fujii et al., 2005). Thus, despite their functional redundancy, specific PIM isoforms are associated with individual disease types, suggesting that isoform specific inhibitors could have activity in these malignancies.
4.3. Clinical efficacy of Proviral Integrations of Moloney virus inhibitors
Since their discovery, the development of small molecule PIM inhibitors as an anticancer strategy has been a priority in both academic and industrial settings. A majority of PIM kinase inhibitors were designed to target the ATP binding pocket of PIM1, and most inhibitors block the activity of all three PIM isoforms, with PIM2 being the least sensitive. However, due to their functional redundancy, inhibiting all PIM isoforms will likely be the most efficacious strategy in the context of cancer therapy. Mice lacking all PIM isoforms display very modest defects in growth and hematopoiesis, suggesting that pan-PIM kinase inhibitors (Mikkers et al., 2004) should not cause severe side-effects. In cell based assays and in vivo models, PIM inhibitors are active in leukemia and lymphoma cell lines, although responses have also been reported in various solid tumor models. To date, hundreds of PIM inhibitors comprising different chemical classes have been described for academic use. Information regarding the structure and selectivity of various PIM inhibitors has been reviewed elsewhere in great detail (Narlik-Grassow et al., 2014). Despite the growing number of PIM inhibitors described in the literature, few have been tested in human clinical trials; as the focus of this review article is on PIM biology and signaling, we chose to highlight compounds that have been tested in the clinical setting.
The first generation PIM-inhibitor, SGI-1776, inhibited all PIM isoforms, and it exhibited good antitumor activity in vitro and in vivo. In preclinical models, SGI-1776 markedly inhibited cMYC, Cyclin D1, and MCL-1, and it was sufficient to restore sensitivity in PIM2-mediated rapamycin resistant cells in vivo (Hospital et al., 2012). A phase 1 clinical trial was initiated in patients with castration-resistant prostate cancer or relapsed/refractory NHL (NCT00848601). However, this trial was terminated prematurely due to cardiotoxicity. It is not known whether this side effect could be associated with the reported cardioprotective role of PIM1 (Fischer et al., 2011).
Second generation Pim-inhibitors, such as LGB321 and LGH447 are generally ATP competitive inhibitors with IC50 values in the low nanomolar range. Preclinical studies in a panel of cancer cell lines demonstrated that LGB321 had limited activity in cell lines derived from solid tumors, whereas it dramatically inhibited hematological cancer cells, including ALL, AML, and in particular, multiple myeloma where IC50 values were in the picomolar range (Garcia et al., 2014). Moreover, LGB321 synergized with cytarabine in the KG-1 AML xenograft model, for which modulation of pharmacodynamics markers predicts efficacy (Garcia et al., 2014). A phase I clinical trial evaluating the LGH447 activity in relapse/refractory myeloma is currently underway (NCT02144038).
AZD1208 is a thiazolidine that is highly selective for all PIM isoforms, with IC50 values in the low nanomolar range. AZD1208 inhibited the growth of 5 of 14 acute myeloid leukemia (AML) cell lines, and its sensitivity correlated with Pim-1 expression and STAT5 activation (Keeton et al., 2014). Moreover, this inhibitor significantly reduced growth in a dose dependent manner in both AML cell lines and xenograft tumor models. AZD1208 decreased phosphorylation of Bad, 4EBP1, p70S6K, and S6, as well as increased cleaved caspase 3 and p27 in human primary AML cells. In addition, this drug was recently reported to dramatically inhibit myc-driven prostate cancer growth and functioned as a radiation sensitizer by enhancing the p53 pathway (Kirschner et al., 2015). AZD1208 was evaluated in phase 1 clinical trials for AML (NCT01489722), as well as advanced solid tumors and malignant lymphoma (NCT01588548).
4.4. Implications for therapeutic resistance
Resistance to targeted and chemotherapeutic drugs is a significant clinical problem for the treatment of cancer patients, and it is frequently linked to the activation of survival pathways and multi-drug efflux transporters. In addition to their role in promoting cell proliferation and survival, PIM kinases mediate therapeutic resistance in multiple cellular contexts through several mechanisms (Fig. 3). Notably, PIM kinases promote the expression and cell surface of multi-drug resistance associated ATP-binding cassette (ABC) proteins. ABC transporters function as efflux pumps for multiple chemotherapeutic drugs, including anthracyclines, taxanes, and vinca alkaloids, and their overexpression is strongly associated with clinical drug resistance (Mahadevan & Shirahatti, 2005). PIM1 directly phosphorylates the G-subfamily ABC transporter, breast cancer resistance protein (BCRP; ABCG2), at Thr362, promoting its multimerization and cell surface translocation (Xie et al., 2008). In addition, PIM isoforms regulate the expression of a multidrug transporter associated therapeutic resistance, p-glycoprotein (Pgp), which is frequently overexpressed in a diverse array of malignancies. PIM1 phosphorylates Pgp at Ser683, protecting it from ubiquitination and proteasomal degradation, as well as enhancing its glycosylation and subsequent cell surface expression. PIM1 knockdown or inhibition decreased Pgp expression and sensitized Pgp-overexpressing cells to doxorubicin (Xie et al., 2010). Thus, PIM1 is a regulator of efflux-mediated drug resistance, and inhibiting Pim-1 provides a novel approach to sensitizing cancer cells to chemotherapies.
Fig. 3.

PIM substrates regulate cellular processes that are critical for tumor progression and therapeutic resistance, making PIM a promising target for cancer therapy.
In the laboratory, Pim kinase inhibitors act synergistically with several targeted chemotherapies in solid tumors. For example, the expression of Bcl-2 family members is associated with resistance to standard therapy. ABT-737, an inhibitor of Bcl-2, Bcl-xL and Bcl-w, has shown promising apoptotic activity in several solid tumor cell lines. However, this compound is incapable of binding to the Bcl-2 like protein, Mcl-1, and increased expression of this protein promotes resistance to ABT-737. A small molecule PIM inhibitor, SMI-4a, was synergistic when combined with ABT-737 in prostate cancer therapy in vitro and in vivo (Song & Kraft, 2012). The synergism of these agents can largely be attributed to the ability of PIM inhibitors to decrease Mcl-1 expression. Overexpression of Mcl-1 blocked the apoptotic activity of ABT-737. Pim inhibitors also transcriptionally increased the levels of the BH3 protein, Noxa, by activating the unfolded protein response (UPR), which increased the expression of the pro-apoptotic transcription factor, CHOP (Song & Kraft, 2012), which inhibited the remaining levels of MCL-1. Through these dual affects, PIM inhibitors effectively enhanced the activity of ABT-737.
Agents targeting multiple proteins involved in the PI3K/Akt/mTOR signaling axis have performed well in clinical trials, and inhibition of mTOR in particular has proven efficacious in solid tumors, such as breast and kidney cancer and sarcomas. Several agents targeting mTOR are routinely used in clinical practice. However, the use of small molecule inhibitors to block this pathway has been challenging, with up-regulation of reciprocal feedback pathways contributing to treatment failure (Faivre et al., 2006). In particular, the upregulation of receptor tyrosine kinases (RTK) is frequently observed in response to Akt inhibition. A recent study by Cen and colleagues demonstrated that GSK690693, a pan-Akt inhibitor, increased the expression of PIM1, as well as multiple RTKs in prostate cancer cells. Moreover, siRNA or chemical inhibitors of PIM kinase activity abrogated Akt inhibitor-induced upregulation of RTKs in prostate cancer cells by controlling cap-independent translation via internal ribosome entry. Combination therapy with Pim and Akt inhibitors synergistically blocked prostate tumor growth in vitro and in vivo (Cen et al., 2013). Similarly, in vitro studies evaluated the efficacy of combined PIM (AZD1208) and Akt (AZD5363) inhibitors in AML. While either agent alone displayed limited activity, a synergistic cytotoxic association was observed with the combination, and this effect was correlated with decreased phosphorylation of mTOR substrates (4EBP1 and S6K) and decreased Mcl-1 expression (Meja et al., 2014). These data define a mechanism by which PIM1 mediates resistance to Akt inhibition and provide further rationale for combining PIM and Akt inhibitors in anticancer therapy. Of particular relevance to solid tumor biology, PIM kinase expression is dramatically enhanced in response to hypoxia through both HIF-1-dependent and -independent mechanisms. Hypoxia is frequently observed in solid tumors due to regions of poor and/or aberrant vasculature, and it is strongly associated with enhanced survival, proliferation, therapeutic resistance, and ultimately, poor patient prognosis in solid tumors (Semenza, 2003; Warfel & El-Deiry, 2014). A recent report demonstrated that HIF-1α induces PIM2 expression via binding to hypoxia-responsive elements (HREs) within the PIM2 promoter. In turn, PIM2 binds to a transactivation domain of HIF-1α and acts as a co-factor to enhance HIF-1-mediated transcription in response to hypoxia. This positive feedback loop between PIM2 and HIF-1α increased glucose metabolism and cell survival in HepG2 cells, suggesting that PIM kinases might be important for full activation of HIF-1 (Yu et al., 2014). Alternatively, PIM1 is up-regulated in response to hypoxia in a HIF-1-independent manner. Hypoxia prevents the ubiquitin-mediated proteasomal degradation of PIM1, although the mechanism underlying this observation remains unclear (Chen et al., 2009a,b).
PIM kinases play an important physiological role in mediating the prosurvival effects associated with hypoxia. For example, overexpression of PIM1 increased NIH3T3 cell transformation exclusively under hypoxic conditions, suggesting that hypoxia-mediated expression of PIM1 is critical for the transformation of solid tumors (Chen et al., 2009a,b). Alternatively, siRNA knockdown or overexpression of a dominant negative PIM1 reduced xenograft tumor growth and angiogenesis, as well as increased the efficacy of chemotherapies in drug-resistant, hypoxic prostate cancer cell lines. Alternatively, forced overexpression of PIM1 is sufficient to drive therapeutic resistance to cisplatin under normoxic conditions (Chen et al., 2009a,b). Thus, given the correlation between hypoxia, PIM kinases and therapeutic resistance, further studies are warranted to elucidate the importance of PIM kinases in regulating cellular responses to hypoxia and determine the utility of PIM kinase inhibitors in this context.
5. Summary and perspectives
In many tumor types, prosurvival kinases represent a sought after target for drug development. While the PI3K/Akt pathway inhibitors have been validated in preclinical and clinical trials, resistance remains a major obstacle to their effective use. Based on their activity in the cell cycle and apoptosis, their frequent overexpression in cancer and their association with enhanced tumor growth and chemoresistance, the PIM kinases represent a promising target for anticancer drug discovery. As PIM inhibitors now are entering the clinic, several questions remain regarding how to most efficiently apply these agents. Growing evidence suggests that PIM kinase inhibitors are most effective when used in combination with chemo- or targeted therapies. While preclinical data indicates that PIM inhibitors can be effective as single agents in some tumor types, in cell and animal models, their antitumor effect is greatly enhanced by the combination with targeted therapies, particularly PI3K/Akt/mTOR inhibitors. Notably, PIM inhibitors have the ability to reverse drug-resistant phenotypes in preclinical models, especially in the context of tumor hypoxia. Because PIM inhibitors have not demonstrated significant toxicity in preclinical models when applied alone, it will be critical to determine the appropriate dosage for combination therapies to maximize target inhibition while minimizing toxicity. As for multiple other chemotherapeutic agents, the results of ongoing clinical trials are needed to determine whether tumor cells will develop resistance to PIM kinases inhibitors. As might be expected due to their overlapping role in the regulation of survival signaling pathways, PIM kinases promote resistance to Akt inhibitors. Thus, the combination of Pim and Akt inhibitors appears to be a promising option for anticancer therapy. To further develop PIM inhibitors, predictive biomarkers that will allow for selection of patient populations that will benefit most from PIM inhibitors are needed. Basic research to improve our knowledge of PIM substrates and PIM-dependent gene expression will help identify biomarkers for Pim inhibitor activity that can be used in the clinic. Future research on PIM kinases and their involvement in mechanisms of drug resistance is needed to recognize the full potential of PIM inhibitors as a strategy for cancer therapy.
Abbreviations
- AML
acute myeloid leukemia
- AMPK
AMP activated protein kinase
- CXCR-4
C–X–C chemokine receptor type 4
- EGFR
epidermal growth factor receptor
- EBV
Epstein–Barr virus
- FLT3
FMS-like tyrosine kinase 3
- HRE
hypoxia-responsive element
- JAK
Janus kinase 2
- KSHV
Kaposi sarcoma-associated herpes virus
- Pgp
p-glycoprotein
- PIM
Proviral Integrations of Moloney virus
- PI3K
phosphatidylinositol 3-kinase
- PDK-1
phosphoinositide dependent kinase-1
- mTOR
mammalian target of rapamycin
- STAT
Signal Transducer and Activator of Transcription
Footnotes
Conflict of interest
The authors disclose no conflicts of interest.
References
- Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, Fox CJ, Thompson CB, Cools J, Schwaller J. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006;66:3828–3835. doi: 10.1158/0008-5472.CAN-05-2309. [DOI] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Aho TL, Sandholm J, Peltola KJ, Mankonen HP, Lilly M, Koskinen PJ. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004;571:43–49. doi: 10.1016/j.febslet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Allen JD, Verhoeven E, Domen J, van der Valk M, Berns A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene. 1997;15:1133–1141. doi: 10.1038/sj.onc.1201288. [DOI] [PubMed] [Google Scholar]
- Bachmann M, Hennemann H, Xing PX, Hoffmann I, Moroy T. The oncogenic serine/threonine kinase Pim-1 phosphorylates and inhibits the activity of Cdc25C-associated kinase 1 (C-TAK1): a novel role for Pim-1 at the G2/M cell cycle checkpoint. J Biol Chem. 2004;279:48319–48328. doi: 10.1074/jbc.M404440200. [DOI] [PubMed] [Google Scholar]
- Bachmann M, Kosan C, Xing PX, Montenarh M, Hoffmann I, Moroy T. The oncogenic serine/threonine kinase Pim-1 directly phosphorylates and activates the G2/M specific phosphatase Cdc25C. Int J Biochem Cell Biol. 2006;38:430–443. doi: 10.1016/j.biocel.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr., Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Baytel D, Shalom S, Madgar I, Weissenberg R, Don J. The human Pim-2 proto-oncogene and its testicular expression. Biochim Biophys Acta. 1998;1442:274–285. doi: 10.1016/s0167-4781(98)00185-7. [DOI] [PubMed] [Google Scholar]
- Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z, Smith CD, Kraft AS. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci U S A. 2011;108:528–533. doi: 10.1073/pnas.1013214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Debreczeni J, Amos AL, Knapp S, Turk BE. Structure and substrate specificity of the Pim-1 kinase. J Biol Chem. 2005;280:41675–41682. doi: 10.1074/jbc.M510711200. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Cen B, Mahajan S, Wang W, Kraft AS. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res. 2013;73:3402–3411. doi: 10.1158/0008-5472.CAN-12-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen B, Mahajan S, Zemskova M, Beharry Z, Lin YW, Cramer SD, Lilly MB, Kraft AS. Regulation of Skp2 levels by the Pim-1 protein kinase. J Biol Chem. 2010;285:29128–29137. doi: 10.1074/jbc.M110.137240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen B, Xiong Y, Song JH, Mahajan S, DuPont R, McEachern K, DeAngelo DJ, Cortes JE, Minden MD, Ebens A, Mims A, LaRue AC, Kraft AS. The Pim-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol Cell Biol. 2014;34:2517–2532. doi: 10.1128/MCB.00147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Kondo S, Wang H, Yeung SC, Lee MH. Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor formation. Am J Pathol. 2009a;175:400–411. doi: 10.2353/ajpath.2009.080972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Yeung SC, Lee MH. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene. 2009b;28:2581–2592. doi: 10.1038/onc.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS. Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res. 2005;3:443–451. doi: 10.1158/1541-7786.MCR-05-0007. [DOI] [PubMed] [Google Scholar]
- Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009;5:e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, Luo Y, Cheng L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–288. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon AC, Pickard A, McDade SS, Sharpe DJ, Moran M, James JA, McCance DJ. AKT in stromal fibroblasts controls invasion of epithelial cells. Oncotarget. 2013;4:1103–1116. doi: 10.18632/oncotarget.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AM, Grinblat B, Bessler H, Kristt D, Kremer A, Schwartz A, Halperin M, Shalom S, Merkel D, Don J. Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma. 2004;45:951–955. doi: 10.1080/10428190310001641251. [DOI] [PubMed] [Google Scholar]
- Cuypers HT, Selten G, Berns A, Geurts van Kessel AH. Assignment of the human homologue of Pim-1, a mouse gene implicated in leukemogenesis, to the pter-q12 region of chromosome 6. Hum Genet. 1986;72:262–265. doi: 10.1007/BF00291892. [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Deneen B, Welford SM, Ho T, Hernandez F, Kurland I, Denny CT. PIM3 proto-oncogene kinase is a common transcriptional target of divergent EWS/ETS oncoproteins. Mol Cell Biol. 2003;23:3897–3908. doi: 10.1128/MCB.23.11.3897-3908.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- Domen J, Von Lindern M, Hermans A, Breuer M, Grosveld G, Berns A. Comparison of the human and mouse PIM-1 cDNAs: nucleotide sequence and immunological identification of the in vitro synthesized PIM-1 protein. Oncog Res. 1987;1:103–112. [PubMed] [Google Scholar]
- Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- Feldman JD, Vician L, Crispino M, Tocco G, Baudry M, Herschman HR. Seizure activity induces PIM-1 expression in brain. J Neurosci Res. 1998;53:502–509. doi: 10.1002/(SICI)1097-4547(19980815)53:4<502::AID-JNR13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Fischer KM, Cottage CT, Konstandin MH, Volkers M, Khan M, Sussman MA. Pim-1 kinase inhibits pathological injury by promoting cardioprotective signaling. J Mol Cell Cardiol. 2011;51:554–558. doi: 10.1016/j.yjmcc.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann M, Nissen MS, Hoover DS, Reeves R, Magnuson NS. Characterization of the proto-oncogene Pim-1: kinase activity and substrate recognition sequence. Arch Biochem Biophys. 1992;298:594–601. doi: 10.1016/0003-9861(92)90454-5. [DOI] [PubMed] [Google Scholar]
- Fujii C, Nakamoto Y, Lu P, Tsuneyama K, Popivanova BK, Kaneko S, Mukaida N. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int J Cancer. 2005;114:209–218. doi: 10.1002/ijc.20719. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14–3–3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, Ison M, Zavorotinskaya T, Dai Y, Lu J, Niu XH, Basham S, Chan J, Yu J, Doyle M, Feucht P, Warne R, Narberes J, Tsang T, Fritsch C, Kauffmann A, Pfister E, Drueckes P, Trappe J, Wilson C, Han W, Lan J, Nishiguchi G, Lindvall M, Bellamacina C, Aycinena JA, Zang R, Holash J, Burger MT. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res. 2014;20:1834–1845. doi: 10.1158/1078-0432.CCR-13-2062. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, Pogacic V, Villa A, Ehret S, Berridge G, Spoo A, Dierks C, Biondi A, Knapp S, Duyster J, Schwaller J. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12–CXCR4-mediated homing and migration. J Exp Med. 2009;206:1957–1970. doi: 10.1084/jem.20082074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu JJ, Wang Z, Reeves R, Magnuson NS. PIM1 phosphorylates and negatively regulates ASK1-mediated apoptosis. Oncogene. 2009;28:4261–4271. doi: 10.1038/onc.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood. 2005;105:4477–4483. doi: 10.1182/blood-2004-09-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hogan C, Hutchison C, Marcar L, Milne D, Saville M, Goodlad J, Kernohan N, Meek D. Elevated levels of oncogenic protein kinase Pim-1 induce the p53 pathway in cultured cells and correlate with increased Mdm2 in mantle cell lymphoma. J Biol Chem. 2008;283:18012–18023. doi: 10.1074/jbc.M709695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DS, Wingett DG, Zhang J, Reeves R, Magnuson NS. Pim-1 protein expression is regulated by its 5′-untranslated region and translation initiation factor elF-4E. Cell Growth Differ. 1997;8:1371–1380. [PubMed] [Google Scholar]
- Hospital MA, Green AS, Lacombe C, Mayeux P, Bouscary D, Tamburini J. The FLT3 and Pim kinases inhibitor SGI-1776 preferentially target FLT3-ITD AML cells. Blood. 2012;119:1791–1792. doi: 10.1182/blood-2011-11-393066. [DOI] [PubMed] [Google Scholar]
- Huttmann A, Klein-Hitpass L, Thomale J, Deenen R, Carpinteiro A, Nuckel H, Ebeling P, Fuhrer A, Edelmann J, Sellmann L, Duhrsen U, Durig J. Gene expression signatures separate B-cell chronic lymphocytic leukaemia prognostic subgroups defined by ZAP-70 and CD38 expression status. Leukemia. 2006;20:1774–1782. doi: 10.1038/sj.leu.2404363. [DOI] [PubMed] [Google Scholar]
- Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao Y, Grondine MR, Kaur S, Wang S, Chen Y, Wu A, Shen M, Gibbons FD, Lamb ML, Zheng X, Stone RM, Deangelo DJ, Platanias LC, Dakin LA, Chen H, Lyne PD, Huszar D. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood. 2014;123:905–913. doi: 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Roh M, Abdulkadir SA. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer. 2010;10:248. doi: 10.1186/1471-2407-10-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, Levis M, Small D. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105:1759–1767. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- Kim O, Jiang T, Xie Y, Guo Z, Chen H, Qiu Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2004;23:1838–1844. doi: 10.1038/sj.onc.1207304. [DOI] [PubMed] [Google Scholar]
- Kirschner AN, Wang J, van der Meer R, Anderson PD, Franco-Coronel OE, Kushner MH, Everett JH, Hameed O, Keeton EK, Ahdesmaki M, Grosskurth SE, Huszar D, Abdulkadir SA. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/dju407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14–3–3 binding partner. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, Tempst P, Pandolfi PP. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HT, Wang N, Wang X, Li SL. Overexpression of Pim-1 is associated with poor prognosis in patients with esophageal squamous cell carcinoma. J Surg Oncol. 2010;102:683–688. doi: 10.1002/jso.21627. [DOI] [PubMed] [Google Scholar]
- Losman JA, Chen XP, Vuong BQ, Fay S, Rothman PB. Protein phosphatase 2A regulates the stability of Pim protein kinases. J Biol Chem. 2003;278:4800–4805. doi: 10.1074/jbc.M208246200. [DOI] [PubMed] [Google Scholar]
- Lu J, Zavorotinskaya T, Dai Y, Niu XH, Castillo J, Sim J, Yu J, Wang Y, Langowski JL, Holash J, Shannon K, Garcia PD. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood. 2013;122:1610–1620. doi: 10.1182/blood-2013-01-481457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Arnold HK, Lilly MB, Sears RC, Kraft AS. Negative regulation of Pim-1 protein kinase levels by the B56beta subunit of PP2A. Oncogene. 2007;26:5145–5153. doi: 10.1038/sj.onc.1210323. [DOI] [PubMed] [Google Scholar]
- Mahadevan D, Shirahatti N. Strategies for targeting the multidrug resistance-1 (MDR1)/P-gp transporter in human malignancies. Curr Cancer Drug Targets. 2005;5:445–455. doi: 10.2174/1568009054863609. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and Pim-1 genes in human T cells. Blood. 1999;93:1980–1991. [PubMed] [Google Scholar]
- Meja K, Stengel C, Sellar R, Huszar D, Davies BR, Gale RE, Linch DC, Khwaja A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br J Haematol. 2014;167:69–79. doi: 10.1111/bjh.13013. [DOI] [PubMed] [Google Scholar]
- Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, Berns A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24:6104–6115. doi: 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Shirogane T, Shinohara A, Iwamatsu A, Hibi M, Hirano T. Regulation of Pim-1 by Hsp90. Biochem Biophys Res Commun. 2001;281:663–669. doi: 10.1006/bbrc.2001.4405. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Kitanaka C, Noguchi K, Muramatsu T, Asai A, Kuchino Y. Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J Biol Chem. 1999;274:18659–18666. doi: 10.1074/jbc.274.26.18659. [DOI] [PubMed] [Google Scholar]
- Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008;68:5076–5085. doi: 10.1158/0008-5472.CAN-08-0634. [DOI] [PubMed] [Google Scholar]
- Moroy T, Verbeek S, Ma A, Achacoso P, Berns A, Alt F. E mu N- and E mu L-myc cooperate with E mu Pim-1 to generate lymphoid tumors at high frequency in double-transgenic mice. Oncogene. 1991;6:1941–1948. [PubMed] [Google Scholar]
- Mumenthaler SM, Ng PY, Hodge A, Bearss D, Berk G, Kanekal S, Redkar S, Taverna P, Agus DB, Jain A. Pharmacologic inhibition of Pim kinases alters prostate cancer cell growth and resensitizes chemoresistant cells to taxanes. Mol Cancer Ther. 2009;8:2882–2893. doi: 10.1158/1535-7163.MCT-09-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34:136–159. doi: 10.1002/med.21284. [DOI] [PubMed] [Google Scholar]
- Narlik-Grassow M, Blanco-Aparicio C, Cecilia Y, Peregrina S, Garcia-Serelde B, Munoz-Galvan S, Canamero M, Carnero A. The essential role of PIM kinases in sarcoma growth and bone invasion. Carcinogenesis. 2012;33:1479–1486. doi: 10.1093/carcin/bgs176. [DOI] [PubMed] [Google Scholar]
- Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA, Skorski T. Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase Pim-1 in the BCR/ABL-mediated leukemogenesis. Blood. 2002;99:4531–4539. doi: 10.1182/blood.v99.12.4531. [DOI] [PubMed] [Google Scholar]
- Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Peltola KJ, Paukku K, Aho TL, Ruuska M, Silvennoinen O, Koskinen PJ. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood. 2004;103:3744–3750. doi: 10.1182/blood-2003-09-3126. [DOI] [PubMed] [Google Scholar]
- Qian KC, Studts J, Wang L, Barringer K, Kronkaitis A, Peng C, Baptiste A, LaFrance R, Mische S, Farmer B. Expression, purification, crystallization and preliminary crystallographic analysis of human Pim-1 kinase. Acta Crystallogr Sect F: Struct Biol Cryst Commun. 2005;61:96–99. doi: 10.1107/S1744309104029963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnelle KM, Boehm AL, Grandis JR. STAT-mediated EGFR signaling in cancer. J Cell Biochem. 2007;102:311–319. doi: 10.1002/jcb.21475. [DOI] [PubMed] [Google Scholar]
- Rainio EM, Ahlfors H, Carter KL, Ruuska M, Matikainen S, Kieff E, Koskinen PJ. Pim kinases are upregulated during Epstein–Barr virus infection and enhance EBNA2 activity. Virology. 2005;333:201–206. doi: 10.1016/j.virol.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Reiser-Erkan C, Erkan M, Pan Z, Bekasi S, Giese NA, Streit S, Michalski CW, Friess H, Kleeff J. Hypoxia-inducible proto-oncogene Pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol Ther. 2008;7:1352–1359. doi: 10.4161/cbt.7.9.6418. [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N. Is Akt the “Warburg kinase”?—Akt–energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005a;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005b;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Saris CJ, Domen J, Berns A. The Pim-1 oncogene encodes two related protein–serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991;10:655–664. doi: 10.1002/j.1460-2075.1991.tb07994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten G, Cuypers HT, Zijlstra M, Melief C, Berns A. Involvement of c-myc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of activation. EMBO J. 1984;3:3215–3222. doi: 10.1002/j.1460-2075.1984.tb02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Pang B, Yeoh KG, Thorn S, Chen CS, Lilly MB, Salto-Tellez M. Potential roles for the PIM1 kinase in human cancer — a molecular and therapeutic appraisal. Eur J Cancer. 2008;44:2144–2151. doi: 10.1016/j.ejca.2008.06.044. [DOI] [PubMed] [Google Scholar]
- Shay KP, Wang Z, Xing PX, McKenzie IF, Magnuson NS. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin–proteasome pathway. Mol Cancer Res. 2005;3:170–181. doi: 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- Song JH, An N, Chatterjee S, Kistner-Griffin E, Mahajan S, Mehrotra S, Kraft AS. Deletion of Pim kinases elevates the cellular levels of reactive oxygen species and sensitizes to K-Ras-induced cell killing. Oncogene. 2014:1–9. doi: 10.1038/onc.2014.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JH, Kraft AS. Pim kinase inhibitors sensitize prostate cancer cells to apoptosis triggered by Bcl-2 family inhibitor ABT-737. Cancer Res. 2012;72:294–303. doi: 10.1158/0008-5472.CAN-11-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, Berns A. Predisposition to lymphomagenesis in Pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell. 1989;56:673–682. doi: 10.1016/0092-8674(89)90589-8. [DOI] [PubMed] [Google Scholar]
- Verbeek S, van Lohuizen M, van der Valk M, Domen J, Kraal G, Berns A. Mice bearing the E mu-myc and E mu-Pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol Cell Biol. 1991;11:1176–1179. doi: 10.1128/mcb.11.2.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bhattacharya N, Mixter PF, Wei W, Sedivy J, Magnuson NS. Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by Pim-1 kinase. Biochim Biophys Acta. 2002;1593:45–55. doi: 10.1016/s0167-4889(02)00347-6. [DOI] [PubMed] [Google Scholar]
- Wang Z, Weaver M, Magnuson NS. Cryptic promoter activity in the DNA sequence corresponding to the Pim-1 5′-UTR. Nucleic Acids Res. 2005;33:2248–2258. doi: 10.1093/nar/gki523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel NA, El-Deiry WS. HIF-1 signaling in drug resistance to chemotherapy. Curr Med Chem. 2014;21:3021–3028. doi: 10.2174/0929867321666140414101056. [DOI] [PubMed] [Google Scholar]
- Warfel NA, Niederst M, Newton AC. Disruption of the interface between the pleckstrin homology (PH) and kinase domains of Akt protein is sufficient for hydrophobic motif site phosphorylation in the absence of mTORC2. J Biol Chem. 2011;286:39122–39129. doi: 10.1074/jbc.M111.278747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, Walz C, Reiter A, Podar K, Royer Y, Constantinescu SN, Tomasson MH, Griffin JD, Gilliland DG, Sattler M. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood. 2008;111:3751–3759. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol. 2010;78:310–318. doi: 10.1124/mol.109.061713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Xu K, Dai B, Guo Z, Jiang T, Chen H, Qiu Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene. 2006;25:70–78. doi: 10.1038/sj.onc.1209058. [DOI] [PubMed] [Google Scholar]
- Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, Nakanishi T, Ross DD, Chen H, Fazli L, Gleave ME, Qiu Y. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349–3356. doi: 10.1074/jbc.M707773200. [DOI] [PubMed] [Google Scholar]
- Yan B, Zemskova M, Holder S, Chin V, Kraft A, Koskinen PJ, Lilly M. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J Biol Chem. 2003;278:45358–45367. doi: 10.1074/jbc.M307933200. [DOI] [PubMed] [Google Scholar]
- Yip-Schneider MT, Horie M, Broxmeyer HE. Transcriptional induction of Pim-1 protein kinase gene expression by interferon gamma and posttranscriptional effects on costimulation with steel factor. Blood. 1995;85:3494–3502. [PubMed] [Google Scholar]
- Yu Z, Zhao X, Ge Y, Zhang T, Huang L, Zhou X, Xie L, Liu J, Huang G. A regulatory feedback loop between HIF-1alpha and PIM2 in HepG2 cells. PLoS One. 2014;9:e88301. doi: 10.1371/journal.pone.0088301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemskova MY, Song JH, Cen B, Cerda-Infante J, Montecinos VP, Kraft AS. Regulation of prostate stromal fibroblasts by the PIM1 protein kinase. Cell Signal. 2014;27:135–146. doi: 10.1016/j.cellsig.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Beharry ZM, Harris TE, Lilly MB, Smith CD, Mahajan S, Kraft AS. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol Ther. 2009;8:846–853. doi: 10.4161/cbt.8.9.8210. [DOI] [PubMed] [Google Scholar]
- Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa WC, Min W. Hsp90–Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene. 2005;24:3954–3963. doi: 10.1038/sj.onc.1208548. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27:4809–4819. doi: 10.1038/onc.2008.123. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang Z, Magnuson NS. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol Cancer Res. 2007;5:909–922. doi: 10.1158/1541-7786.MCR-06-0388. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hamza MS, Leong HS, Lim CB, Pan YF, Cheung E, Soo KC, Iyer NG. Kruppel-like factor 5 modulates p53-independent apoptosis through Pim1 survival kinase in cancer cells. Oncogene. 2008;27:1–8. doi: 10.1038/sj.onc.1210625. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-over-expressing cells. Nat Cell Biol. 2001a;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001b;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J Immunol. 2002;168:744–754. doi: 10.4049/jimmunol.168.2.744. [DOI] [PubMed] [Google Scholar]
- Zhukova Iu N, Alekseeva MG, Zakharevich NV, Shtil AA, Danilenko VN. The Pim family of protein kinases: structure, functions and roles in hematopoietic malignancies. Mol Biol. 2011;45:755–764. [PubMed] [Google Scholar]
- Zippo A, De Robertis A, Serafini R, Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol. 2007;9:932–944. doi: 10.1038/ncb1618. [DOI] [PubMed] [Google Scholar]