

SUMMARY
Maternal obesity impairs offspring health, but the responsible mechanisms are not fully established. To address this question, we fed female mice a high-fat/high-sugar diet from before conception until weaning and then followed the outcomes in the next three generations of offspring, all fed a control diet. We observed that female offspring born to obese mothers had impaired peripheral insulin signaling that was associated with mitochondrial dysfunction and altered mitochondrial dynamic and complex proteins in skeletal muscle. This mitochondrial phenotype persisted through the female germline and was passed down to the second and third generations. Our results indicate maternal programing of metabolic disease can be passed through the female germline and that the transfer of aberrant oocyte mitochondria to subsequent generations may contribute to the increased risk for developing insulin resistance.
eTOC blurb
Saben et al demonstrate that maternal diet-induced metabolic syndrome in an inbred mouse model results in transgenerational inheritance of aberrant mitochondria. Abnormal expression of mitochondrial ETC Complex and dynamic proteins are seen in F1-F3, despite the fact that they are eating a regular diet. The transmission appears to be germline and through aberrant oocytes.
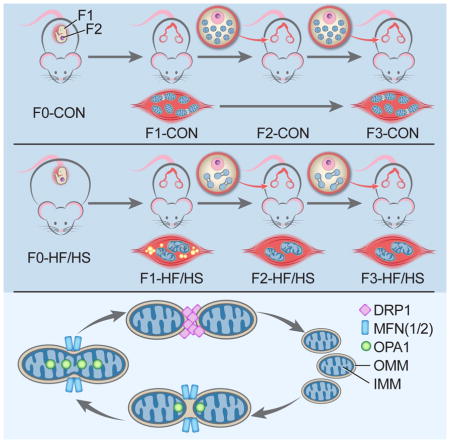
INTRODUCTION
The Developmental Origins of Health and Disease Hypothesis states that exposures during embryonic and fetal life are critical to the developmental patterning of tissues. Maternal obesity and metabolic disease are two of the most prominent of these potential exposures, as over two-thirds of reproductive-age women in the US are overweight or obese (Flegal et al., 2012). Offspring (first generation, F1) born to obese mothers are at an increased risk for being born large for gestational age (Carlsen et al., 2014) and for developing obesity, cardiovascular disease, and diabetes in adulthood (Tanvig, 2014), thus potentiating the obesity epidemic.
The grandchildren (F2) and great grandchildren (F3) of obese women may also inherit risk of metabolic disease even if they are not directly exposed to the metabolic insult. This type of inheritance, occurs when phenotypes acquired from an initial exposure (in the F1) are transmitted to the next generations (F2 and F3) in the absence of any continued exposure (Skinner, 2011). To be considered transgenerational, the inherited traits must be apparent in the F3 generation because the F1 embryo and F2 primordial germ cells are directly exposed to a given environmental factor in utero. Evidence exists to suggest that the transmittance of disease risk can penetrate through both the maternal (Ponzio et al., 2012) and paternal germlines (Dunn and Bale, 2011; Song et al., 2014). However, a recent study showed that associations between maternal and offspring BMI persisted through three generations, but similar associations did not exist between paternal and offspring BMI (Murrin et al., 2012), suggesting a stronger relationship between maternal BMI and offspring adiposity.
The maternal but not paternal transmission of obesity risk may be explained by the fact that mitochondria, which regulate metabolism, apoptosis, cellular redox homeostasis, and numerous biochemical pathways, are maternally inherited. Although the contribution of mitochondrial dysfunction to development of metabolic disease remains in debate (Montgomery and Turner, 2015), it is clear that defects in mitochondrial health are associated with obesity, insulin resistance, diabetes, and cardiovascular disease (Crescenzo et al., 2014; Kelley et al., 2002; Montgomery and Turner, 2015). Mitochondrial health is preserved through fission and fusion, collectively called mitochondrial dynamics. Importantly, an imbalance in mitochondrial dynamics has been associated with metabolic disease (Galloway and Yoon, 2013).
We previously presented evidence that diet-induced metabolic syndrome alters mitochondrial dynamics in the oocyte. Briefly, we demonstrated that female mice with diet-induced metabolic syndrome had abnormal oocyte mitochondrial morphology characterized by fewer, more disarrayed cristae, decreased electron density of the matrix, increased swelling, and more vacuoles (Luzzo et al., 2012). Additionally, oocyte mitochondria from mice fed a high fat-high sugar (HF/HS) were less round and more dumbbell shaped than their chow-fed counterparts, suggesting a defect in mitochondrial dynamics (Boudoures et al., 2016). These defects in mitochondrial morphology were accompanied by impaired beta oxidation (Boudoures et al., 2016), decreased mitochondrial membrane potential (Reynolds et al., 2015), and altered ATP and citrate levels (Boudoures et al., 2016; Luzzo et al., 2012; Reynolds et al., 2015). Adult offspring bore to obese mothers developed characteristics of metabolic syndrome (Jungheim et al., 2010), suggesting that the maternal germline can transmit changes to offspring metabolic function.
To our knowledge, the transgenerational inheritance of altered mitochondrial phenotypes through the maternal germline has not been explored. In this study, we examined whether maternal diet-induced metabolic syndrome in an inbred mouse model can cause transgenerational inheritance of aberrant mitochondria. Our data show that mice with diet-induced metabolic syndrome before and during pregnancy can transmit impaired mitochondrial dynamic and complex proteins through the female germline to the F3 generation. Importantly, our study indicates that oocyte mitochondria passed from mother to offspring may carry information that programs mitochondrial dysfunction throughout the entire organism.
RESULTS
Exposure to maternal metabolic syndrome induces peripheral insulin resistance in first generation (F1) female offspring
Female C57BL/6 mice fed a high fat/high sugar diet (F0-HF/HS) were obese (Fig. 1A and B), glucose intolerant (Fig. 1C), and had higher fasting levels of circulating glucose (Fig. 1C), triglycerides (Fig. 1E), and cholesterol (Fig. 1F) than chow-fed controls (F0-Con). To test the effects of maternal metabolic syndrome on risk for developing metabolic disease in the offspring, we examined glucose disposal and insulin signaling in the first generation of female offspring bore to HF/HS-fed mice (F1-HF/HS). Although these mice (which were fed control chow after weaning) were not obese (Fig. 1G), they showed slight but significant impairments in glucose tolerance (Fig. 1H) and had significantly higher fasting insulin levels (Fig. 1I) than F1-Con mice. Consistent with the hyperinsulinemia, F1-HF/HS offspring had impaired basal insulin signaling in their visceral adipose (S1) and mixed fiber skeletal muscle (Fig. 1J) as indicated by decreased tyrosine phosphorylation of IRS-1 (Tyr895) and decreased serine phosphorylation of AKT (Ser473).
Figure 1. Maternal metabolic syndrome leads to glucose intolerance and altered insulin signaling in the first generation (F1) female offspring.

Metabolic parameters were measured in F0-HF/HS and F0-Con mice prior to mating (A–F) and in the F1 female offspring (G–J). Body weight (A and G), body composition presented as percent fat mass (FM) and fat-free mass (FFM) (B), glucose tolerance test (C and H), and serum insulin (D and I), triglyceride (E), and cholesterol (F) were measured in mice after a 6 h fast. Western blot analysis of tyrosine phosphorylation of IRS1(tyr895) and serine phosphorylation of AKT(ser473) in F1 mixed fiber muscle (J). All data are expressed as mean ± SEM. *P < 0.05 by Student’s T-test.
Mitochondrial dysfunction and imbalanced mitochondrial dynamics occur in F1-HF/HS skeletal muscle
We hypothesized that mitochondrial dysfunction was involved in the altered insulin signaling observed in F1-HF/HS skeletal muscle. So, we examined mitochondria morphology and function in soleus (slow-twitch, oxidative, Type I/IIA) and lateral gastrocnemius (fast-twitch, glycolytic, Type IIX/IIB) (Bloemberg and Quadrilatero, 2012) muscle to determine whether or not specific muscle fiber types were affected differently by exposure to maternal HF/HS diet. Previous studies have indicated that mitochondrial dynamics are dependent on the oxidative capacity of the different muscle fibers such that fusion events are more common in oxidative fibers, creating long strings of connected mitochondria (Mishra et al., 2015). Conversely, mitochondria in glycolytic fibers have a more punctate appearance. Ultrastructure analyses of the F1-Con soleus (Fig. 2A and S2A) and lateral gastrocnemius (Fig. 2F and S2B) were consistent with these morphologic phenotypes. However, F1-HF/HS mice had significantly larger mitochondria in both the soleus (Fig. 2A and S2C) and lateral gastrocnemius muscles (Fig. 2F and S2D) than did F1-Con mice.
Figure 2. Mitochondrial function and morphology are altered in F1-HF/HS skeletal muscles.

Soleus (A–E) and lateral gastrocnemius (F–H) were isolated from eight-week old F1 mice. Representative transmission electron microscopy (TEM) images of soleus (A) and lateral gastrocnemius (F). Arrowheads depict lipid droplets, star indicates mitochondria undergoing fission, and arrows denote abnormal inner mitochondrial membrane morphology. Lipid peroxidation content measured by malondialdehyde (MDA) in soleus muscle (B). Morphological analysis of TEM images lipid content in soleus muscle (C). HADHA activity (D) and mitochondrial respiration (E) in soleus muscle. Western blot analysis of DRP1, P-DRP1(ser616), P-DRP1/DRP1 (Ratio) and OPA1 in lateral gastrocnemius (G) and electron transport chain complex content (CI = NDUFB8, CII = total complex, CIII = Core protein 2, CIV = subunit I and CV = alpha subunit) (H). All data are expressed as mean ± SEM. *P < 0.05 by Student’s T-test.
In the F1-HF/HS soleus, mitochondria appeared disorganized, and macro-mitochondria were almost always associated with lipid droplets (Fig. 2A, S2A). Image analysis confirmed that F1-HF/HS soleus contained significantly more intramuscular lipid than F1-Con soleus did (Fig. 2C). This observation led us to hypothesize that fatty acid oxidation was impaired in the F1-HF/HS soleus. Consistent with this idea, the activity of hydroxyacyl-coenzyme A dehydrogenase, type II, the rate limiting enzyme in beta-oxidation, was lower in F1-HF/HS soleus than in F1-Con soleus (Fig. 2D). Consistent with elevated lipid and mitochondrial dysfunction, soleus muscle from F1-HF/HS contained significantly more lipid peroxide products than the F1-Con muscle (Fig. 2B), suggesting a state of oxidative stress. Furthermore, ADP-dependent (state 3) oxygen consumption was significantly lower in F1-HF/HS soleus than in F1-Con soleus (Fig. 2E, S3A), indicating impairments in mitochondrial respiration that were not a result of decreased mitochondrial content (S2E). Consistent with impaired mitochondrial function we also observed a trending lower (P = 0.06) RCR in the F1-HF/HS offspring (S3B).
Mitochondria in the F1-HF/HS lateral gastrocnemius were arranged abnormally. Elongated mitochondria that seemed to stretch longitudinally across the muscle fiber (S2B) were observed. Additionally, mitochondria in the lateral gastrocnemius of F1-HF/HS mice showed a very distinct inner mitochondrial membrane (IMM) phenotype (Fig. 2F). Cristae appeared bloated and disorganized, and the electron density of the inner mitochondrial matrix was almost completely absent (Fig 2F). Some mitochondria contained onion-shaped cristae and vacuoles (Fig. 2F).
The increased mitochondrial size and abnormal cristae led us to hypothesize that mitochondrial dynamics were altered in the F1-HF/HS muscle. Thus, we examined the protein levels of the four main GTPases required for mitochondrial fusion (MFN1, MFN2, OPA1) and fission (DRP1). Total levels of the mitofusins (MFN1 and MFN2) were equivalent between F1-HF/HS and F1-Con gastrocnemius (S2), indicating that increased outer mitochondrial membrane (OMM) fusion could not explain the enlarged mitochondria observed in F1-HF/HS muscle. Conversely, activity of DRP1 was significantly reduced in F1-HF/HS lateral gastrocnemius muscle as measured by a decrease in the ratio of phosphorylated-DRP1 (ser616) to total DRP1 levels (Fig. 2G), suggesting that mitochondrial fission was impaired. We next examined expression of Optic atrophy 1 (OPA1), which regulates IMM fusion and cristae formation (Frezza et al., 2006), and found that F1-HF/HS muscle contained significantly less total OPA1 than F1-Con muscle did (Fig. 2G). Finally, we observed a significant reduction in the levels of electron transport chain complex II (succinate dehydrogenase), complex III-core protein 2 and the alpha subunit of complex V in the F1-HF/HS lateral gastrocnemius muscle (Fig. 2H). A reduction in these subunits suggests impairment in complex assembly, which has been associated with altered cristae formation (Cogliati et al., 2013).
Mitochondrial dysfunction and altered mitochondrial dynamics occur in the F1-HF/HS oocytes
To determine whether or not abnormal mitochondria could be passed to subsequent generations, we next examined the mitochondria in oocytes of F1 mice (F1 oocytes). Mitochondrial morphology in oocytes differs from that of somatic cells; they are characteristically round to oval-shaped, have a dense matrix and sparse circular cristae, and occasionally contain regular-shaped vacuoles (Wakai et al., 2014) (Han et al., 2008) as observed in F1-Con oocytes (Fig. 3A). We observed characteristically poor oocyte mitochondrial morphology (Han et al., 2008) in F1-HF/HS oocytes; mitochondria were significantly larger (Fig. 3B) and less round (Fig. 3C) than those of F1-Con oocytes, and we observed a number of mitochondria containing irregular vacuoles inclosing lamellar membranes (Fig. 3A).
Figure 3. Mitochondrial function and morphology are altered in F1-HF/HS oocytes.
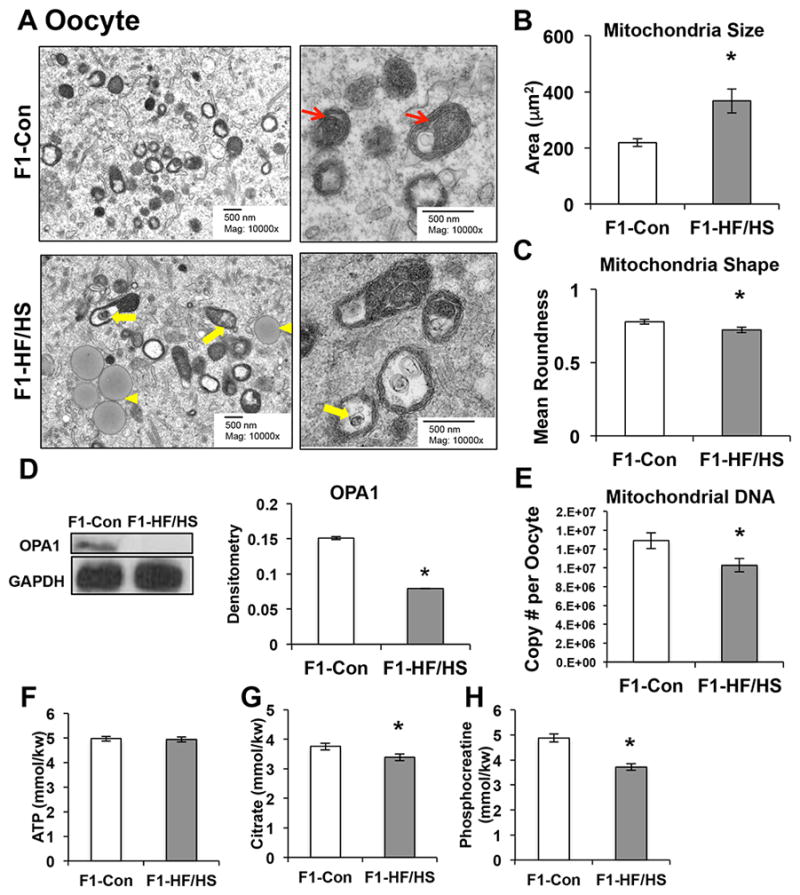
Oocytes were collected from eight-week old F1 mice. Mitochondrial morphologies were analyzed from TEM images (A-C). Representative low-magnification TEM images of mitochondria for F1-Con and F1-HF/HS oocytes (A: Left). High-magnification (A: Right) images depict normal mitochondria in F1-Con (upper right panel) and abnormal mitochondria (lower right panel) in F1-HF/HS oocytes. Yellow arrowheads depict lipid droplet, yellow arrows denote abnormal inner mitochondrial membrane morphology, and red arrows indicate normal circular cristae. TEM image analysis for mitochondrial size (B) and mean roundness (C). Western blot analysis of OPA1 (D) via western blotting. Mitochondrial DNA copy number (20 single oocytes, n = 6 mice) (E). mice) ATP (F), citrate (G), and phosphocreatine (H) from 60 oocytes, n = 4 per group. All data are expressed as mean ± SEM. *P < 0.05 by Student’s T-test.
Given these findings and those observed in the skeletal muscle of F1-HF/HS mice, we hypothesized that expression of mitochondrial dynamics proteins would be altered in the oocytes of F1-HF/HS mice. Consistent with this hypothesis, F1-HF/HS oocytes expressed significantly less OPA1 protein than F1-Con oocytes (Fig. 3D). Chronic OPA1 depletion has been shown to significantly reduce mitochondrial DNA (mtDNA) copy number (Cogliati et al., 2013), which may impair cellular metabolism independent of changes to mitochondrial morphology. Thus, it was not surprising that F1-HF/HS oocytes had significantly reduced mtDNA copy number (Fig. 3E), intracellular citrate (Fig. 3G), and phosphocreatine (Fig. 3H). However, ATP levels were not affected (Fig. 3F).
Our findings in this section suggest that maternal HF/HS diet prior to and during gestation promotes mitochondrial dysfunction in F1 female offspring tissues. These defects are transmitted to the F1 oocytes, which have abnormal mitochondrial morphologies, decreased OPA1 expression, and impaired mitochondrial metabolism.
F2- and F3-HF/HS offspring inherit defects in mitochondrial dynamics that penetrate through the female germline and somatic tissue
The drastic reduction in OPA1 protein levels in F1 oocytes led us to suspect that programing of impaired mitochondrial dynamics proteins may occur in the F2 generation. Accordingly, the mitochondria in F2-HF/HS lateral gastrocnemius muscle had almost identical morphologic phenotypes as observed in the F1-HF/HS gastrocnemius (compare Fig. 4A to Fig. 2F). Specifically, we observed enlarged mitochondria (S4) containing aberrant IMMs and matrix that was significantly less electron dense than in F2-Con muscle (Fig. 4A). Additionally, we observed lower DRP1 activity and OPA1 expression in F2-HF/HS muscle than in F2-Con muscle (Fig. 4B). Similarly, we observed a significant reduction in all the electron transport chain complexes in F2-HF/HS lateral gastrocnemius muscle (Fig. 4C), suggesting a reduction in complex assembly leading to impaired cristae formation. Furthermore, F2-HF/HS oocytes had significantly lower levels of total DRP1, P-DRP1 (ser6161), and OPA1 protein than F2-Con oocytes (Fig. 4D), indicating that mitochondrial dynamics were likely impaired in the F2-HF/HS oocytes.
Figure 4. Altered mitochondrial morphology and dynamics and complex proteins are transferred to F3-HF/HS mice through the F2-HF/HS female germline.

Lateral gastrocnemius muscle was isolated from eight-week old F2 (A–C) and F3 (D–G) offspring. Oocytes were collected from the F2 mice (D). Representative transmission electron microscopy (TEM) images of lateral gastrocnemius muscle (F2: A, F3: E). Star indicates mitochondria undergoing fission, and arrows denote abnormal inner mitochondrial membrane morphology. Western blot analysis DRP1, P-DRP1(ser616), and OPA1 in lateral gastrocnemius muscle (F2: B, F3: F) and F2-oocytes (C) and electron transport chain complex content (CI = NDUFB8, CII = total complex, CIII = Core protein 2, CIV = subunit I and CV = alpha subunit) (F2:C, F3:G). All data are expressed as mean ± SEM. *P < 0.05 by Student’s T-test.
If the mitochondrial effects we observed were truly transgenerational, we would detect them in the mice whose great grandmothers had diet-induced metabolic syndrome (F3-HF/HS). Indeed, we found that the lateral gastrocnemius muscle of F3-HF/HS mice had significantly larger mitochondria than did that of F3-Con mice (Fig. 4E and S5). However, the IMM phenotype was less severe in F3-HF/HS than in the F1 and F2 generations. However, cristae formation appeared abnormal, and in some cases cristae were lacking in F3-HF/HS muscle mitochondria (Fig. 4E). Levels of both total DRP1 and P-DRP1 (ser616) were lower in F3-HF/HS than in F3-Con muscle, but the ratio of P-DRP1 to total DRP1 was not affected (Fig. 4F). This reduced level of DRP1 likely contributed to the enlarged mitochondria phenotype (S4). Finally, OPA1 protein levels were lower in F3-HF/HS than in F3-Con lateral gastrocnemius muscle, even though the IMM morphology was not as malformed as it was in the F1- and F2-HF/HS muscle (Fig. 4E). Consistent with a milder IMM phenotype in the F3-HF/HS offspring, we did not observe any differences in the electron transport chain complexes between F3-Con and F3-HF/HS offspring (Fig. 4G). Finally, it is possible to hypothesize that the mitochondrial phenotype was merely a result of inherited hyperinsulinemia, however these mitochondrial defects occurred in the absence of altered circulating insulin (0.33 ± 0.09 and 0.35 ± 0.05, F3-Con and F3-HF/HS respectively).
Together, these findings indicate that maternal metabolic syndrome programs defects in glycolytic muscle fiber mitochondria that are characterized by impaired fission and decreased OPA1 expression that persists across three generations of offspring and is transmitted through the female germline.
DISCUSSION
Here, we used an inbred mouse model to examine the impact of maternal metabolic syndrome on health of the ensuing generations. We found that, despite consuming a normal diet, the first, second, and third generations of female offspring developed mitochondrial dysfunction and abnormal mitochondrial morphology in their skeletal muscle. Moreover, the mitochondrial dynamic proteins DRP-1 and OPA1 were mis-expressed in the skeletal muscles of F1, F2, and F3 mice, suggesting an imbalance of fission and fusion. Mitochondrial abnormalities were also present in the germ cells of F1 and F2 females, suggesting that transmission of the mitochondrial phenotype occurred through the maternal germline.
Transgenerational inheritance occurs when an environmental exposure alters the germline epigenome, resulting in altered gene expression that persists through generations that were not directly exposed (i.e., F3) (Skinner, 2011). A great deal of evidence supports the notion that early life exposure to maternal obesity alters the nuclear epigenome of the exposed offspring and may promote metabolic disease (Zheng et al., 2014). However, given that mitochondrial DNA is inherited from the mother, effects of maternal obesity on germline mitochondria must also be considered as a mechanism for transmission of metabolic dysfunction. Although epigenetic modifications in mtDNA have not been explored in this context, two types of epigenetic modifications have been detected in mtDNA. First, studies in mouse oocytes demonstrated that methylation occurring at both CpG and non-CpG sites in mtDNA positively correlates with gene expression (Kobayashi et al., 2012). Second, the recently discovered epigenetic mark hydroxymethylated cytosine (5hmC) is widely distributed in all types of tissues with varying degrees of abundance and the highest density of 5hmC was found in the mtDNA (Sun et al., 2013). Although we did not examine the epigenetic mechanisms leading to inheritance of abnormal mitochondrial phenotypes, our data showing altered mitochondrial metabolism, morphology, and OPA1 expression in offspring germ cells suggest that both modification of nuclear-encoded mitochondrial genes and altered mtDNA play a role.
Maintaining normal IMM structure is essential for mitochondrial function, as abnormal cristae formation is associated with decreased membrane potential (Bornhovd et al., 2006), altered metabolite diffusion, and impaired regulation of apoptosis (Mannella et al., 2013). Although the molecular regulators of IMM topology are still being uncovered, it is clear that OPA1 plays a dominant role (Mannella et al., 2013). Here, decreased OPA1 expression in F1-, F2-, and F3-HF/HS offspring may explain the bloated cristae phenotype, as ablation of OPA1 leads to decreased mtDNA copy number, disorganized cristae (Frezza et al., 2006), and impaired respiratory chain supercomplex assembly (Cogliati et al., 2013). Moreover, studies from Cogliati et al. showed a functional relationship between respiratory chain supercomplex assembly and respiratory capacity that are negatively affected by acute OPA1 depletion (Cogliati et al., 2013), suggesting a link between decreased oxygen consumption and lower OPA1 levels in F1-HF/HS skeletal muscle. Our findings that the F1 and F2-HF/HS exposed offspring have altered expression of numerous complex subunits supports the idea that impaired supercomplex assembly is an underlying mechanism driving the extreme cristae phenotype in these mice. Importantly, mutations in mtDNA, mtDNA depletion, or epigenetic modification to mtDNA can lead to altered levels of complex assembly, suggesting a key role for the inheritance of aberrant mitochondria in programming mitochondrial dysfunction throughout generations.
Despite its importance, the decreased OPA1 level cannot account for all of the changes to mitochondrial morphology we observed in HF/HS offspring. Whereas OPA1 knockdown causes extensive mitochondrial fragmentation (Frezza et al., 2006), we observed enlarged mitochondria, suggesting that fission may also be impaired. Consistent with this idea, cells lacking both OPA1 and Drp1 have large mitochondria with disrupted cristae structures or onion-like inner membrane structures (Otera et al., 2010). We detected decreased activation of Drp1 in F1- and F2-HF/HS muscle and decreased total Drp1 protein in F3-HF/HS muscle, suggesting a reduction in mitochondrial fission. Fission is essential for targeting and degrading damaged mitochondria through mitophagy (Rambold et al., 2011), and yeast lacking the homologues of both Drp1 and OPA1 (Dnm1 and Mgm1, respectively) have decreased mitophagy and a shorter replicative lifespan (Bernhardt et al., 2015). Moreover, the mitophagy proteins PINK and Parkin promote mitochondrial fission (Poole et al., 2008), suggesting a complex crosstalk between mitophagy and mitochondrial fission. Interestingly, impaired Parkin action results in a similar mitochondrial phenotype observed in this study enlarged (Drew et al., 2014). Although we did not address whether or not mitophagy was altered following exposure to maternal metabolic syndrome, this may be one mechanism by which damaged mitochondria are passed from one generation to the next. Future work will be focused on addressing this possibility.
In conclusion, our study demonstrates that early life exposure to maternal metabolic syndrome programs impaired mitochondrial health that is transmitted transgenerationally through the female germline. Importantly, the F1, F2, and F3 animals in our study all consumed control diets. In humans, in which the diets of children closely parallel those of their parents, the effects of maternal metabolic syndrome may be greater than in this mouse model.
EXPERIMENTAL PROCEDURES
Mouse breeding scheme, feeding paradigm, and metabolic analysis
All procedures in this study were approved by the Animal Studies Committee at Washington University School of Medicine and conformed to National Institutes of Health guidelines. Four-week-old female C57Bl/6 mice were fed either a high-fat/ high-sugar (HF/HS) (Test Diet 58R3; 59% fat, 26% carbohydrates [17% sucrose] and 15% protein) or standard chow (Con) (PicoLab Rodent diet 20; 13% fat, 62% carbohydrates [3.2% sucrose] and 25% protein for six weeks (Boudoures et al., 2016; Reynolds et al., 2015). Con- and HF/HS-fed F0 mice were mated with chow-fed male mice to produce the first generation. All offspring (first generation (F1), second generation (F2), and third generation (F3)) were weaned onto chow diets and mated with chow-fed males. Serum, skeletal muscle (soleus and lateral gastrocnemius), and germinal vesicle (GV)-stage oocytes were collected in non-mated F1, F2, and F3 eight-week old mice as described in supplemental experimental procedures. Denuded GV oocytes were pooled from three animals per litter for each dietary exposure. Skeletal muscle and oocytes from 6–8 litters were used for the experiments described below. All metabolic parameters were determined as described in supplemental experimental procedures.
Protein Isolation and Western Blotting
Lateral gastrocnemius tissue lysates from individual offspring bore to F0-Con or F0-HF/HS dams (n = 5–6 per group) were prepared in RIPA buffer. Oocyte proteins were prepared by boiling a pool of 200 GV oocytes (from 3 offspring per litter, from 4 litters per group) for five minutes in 2X laemmli buffer (Bio-Rad #161-0737) plus 2-Mercaptoethanol (Sigma). Western blotting was performed following standard procedures. See supplemental experimental procedures for details.
Transmission Electron Microscopy
Soleus, lateral gastrocnemius, and GV-stage oocytes were fixed and stained following standard protocols. See supplemental experimental procedures for details. Ten images from six offspring selected from six litters per group were analyzed by three independent, blinded observers for the average mitochondrial size, mitochondrial shape, and the number of lipid droplets per image.
Hydroxyacyl-coenzyme A dehydrogenase, type II (HADHA) Enzymatic Activity
Soleus muscles were collected from eight offspring selected from eight litters per group and homogenized with a glass homogenizer in enzyme extraction buffer (20mM Pi Buffer pH 7.4 (16mM Na2HPO4 Dibasic/4mM NaH2PO4 Monobasic), 0.02% BSA fraction V protease free, 0.5mM EDTA pH 7.0, 5mM 2-Mercaptoethanol, and 25% Glycerol). HADHA enzyme activity was measured by enzyme-linked cycling assays as previously described (Henriksson et al., 1986).
Metabolite Microanalytic Assays
GV-stage oocytes (pooled from 3 offspring per litter, from 4 litters per group) were frozen on a glass slide by dipping in isopentane equilibrated with liquid nitrogen. After freeze-drying overnight under vacuum at 35 °C, the oocytes were extracted in a nanoliter volume under oil. Adenosine triphosphate [ATP], citrate, and phosphocreatine were measured by performing an enzyme-linked assay as previously described (Chi et al., 1988).
MDA Quantification
Soleus lipid peroxidation was assessed using the OxiSelect TBARS Assay Kit (Cell Biolabs, Inc, San Diego, CA) per manufacturer’s instructions.
Preparation of permeabilized muscle fibers and high-resolution respirometry
Soleus muscle fibers (n = 4 offspring from 4 separate litters per group) were isolated and permeabilized with saponin. Mitochondrial respiration was measured using an Oxygraph 2K (OROBOROS Instruments, Innsbruck, Austria). Cytochrome C was used as a control for permeabilization. See supplemental experimental procedures for details.
mtDNA copy number in GV oocytes
Quantitative real-time PCR was used to determine mitochondrial DNA (mtDNA) abundance in single oocytes. See supplemental experimental procedures for details.
Statistical methods
Data are expressed as means ±SEM. All experiments were performed in triplicate. Differences between control and experimental values were compared using appropriate parametric and non-parametric tests for continuous variables, including the student’s t-test for normally distributed data and the Mann Whitney U test for non-normally distributed data. Significance was defined as P <0.05.
Supplementary Material
Highlights.
Inbred mice fed a high fat/high sucrose (HF/HS) diet develop metabolic syndrome
F1, F2, and F3 offspring from HF/HS fed dams develop mitochondrial dysfunction
Proteins involved in mitochondrial dynamics and ETC are misexpressed in F1-F3 pups
Mitochondrial changes are seen in F1-F2 oocytes suggesting germline transmission
Acknowledgments
Grants supporting this research: KHM: R01HD065435, JLS: T32HD049305
The authors thank Deborah J. Frank, PhD, Obstetrics and Gynecology, Washington University in St. Louis, for critical reading and editing of this manuscript. We acknowledge the Diabetes Research Center, Diabetes Models Phenotyping Core, for the serum metabolite analysis and body composition analysis (Supported by NIH grant P30 DK020579); and the Nutrition Obesity Research Center, Adipocyte Biology and Molecular Nutrition Core (Supported by NIH grant P30 DK056341) for the studies measuring mitochondrial respiration using the OROBOROS instrument. We also would like to thank the molecular microbiology imaging facility for the Electron Microscopy service.
Footnotes
Disclosure Statement: The authors have nothing to disclose
AUTHOR CONTRIBUTIONS
Conceptualization, J.L.S and K.H.M.; Methodology, J.L.S. and K.H.M.; Investigation, J.L.S., Z.A.A., A.T., A.D., W.Z., M.M.C., and S.S.; Writing – Original Draft, J.L.S.; Writing – Review & Editing, J.L.S. and K.H.M.; Funding Acquisition, K.H.M; Resources, K.H.M; Supervision, J.L.S and K.H.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernhardt D, Muller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015;5:7885. doi: 10.1038/srep07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D, Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One. 2012;7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhovd C, Vogel F, Neupert W, Reichert AS. Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J Biol Chem. 2006;281:13990–13998. doi: 10.1074/jbc.M512334200. [DOI] [PubMed] [Google Scholar]
- Boudoures AL, Chi M, Thompson A, Zhang W, Moley KH. The effects of voluntary exercise on oocyte quality in a diet-induced obese murine model. Reproduction. 2016;151:261–270. doi: 10.1530/REP-15-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen EM, Renault KM, Norgaard K, Nilas L, Jensen JE, Hyldstrup L, Michaelsen KF, Cortes D, Pryds O. Newborn regional body composition is influenced by maternal obesity, gestational weight gain and the birthweight standard score. Acta Paediatr. 2014;103:939–945. doi: 10.1111/apa.12713. [DOI] [PubMed] [Google Scholar]
- Chi MM, Manchester JK, Yang VC, Curato AD, Strickler RC, Lowry OH. Contrast in levels of metabolic enzymes in human and mouse ova. Biol Reprod. 1988;39:295–307. doi: 10.1095/biolreprod39.2.295. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. Mitochondrial efficiency and insulin resistance. Front Physiol. 2014;5:512. doi: 10.3389/fphys.2014.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, et al. HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes. 2014;63:1488–1505. doi: 10.2337/db13-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152:2228–2236. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. Jama. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Galloway CA, Yoon Y. Mitochondrial morphology in metabolic diseases. Antioxid Redox Signal. 2013;19:415–430. doi: 10.1089/ars.2012.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Vassena R, Chi MM, Potireddy S, Sutovsky M, Moley KH, Sutovsky P, Latham KE. Role of glucose in cloned mouse embryo development. Am J Physiol Endocrinol Metab. 2008;295:E798–809. doi: 10.1152/ajpendo.00683.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J, Chi MM, Hintz CS, Young DA, Kaiser KK, Salmons S, Lowry OH. Chronic stimulation of mammalian muscle: changes in enzymes of six metabolic pathways. Am J Physiol. 1986;251:C614–632. doi: 10.1152/ajpcell.1986.251.4.C614. [DOI] [PubMed] [Google Scholar]
- Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology. 2010;151:4039–4046. doi: 10.1210/en.2010-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, Schedl T, Moley KH. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7:e49217. doi: 10.1371/journal.pone.0049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella CA, Lederer WJ, Jafri MS. The connection between inner membrane topology and mitochondrial function. J Mol Cell Cardiol. 2013;62:51–57. doi: 10.1016/j.yjmcc.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Varuzhanyan G, Pham AH, Chan DC. Mitochondrial Dynamics Is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015 doi: 10.1016/j.cmet.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect. 2015;4:R1–R15. doi: 10.1530/EC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrin CM, Kelly GE, Tremblay RE, Kelleher CC. Body mass index and height over three generations: evidence from the Lifeways cross-generational cohort study. BMC Public Health. 2012;12:81. doi: 10.1186/1471-2458-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzio BF, Carvalho MH, Fortes ZB, do Carmo Franco M. Implications of maternal nutrient restriction in transgenerational programming of hypertension and endothelial dysfunction across F1-F3 offspring. Life Sci. 2012;90:571–577. doi: 10.1016/j.lfs.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Lippincott-Schwartz J. Fuse or die: Shaping mitochondrial fate during starvation. Commun Integr Biol. 2011;4:752–754. doi: 10.4161/cib.17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds KA, Boudoures AL, Chi MM, Wang Q, Moley KH. Adverse effects of obesity and/or high-fat diet on oocyte quality and metabolism are not reversible with resumption of regular diet in mice. Reprod Fertil Dev. 2015 doi: 10.1071/RD14251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Res C Embryo Today. 2011;93:51–55. doi: 10.1002/bdrc.20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Wu N, Wang S, Gao M, Song P, Lou J, Tan Y, Liu K. Transgenerational impaired male fertility with an Igf2 epigenetic defect in the rat are induced by the endocrine disruptor p,p′-DDE. Hum Reprod. 2014;29:2512–2521. doi: 10.1093/humrep/deu208. [DOI] [PubMed] [Google Scholar]
- Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, Guan S, Wang H, Sun D, Cheng X, Zhu Z, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–576. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanvig M. Offspring body size and metabolic profile - effects of lifestyle intervention in obese pregnant women. Dan Med J. 2014;61:B4893. [PubMed] [Google Scholar]
- Wakai T, Harada Y, Miyado K, Kono T. Mitochondrial dynamics controlled by mitofusins define organelle positioning and movement during mouse oocyte maturation. Mol Hum Reprod. 2014;20:1090–1100. doi: 10.1093/molehr/gau064. [DOI] [PubMed] [Google Scholar]
- Zheng J, Xiao X, Zhang Q, Yu M. DNA methylation: the pivotal interaction between early-life nutrition and glucose metabolism in later life. Br J Nutr. 2014;112:1850–1857. doi: 10.1017/S0007114514002827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.