

Abstract
The potent and selective KOR antagonist JDTic was derived from the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine class of pure opioid antagonists. In previous studies we reported that compounds that did not have a hydroxyl on the 3-hydroxyphenyl group and did not have methyl groups at the 3- and 4-position of the piperidine ring were still potent and selective KOR antagonists. In this study we report JDTic analogs 2, 3a-b, 4a-b, and 5, where the 3-hydroxyphenyl ring has been replaced by a 2-, 3-, or 4-pyridyl or 3-thienyl group and do not have the 3-methyl or 3,4-dimethyl groups, remain potent and selective KOR antagonists. Of these, (3R)-7-hydroxy-N-(1S)-2-methyl-[4-methyl-4-pyridine-3-yl-carboxamide (3b) had the best overall binding potency and selectivity in a [35S] GTPγS functional assay, with a Ke = 0.18 nM at the KOR and 273- and 16,700-fold selectivity for the KOR relative to the MOR and DOR, respectively. Calculated physiochemical properties for 3b suggest that it will cross the blood-brain barrier.
Keywords: JDTic, Opioids, Kappa antagonist, ADME properties
Graphical abstract
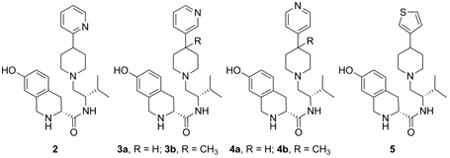
1. Introduction
During the last 15-20 years considerable effort has been devoted by academic institutions, research institutions, and pharmaceutical companies to the development of potent and selective kappa opioid receptor (KOR) antagonists (see ref 1 for a review).[1] Animal behavioral studies have suggested that potent and selective KOR antagonists have potential as pharmacotherapies for treating mood disorders such as depression, anxiety, and substance abuse (nicotine, alcohol, cocaine, and opiates).
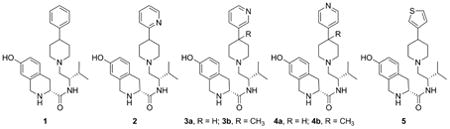
In our early studies we showed that (3R)-1,2,3,4-tetrahydro-7-hydroxy-N-[(1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl]-3-isoquinolinecarboxamide (JDTic) was a potent and selective KOR antagonist.[2, 3] In more recent studies we reported that JDTic analog 1 (Figure 1), where both the 3- and 4-methyl groups were removed from JDTic, was still a potent and selective KOR antagonist.[4] In addition we reported that JDTic analogs with the hydroxyl group removed from the 4-(3-hydroxyphenyl) moiety were still potent and selective KOR antagonists.[5] The fact that 1 is a potent and selective KOR antagonist suggest that replacement of the 4-phenyl group in 1 with heterocyclic group could also lead to potent and selective KOR antagonist. In this study we report the synthesis, [35S]GTPγS in vitro binding and calculated physiochemical properties of compounds 2, 3a–b, and 4a–b which have a 2-, 3-, or 4-pyridyl group or 5 which has a thiophene ring replacing the phenyl ring in previously reported JDTic analogs (Figure 1).
Figure 1.

Structures of JDTic, 1, 2, 3a-b, 4a-b, and 5.
2. Chemistry
The known piperidines 9a,[6] 9b,[7] 9c,[8] and 9d were prepared according to the route shown in Scheme 1, affording material consistent with literature characterization. Beginning with vinyl triflate 6, Negishi or Suzuki cross-coupling afforded the pyridyl and thienyl tetrahydropyridine derivatives 7a–d. Catalytic hydrogenation yielded the saturated N-Boc-piperidines 8a–d. Treatment of 8a–d with hydrogen chloride in dioxane deprotected the Boc to afford the amine hydrochloride salts 9a–d, which were used directly in the next step.
Scheme 1a.

aReagents: a) Suzuki or Negishi coupling; b) H2, Pd/C; c) 4M HCl in dioxane, CH3CN.
Amines 9e–f were prepared according to the route shown in Scheme 2. Knoevenagel condensation of the acetylpyridines 10a–b with ethyl cyanoacetate using ammonium acetate and acetic acid followed by condensation with 2-cyanoacetamide gave the intermediates 11a–b. Hydrolysis and decarboxylation of 11a–b using sulfuric acid gave glutaric acids which were not isolated. Upon heating the crude diacids neat with urea as a melt, the glutarimides 12a–b were produced. Reduction of the imides with diborane in tetrahydrofuran afforded piperidines 9e–f.
Scheme 2a.

aReagents: a) (1) Ethyl cyanoacetate, NH4OAc, HOAc, (2) NaOEt, 2-cyanoacetamide; b) (1) H2SO4, (2) urea; c) BH3•S(CH3)2, THF.
Coupling of piperidines 9a–f with N-Boc-L-valine using 3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC•HCl) with catalytic N-hydroxybenzotriazole (HOBt) or 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) in acetonitrile with triethylamine afforded the desired Boc-protected amides, which were deprotected with 4M hydrogen chloride in dioxane or hydrochloric acid in aqueous methanol and were directly reduced with diborane or borane dimethylsulfide in tetrahydrofuran to yield the amines 13a–f (Scheme 3). The amines 13a–f were coupled with (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic (7-hydroxy-Boc-D-Tic-OH) using EDC•HCl with a catalytic amount of HOBt in dichloromethane to give the Boc-protected intermediates. The desired final products 2, 3a–b, 4a–b, and 5 resulted from treatment of an acetonitrile solution of the Boc-intermediates with 4M hydrogen chloride in dioxane.
Scheme 3a.

aReagents: a) (1) N-Boc-L-valine, EDC•HCl, HOBt, Et3N, CH3CN, (2) HCl, (3) BH3, THF; b) (1) 7-hydroxy-Boc-D-Tic, EDC•HCl, HOBt, Et3N, CH2Cl2, (2) 4M HCl in dioxane.
3. Results and Discussion
In order to determine the effect of KOR potency and selectivity by replacing the phenyl ring in previously reported 1 with a 2-, 3-, or 4-pyridyl group or a thiophene ring, compounds 2, 3a–b, 4a–b, and 5 were synthesized and first evaluated at 10 μM for intrinsic activity in the [35S]GTPγS binding assay at all three opioid receptors. As none of these compounds displayed measurable intrinsic activity at this concentration, the compounds were evaluated for antagonist potency and selectivity at the opioid receptors. Compounds 3b and 4b were synthesized and tested for their KOR [35S]GTPγS binding potency in order to determine the effect of adding a 4-methyl group to 3a and 4a.
Compounds 2, 3a, and 4a with Ke values at the KOR of 0.43, 0.51, and 0.51 nM, respectively, were highly potent KOR antagonists but were slightly less potent than 1 which had a Ke = 0.051 nM at the KOR (Table 1). Similar to 1, all three compounds were highly selective for the KOR relative to the DOR. Compounds 2, 3a, and 4a were 33-, 89-, and 112-fold selective for the KOR relative to the MOR, compared to 77-fold selectivity for KOR relative to MOR for 1.
Table 1. Inhibition of agonist-stimulated [35S]GTPγS binding in cloned human μ, δ, and κ opioid receptors.
| |||||
|---|---|---|---|---|---|
| Compound | Ke (nM)a | μ/κ | δ/κ | ||
| μ, DAMGO | δ, DPDPE | κ, U69,593 | |||
| JDTic | 25 ± 4 | 74 ± 2 | 0.02 ± 0.01 | 1,250 | 3,800 |
| 1 | 3.96 ± 1.1 | 281 ± 44 | 0.051 ± 0.01 | 77 | 5,510 |
| 2 | 14.5 ± 2.6 | 590 ± 130 | 0.43 ± 0.12 | 33 | 1,372 |
| 3a | 45.4 ± 15 | 1140 ± 70 | 0.51 ± 0.08 | 89 | 2,235 |
| 3b | 49.2 ± 14 | >3000 | 0.18 ± 0.01 | 273 | >16,700 |
| 4a | 57.1 ± 15 | 690 ± 250 | 0.51 ± 0.14 | 112 | 1,353 |
| 4b | 154 ± 56 | 2,510 ± 490 | 1.75 ± 0.65 | 88 | 1,434 |
| 5 | 3.93 ± 0.73 | 118 ± 27 | 0.12 ± 0.02 | 33 | 983 |
Ke values are the mean ± SEM of at least three independent experiments performed in duplicates.
The addition of a 4-methyl substituent to 3a to give 3b resulted in a significant increase in both KOR potency and selectivity for the KOR relative to both the MOR and DOR. Compound 3b has a Ke = 0.18 nM at the KOR and is 273- and >16,700-fold selective for the KOR relative to the MOR and DOR, respectively, whereas compound 3a has a Ke = 0.51 nM at the KOR and has 89- and 2,235-fold selectively for KOR relative to MOR and DOR, respectively. Compound 4b has a Ke = 1.75 nM at the KOR and is 88- and 1,434 fold selective for the KOR relative to the MOR and DOR, respectively.
Compound 5, which has a thiophene ring in place of a phenyl ring in 1 has a Ke value of 0.12 nM at KOR and is 32.5- and 983-fold selective for KOR relative to the MOR and DOR. Thus, 5 is less potent and less selective than 1.
In order to determine if the compounds would be predicted to cross the blood/brain barrier, their topological polar surface area (TPSA), clogP, and derived logBB values were calculated and compared to JDTic and 1 (Table 2). In general, CNS compounds that have TPSA values less than 76 Å2[9], clogP values in the range of 2-4[10], and derived logBB values greater than -1[11] are predicted to cross the blood/brain barrier. Compounds 3a–b and 4a–b have TPSA values of 77.49 Å2 compared to 84.83 Å2 for JDTic. Compound 5 has a TPSA value of 64.6, which is identical to that of 1. JDTic, 3a–b, 4a–b and 5 have clogP values in the range of 2–4. All of the compounds have logBB values greater than -1 and thus, would be predicted to penetrate the brain. In a study of marketed CNS drugs Wager and co-workers determined that 74% of CNS drugs displayed a CNS MPO (Multi Parameter Optimization) score greater than or equal to four.[12] All of the compounds in this study had a CNS MPO score greater than or equal to four. JDTic and 1 had CNS MPO scores of 3.1 and 3.8, respectively.
Table 2. Calculated physiochemical data for compounds 2, 3a–b, 4a–b, and 5 compared to JDTic and compound 1.
| Compound | TPSA | cLogP | ClarkBB | CNS MPO |
|---|---|---|---|---|
| JDTic | 84.83 | 3.60 | -0.57 | 3.1 |
| 1 | 64.60 | 3.53 | -0.28 | 3.8 |
| 2 | 77.49 | 2.56 | -0.62 | 4.0 |
| 3a | 77.49 | 2.42 | -0.64 | 4.2 |
| 3b | 77.49 | 2.64 | -0.61 | 4.1 |
| 4a | 77.49 | 2.40 | -0.64 | 4.2 |
| 4b | 77.49 | 2.63 | -0.61 | 4.1 |
| 5 | 64.60 | 3.45 | -0.29 | 4.1 |
4. Conclusion
Compounds 2, 3a–b, 4a–b, and 5, which have either a pyridine or thiophene ring in place of the phenyl groups in JDTic analogs were synthesized and evaluted for their [35S] GTPγS binding potency at the MOR, DOR, and KOR. All of the compounds had subnanomolar Ke values at the KOR in the [35S] GTPγS binding assay except 4b which has a Ke = 1.75 nM at the KOR. Compounds 3a–b and 4a–b were as selective or more selective for the KOR relative to the MOR than previously reported 1. All of the compounds were highly selective for the KOR relative to the DOR. Compound 3b with a Ke = 0.18 nM at the KOR with 273- and >16,700-fold selectively for the KOR relative to the MOR and DOR respectively, had the best overall antagonist potency and selectivity in the [35S] GTPγS binding assays. The calculated logBB values suggest that the compounds will penetrate the blood-brain barrier and the calculated CNS MPO values are above 4 and thus are in the range for potential useful CNS drugs.
5. Materials and Methods
Melting points were determined using a MEL-TEMP II capillary melting point apparatus and are uncorrected. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained on a Varian Avance DPX-300 MHz NMR spectrometer or a Bruker Unity Inova 500 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) with reference to internal solvent. Mass spectra (MS) were run on a Perkin-Elmer Sciex API 150 EX mass spectrometer equipped with APCI (atmospheric pressure chemical ionization) or ESI (turbospray) sources or on a Hewlett Packard 5989A instrument by electron impact. Elemental analyses were performed by Atlantic Microlab Inc., Atlanta, GA. Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Analytical thin-layer chromatography (TLC) was carried out using EMD silica gel 60 F254 TLC plates. TLC visualization was achieved with a UV lamp or in an iodine chamber. Flash column chromatography was done on a CombiFlash Companion system using ISCO prepacked silica gel columns or using EM Science silica gel 60A (230-400 mesh). Solvent system: CMA80=80:18:2 CHCl3:MeOH:conc. NH4OH. Unless otherwise stated, reagent-grade chemicals were obtained from commercial sources and were used without further purification. All moisture- and air-sensitive reactions and reagent transfers were carried out under dry nitrogen.
5.1. (3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-pyridin-2-ylpiperidin-1-yl)methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (2) Trihydrochloride
To a solution of 13a (200 mg, 0.56 mmol) in dichloromethane (25 mL) was added (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (7-hydroxy-Boc-D-Tic) (176 mg, 0.60 mmol) and triethylamine (0.35 mL, 0.6 mmol) followed by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC•HCl) (230 mg, 1.2 mmol) and hydroxybenzotriazole (HOBt) (8.1 mg, 0.06 mmol). The reaction mixture was stirred overnight at ambient temperature and then treated with 20 mL of saturated sodium bicarbonate and extracted with 25 mL dichloromethane. The combined organic layers were dried over sodium sulfate, filtered and evaporated. The resulting residue was subjected to chromatography on silica eluting with CMA 80-chloroform (1:1) provided 225 mg (77%) of the Boc-intermediate as a white solid. This material was dissolved in acetonitrile and treated with HCl (4M in dioxane, 1 mL) and stirred for 5 min. The solvent was evaporated to afford a white precipitate which was subjected to chromatography on silica eluting with CMA 80-chloroform (1:1) to provide 60 mg (33%) of 2 as the free base: 1H NMR (300 MHz, METHANOL-d4) δ 8.81 (d, J = 5.27 Hz, 1H), 8.61 (s, 1H), 8.10 (d, J = 8.10 Hz, 1H), 8.00 (d, J = 6.59 Hz, 1H), 7.13 (d, J = 8.48 Hz, 1H), 6.78 (dd, J = 2.45, 8.48 Hz, 1H), 6.68 (d, J = 2.26 Hz, 1H), 4.18– 4.50 (m, 5H), 3.73 (br. s., 1H), 3.04–3.59 (m, 7H), 2.68–2.93 (m, 1H), 2.44–2.66 (m, 1H), 2.21–2.41 (m, 2H), 1.81–1.99 (m, 1H), 1.06 (s, 6H); 13C NMR (75 MHz, METHANOL-d4) δ 171.3, 159.2, 158.1, 148.2, 143.3, 131.2, 129.7, 126.9, 126.5, 121.9, 117.0, 113.7, 61.7, 57.9, 55.7, 52.4, 50.6, 45.5, 39.3, 32.3, 29.9, 29.0, 28.9, 19.9, 18.3; MS (ESI) m/z 423.5 (M+H)+. A solution of the free base in CH2Cl2 was treated with HCl (2 M in ether) to afford the trihydrochloride salt (2•3HCl): mp 225–229 °C (dec); [α]25D = +69.2 (c 1.08, MeOH). Anal. Calcd for C25H37Cl3N4O2•2H2O: C, 52.87; H, 7.28; N, 9.86. Found: C, 52.95; H, 7.21; N, 10.12.
5.2. (3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-pyridin-3-ylpiperidin-1-yl)methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (3a) Trihydrochloride
Compound 3a was prepared according to procedure analogous to that of 2 from (2S)-3-methyl-1-(4-pyridin-3-ylpiperidin-1-yl)butan-2-amine (13b) (200 mg, 0.56 mmol), which in turn was prepared from amine 9b according to procedure analogous to that of 13a, to afford 109 mg (60%) of the 3a free base. The free base was converted to the trihydrochoride salt (3a•3HCl): 1H NMR (300 MHz, METHANOL-d4) δ 8.90 (s, 1H), 8.81 (d, J = 5.65 Hz, 1H), 8.61–8.74 (m, 1H), 8.11 (dd, J = 5.93, 8.01 Hz, 1H), 7.11 (d, J = 8.29 Hz, 1H), 6.75 (dd, J = 2.26, 8.29 Hz, 1H), 6.67 (d, J = 2.07 Hz, 1H), 4.25– 4.48 (m, 4H), 4.17 (d, J = 11.68 Hz, 1H), 3.70 (d, J = 11.49 Hz, 1H), 2.99–3.55 (m, 8H), 2.57–2.77 (m, 1H), 2.33–2.56 (m, 1H), 2.17 (d, J = 9.42 Hz, 2H), 1.77–2.01 (m, 1H), 1.03 (t, J = 5.75 Hz, 6H); 13C NMR (126 MHz, METHANOL-d4) δ 171.2, 158.0, 146.8, 145.8, 141.7, 141.4, 131.2, 129.7, 128.7, 121.9, 116.9, 113.7, 61.5, 57.8, 56.1, 52.8, 50.5, 45.4, 37.9, 32.3, 30.0, 29.9, 19.9, 18.4; MS (ESI) m/z 423.5 (M+H)+; mp 223–227 °C; [α]25D = +70.0 (c 0.95, MeOH). Anal. Calcd for C25H37Cl3N2O2•1.5H2O: C, 53.72; H, 7.21; N, 10.02. Found: C, 53.57; H, 7.37; N, 9.83.
5.3. (3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-methyl-4-pyridin-3-ylpiperidin-1-yl)methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (3b) Trihydrochloride
Compound 3b was prepared according to procedure analogous to that of 2 from amine 13e (117 mg, 0.45 mmol) to afford the 3b freebase: 1H NMR (300 MHz, CHLOROFORM-d) δ 8.50–8.65 (m, 1H), 8.43 (dd, J = 1.32, 4.71 Hz, 1H), 7.54–7.71 (m, 1H), 7.19–7.33 (m, 1H), 7.11 (d, J = 9.42 Hz, 1H), 6.82–6.97 (m, 1H), 6.60 (dd, J = 2.35, 8.19 Hz, 1H), 6.38–6.51 (m, 1H), 4.03–4.18 (m, 1H), 3.71–3.81 (m, 2H), 3.38 (dd, J = 5.27, 10.17 Hz, 1H), 2.94 (dd, J = 5.27, 16.39 Hz, 1H), 2.20–2.81 (m, 7H), 1.99– 2.19 (m, 2H), 1.67–1.92 (m, 3H), 1.20–1.25 (m, 3H), 0.79–0.99 (m, 6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 173.2, 155.2, 147.6, 146.7, 144.3, 136.9, 133.9, 130.4, 125.0, 123.5, 114.1, 112.4, 60.0, 56.6, 50.3, 49.9, 47.5, 36.4, 36.2, 35.1, 31.2, 29.6, 19.1, 17.9. The free base was converted to the trihydrochloride salt affording 62.5 mg (25% over two steps) 3b•3HCl as a white powder: MS (ESI) m/z 437.7 (M+H)+, mp 215–219 °C (fusion), [α]25D +69. (c 0.10, CH3OH). Anal. Calcd for C26H39Cl3N4O2•H2O: C, 53.66; H, 7.45; N, 9.63. Found: C, 53.49; H, 7.41; N, 9.50.
5.4. (3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-pyridin-4-ylpiperidin-1-yl)methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4a) Trihydrochloride
Compound 4a was prepared according to procedure analogous to that of 2 from amine 13c (280 mg, 1.13 mmol) to afford 113 mg (24%) of 4a as the free base: 1H NMR (300 MHz, METHANOL-d4) δ 8.85 (d, J = 6.59 Hz, 2H), 8.12 (d, J = 6.59 Hz, 2H), 7.13 (d, J = 8.48 Hz, 1H), 6.78 (dd, J = 2.45, 8.29 Hz, 1H), 6.68 (d, J = 2.26 Hz, 1H), 4.30–4.45 (m, 4H), 4.23 (br. s., 1H), 3.71 (br. s., 1H), 3.04–3.54 (m, 7H), 2.63–2.87 (m, 1H), 2.37–2.61 (m, 1H), 2.08–2.32 (m, 2H), 1.91 (d, J = 6.59 Hz, 1H), 1.06 (t, J = 6.40 Hz, 6H); 13C NMR (75 MHz, METHANOL-d4) δ 171.3, 166.5, 158.1, 143.0, 131.2, 129.6, 127.3, 121.9, 117.0, 113.7, 61.7, 57.9, 56.1, 52.7, 50.5, 45.5, 40.9, 32.3, 29.9, 29.6, 29.5, 19.9, 18.3; MS (ESI) m/z 423.4 (M+H)+. The free base was converted to the trihydrochloride salt (4a•3HCl): mp 238–241 °C (dec); [α]25D = +60.0 (c 0.98, MeOH). Anal. Calcd for C25H37Cl3N4O2•H2O: C, 54.60; H, 7.15; N, 10.19. Found: C, 54.31; H, 7.16; N, 9.91.
5.5. (3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-methyl-4-pyridin-4-ylpiperidin-1-yl)methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4b) Trihydrochloride
Compound 4b was prepared according to procedure analogous to that of 2 from amine 13f (78 mg, 0.30 mmol) to afford the 4b freebase: 1H NMR (300 MHz, METHANOL-d4) δ 8.44 (dd, J = 1.60, 4.62 Hz, 2H), 7.43 (dd, J = 1.60, 4.62 Hz, 2H), 6.91 (d, J = 8.10 Hz, 1H), 6.52–6.68 (m, 1H), 6.48 (br. s., 1H), 3.79–4.16 (m, 3H), 3.44–3.65 (m, 1H), 2.72–3.00 (m, 2H), 2.63 (br. s., 1H), 2.19–2.56 (m, 5H), 1.94–2.17 (m, 2H), 1.65–1.91 (m, 3H), 1.05–1.35 (m, 4H), 0.75–1.04 (m, 6H); 13C NMR (75 MHz, METHANOL-d4) δ 175.4, 161.2, 156.8, 150.1, 137.3, 130.8, 125.6, 123.2, 114.9, 113.2, 61.4, 58.0, 52.5, 51.3, 51.2, 47.7, 37.5, 37.2, 37.1, 32.4, 32.1, 19.9, 17.9. The free base was converted to the trihydrochloride salt (4b•3HCl) affording 19.2 mg (11% over two steps) of a white powder: MS (ESI) m/z 437.5 (M+H)+; mp 220–224 °C (fusion); [α]25D +75. (c 0.21, CH3OH). Anal. Calcd for C26H39Cl3N4O2•3.25H2O: C, 51.66; H, 7.59; N, 8.85. Found: C, 51.88; H, 7.42; N, 9.27.
5.6 (3R)-7-Hydroxy-N-[(1S)-2-methyl-1-{[4-(3-thienyl)piperidin-1-yl]methyl}propyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (5)
Compound 5 was prepared according to procedure analogous to that of 2 from amine 13d (680 mg, 2.7 mmol) to afford the 5 free base as a viscous, colorless oil (220 mg, 19%): 1H NMR (300 MHz,CHLOROFORM-d) δ 7.35 (br. s., 1H), 7.14 (dd, J = 2.83, 4.71 Hz, 1H), 6.68–6.90 (m, 3H), 6.53 (d, J =7.91 Hz, 1H), 6.39 (br. s., 1H), 4.12 (br. s., 1H), 3.62 (br. s., 2H), 3.21–3.45 (m, 2H), 3.06 (br. s., 1H),2.84 (t, J = 15.16 Hz, 2H), 2.29–2.67 (m, 4H), 2.19 (br. s., 1H), 1.59–2.01 (m, 5H), 0.86 (d, J = 6.03 Hz,6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 172.5, 154.1, 144.8, 135.5, 129.3, 125.6, 124.6, 123.7, 118.2, 113.1, 111.3, 58.4, 55.8, 54.3, 51.4, 48.7, 46.7, 35.5, 30.4, 28.9, 18.2, 17.0. The free base was converted to the trihydrochloride salt (5•3HCl): MS (ESI) m/z 428.2 (M+H)+; mp 196–200 °C (fusion); [α]25D = +75.2 (c 1.0, MeOH). Anal. Calcd for C24H36Cl2N3O2S•H2O: C, 55.11; H, 7.23; N, 8.03.Found: C, 55.09; H, 7.00; N, 7.80.
5.7. tert-Butyl 4-thiophen-3-ylpiperidine-1-carboxylate (8d)
A mixture of 6 (3.2 g, 9.6 mmol), thiophene-3-boronic acid (1.72 g, 13.5 mmol), tetrakis(triphenylphosphine)palladium (1.1 g, 0.96 mmol), cesium carbonate (9.4 g, 28.6 mmol), dimethoxyethane (34 mL) and water (16 mL) was stirred at 80 °C under nitrogen overnight. The cooled mixture was partitioned between water and EtOAc. The aqueous was further extracted with EtOAc (100 mL). The combined organic layers were dried (Na2SO4), filtered and evaporated to obtain intermediate tert-butyl 4-thiophen-3-yl-3,6-dihydropyridine-1(2H)-carboxylate 7d: 1H NMR (300 MHz, CHLOROFORM-d) δ 7.10–7.26 (m, 2H), 7.05 (br. s., 1H), 5.98 (br. s., 1H), 3.99 (d, J = 2.64 Hz, 2H), 3.55 (t, J = 5.75 Hz, 2H), 2.44 (br. s., 2H), 1.36–1.46 (m, 9H). Intermediate 7d was hydrogenated with palladium hydroxide on carbon (1.0 g, 20 wt%) in ethanol under H2 (50 psi) at room temperature for 2 h. The solids were filtered and solvent evaporated to provide 1.25 g (35% over 2 steps) of 8d: 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17–7.23 (m, 1H), 6.80–6.97 (m, 2H), 3.92–4.27 (m, 2H), 2.71 (d, J = 12.06 Hz, 3H), 1.82 (br. s., 2H), 1.52 (br. s., 2H), 1.36–1.43 (m, 9H).
5.8. 4-Thiophen-3-ylpiperidine (9d)
A solution of 8d (1.25 g, 4.67 mmol) in acetonitrile (20 mL) was treated with 4M HCl in dioxane (4 mL) and the mixture stirred for 1 h at room temperature. The solvent was evaporated to obtain 0.86 g (90%) of 9d as the hydrochloride salt: 1H NMR (300 MHz, DMSO-d6) δ 8.66–9.67 (m, 2H), 7.40–7.59 (m, 1H), 7.14–7.28 (m, 1H), 6.97–7.11 (m, 1H), 3.14–3.39 (m, 2H), 2.81–3.06 (m, 3H), 1.93–2.12 (m, 2H), 1.67–1.91 (m, 2H).
5.9. 3-(4-Methylpiperidin-4-yl)pyridine (9e)
A solution of 12a (3.95 g, 19.3 mmol) in THF (50 mL) was treated with BH3•SMe2 (10 mL, 100 mmol) at 0°C, then heated to reflux for 12 h. The reaction mixture was quenched by addition of EtOAc, then methanol. The concentrated residue was dissolved in aq. HCl (2.0 M, 10 mL). The pH was adjusted with NaOH (2 M, 12 mL) and the resulting aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organics were washed with brine, dried (Na2SO4) and concentrated. The resulting residue was subjected to chromatography on silica gel eluting with a gradient up to 100% CMA80 in CH2Cl2 to afford 567 mg (17%) of piperidine 9e: 1H NMR (300 MHz, CHLOROFORM-d) δ 8.54–8.67 (m, 1H), 8.44 (d, J = 3.77 Hz, 1H), 7.56–7.71 (m, 1H), 7.18–7.33 (m, 1H), 3.57 (br. s., 1H), 2.89–3.04 (m, 1H), 2.74–2.89 (m, 1H), 1.52–2.70 (m, 7H), 1.28 (d, J = 7.72 Hz, 2H).
5.10. 4-(4-Methylpiperidin-4-yl)pyridine (9f)
Compound 9f was prepared according to procedure analogous to that of 9e from glutarimide 12b (652 mg, 3.2 mmol) to afford 100 mg (18%) of piperidine 9f: 1H NMR (300 MHz, CDCl3) δ 8.39–8.63 (m, 2H), 7.25 (dd, J = 1.9, 4.1 Hz, 2H), 2.86–3.02 (m, 2H), 2.71–2.86 (m, 2H), 1.93–2.12 (m, 2H), 1.60– 1.79 (m, 2H), 1.17–1.31 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 158.4, 149.7, 121.1, 42.6, 37.2, 36.6, 28.7; MS (ESI) m/z 177.3 (M+H)+.
5.11. 4-Methyl-2,6-dioxo-4-(pyridin-3-yl)piperidine-3,5-dicarbonitrile (11a)
A solution of 3-acetylpyridine (10a) (10.0 g, 82.5 mmol, ethyl cyanoacetate (9.33 g, 82.5 mmol) and ammonium acetate (1.3 g, 16.9 mmol) was refluxed in glacial acetic acid (4.0 mL) and benzene (16.5 mL) with a Dean-Stark apparatus for 12 h. Ethyl acetate was added to the cooled solution. The solution was washed with aq. NaHCO3 then brine, dried (Na2SO4) and concentrated. The residue was eluted through a plug of silica gel using ethyl acetate to afford 16.32 g (92%) of the Knoevenagel product. The oil was combined with 2-cyanoacetamide (7.36 g, 87.5 mmol) in a solution of NaOH (3.5 g, 87.5 mmol) in ethanol (60 mL). Within 5 minutes, a clear solution had formed which was left to stir for 12 h, after which aq. NaHSO4 (1 M, 90 mL) was added. The resulting solids were collected by filtration and the mother liquor was concentrated to approximately half the original volume of kept in a refrigerator overnight. A second filtration yielded more solids, which were combined with the first crop and dried under vacuum to afford 11.6 g (55% over two steps) of 11a: 1H NMR (300 MHz, DMSO-d6) δ 12.40 (br. s., 1H), 8.48–9.02 (m, 2H), 8.08 (d, J = 6.59 Hz, 1H), 7.57 (dd, J = 4.71, 7.91 Hz, 1H), 5.42 (s, 2H), 1.72 (br. s., 3H).
5.12. 4-Methyl-2,6-dioxo-4-(pyridin-4-yl)piperidine-3,5-dicarbonitrile (11b)
A solution of 4-acetylpyridine (10b) (10.0 g, 82.5 mmol), ethyl cyanoacetate (9.33 g, 82.5 mmol) and ammonium acetate (1.3 g, 16.9 mmol) was refluxed in glacial acetic acid (4.0 mL) and benzene (16.5 mL) with a Dean-Stark apparatus for 12 h. Ethyl acetate was added to the cooled solution. The solution was washed with aq. NaHCO3 then brine, dried (Na2SO4) and concentrated. The residue was eluted through a plug of silica gel using ethyl acetate to afford 13.76 g of an amber oil. The oil was combined with cyanoacetamide (5.35 g, 63.6 mmol) in methanol (200 mL) at 0 °C. Ammonia gas was bubbled in to the solution until it was saturated, then the dark red solution was left to warm to room temperature and stir 1 week. The solution was brought to reflux for 1 h, at which point the solution became cloudy. The solution was concentrated to a thick oil which was dissolved in EtOAc. Diethyl ether was slowly added, causing a precipitate to form. Filtration afforded 8.81 g (54%) of 11b as a pale yellow powder. 1H NMR (300 MHz, DMSO-d6) δ 9.39 (d, J = 4.1 Hz, 1H), 8.50 (ddd, J = 1.6, 3.2, 4.6 Hz, 2H), 7.20–7.60 (m, 2H), 4.31–4.74 (m, 1H), 1.28–1.79 (m, 3H); MS (ESI) m/z 253.3 (M–H)-.
5.13. 4-Methyl-4-(pyridin-3-yl)piperidine-2,6-dione (12a)
Sulfuric acid (6 mL) was slowly added to a slurry of 11a (11.6 g, 11.8 mmol) in water (6 mL). The resulting solution was refluxed for 12 h, cooled, quenched with 50% NaOH (6 mL), diluted with methanol (40 mL) then CH2Cl2 (160 mL). The white solids were filtered and washed with 10% methanol in CH2Cl2. The orange filtrate was concentrated to afford 11.03 g of crude material, which was combined with urea (7.1 g, 0.12 mol) and heated as a melt for 3 h until no further gas evolution was observed. The mixture was partitioned between aq. NaHCO3 and EtOAc, and the resulting emulsion was filtered through Celite. The aqueous layer was further extracted with EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. The resulting residue was eluted through a plug of silica gel using 5–7.5% MeOH in CH2Cl2 as the eluent. The concentrated residue, upon trituration with EtOAc, afforded 3.95 g (42% over two steps) of 12a as a solid: 1H NMR (300 MHz, DMSO-d6) δ 10.80 (br. s., 1H), 8.63 (d, J = 2.64 Hz, 1H), 8.36–8.52 (m, 1H), 7.74–7.90 (m, 1H), 7.37 (dd, J = 4.71, 8.10 Hz, 1H), 2.98–3.17 (m, 1H), 2.75–2.94 (m, 1H), 1.33 (br. s., 1H).
5.14. 4-Methyl-4-(pyridin-4-yl)piperidine-2,6-dione (12b)
Sulfuric acid (1.5 mL) was slowly added to a slurry of 11b (3.0 g, 11.8 mmol) in water (1.5 mL). The resulting solution was refluxed for 12 h, cooled, quenched with 50% NaOH (1.5 mL), diluted with methanol (10 mL) then CH2Cl2 (40 mL). The white solids were filtered and washed with 10% methanol in CH2Cl2. The orange filtrate was concentrated to afford 2.23 g (85%) of 3-methyl-3-(pyridin-4-yl)pentanedioic acid. 1H NMR (300 MHz, CD3OD) δ 8.23 – 8.62 (m, 2H), 7.30 – 7.62 (m, 2H), 2.70 – 3.01 (m, 4H), 1.58 (br. s., 3H). The 3-methyl-3-(pyridin-4-yl)pentanedioic acid (2.23 g, 10.0 mmol) was combined with urea (2.0 g, 33 mmol) and heated as a melt for 1 h. The resulting material was subjected to chromatography on silica gel. Although the product had Rf =0.3 in EtOAc, the solubility in pure EtOAc was limited, so a gradient up to 10% methanol in EtOAc was used to afford 652 mg (32%) of 12b as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.59 (dd, J = 1.60, 4.4 Hz, 2H), 7.25 (dd, J = 1.6, 4.4 Hz, 2H), 2.97–3.19 (m, 2H), 2.80 (d, J = 17.0 Hz, 2H), 1.45 (s, 3H); MS (ESI) m/z 205.3 (M+H)+.
5.15. (2S)-3-methyl-1-(4-pyridin-2-ylpiperidin-1-yl)butan-2-amine (13a)
A solution of 9a (4.66 g, 19.8 mmol) in acetonitrile (150 mL) was treated with N-Boc-L-valine (4.55 g, 21.0 mmol), HBTU (7.98 g, 21.0 mmol) and triethylamine (8.4 g, 84 mmol). The reaction mixture was stirred overnight at ambient temperature then evaporated. The residue was partitioned between sat. aq. NaHCO3 (100 mL) and EtOAc (100 mL). The aqueous was extracted further with EtOAc (2 × 100 mL). The combined organic layer was dried (Na2SO4) then evaporated to obtain the crude Boc intermediate which was subjected to silica gel chromatography eluting with a gradient up to 10% MeOH in CH2Cl2: MS (ESI) m/z 362.3 (M+H)+. The purified Boc intermediate was dissolved in acetonitrile (100 mL), treated with 4M HCl in dioxane (20 mL) and stirred for 2 h. The reaction was evaporated to obtain a solid which was subjected to chromatography on silica gel using a gradient of 10-50% CMA80 in CHCl3 to afford 5.5 g (83% over two steps) of the intermediate amide (2S)-3-methyl-1-oxo-1-(4-pyridin-2-ylpiperidin-1-yl)butan-2-amine as an off white solid: MS (ESI) m/z 262.2 (M+H)+. A solution of the intermediate amide (5.5 g, 21 mmol) in THF (40 mL) at 0 °C was treated with borane dimethyl sulfide complex (3.3 mL, 33 mmol). The reaction mixture was warmed to ambient temperature then heated at reflux overnight. The reaction was cooled in an ice bath, treated with 20 mL methanol then warmed to ambient temperature over 1 h. The resulting solution was again cooled in an ice bath, treated with 2M HCl in ether (100 mL) and then heated at reflux for 1 h. The solvent was evaporated and the residue was subjected to chromatography on silica gel eluting with a gradient of CMA80 in CHCl3 to afford 2.2 g (42%) of 13a as a white solid.
5.16. (2S)-3-methyl-1-(4-pyridin-4-ylpiperidin-1-yl)butan-2-amine (13c)
Compound 13c was prepared according to the general procedure of 13a from the amine 9c (1.42 g, 5.9 mmol) to afford 0.77 g (53% yield) of the desired 13c free base: 1H NMR (300 MHz, METHANOL-d4) δ 8.42–8.52 (m, 2H), 7.35–7.46 (m, 2H), 3.21 (q, J = 7.35 Hz, 2H), 3.00 (d, J = 11.11 Hz, 1H), 2.42– 2.77 (m, 3H), 2.19 (dt, J = 4.33, 10.83 Hz, 1H), 1.84–1.97 (m, 4H), 1.32 (t, J = 7.35 Hz, 2H), 0.98–1.11 (m, 6H).
5.17. (2S)-3-Methyl-1-[4-(thiophen-3-yl)piperidin-1-yl]butan-2-amine (13d)
A solution of 4-(thiophen-3-yl)piperidine (9d) (850 mg, 4.17 mmol) in CH3CN (30 mL) was treated with N-Boc-L-valine(1.08 g, 5.0 mmol), HBTU (1.9 g, 5.0 mmol) and NEt3 (1.5 g, 15 mmol). The reaction mixture was stirred overnight at ambient temperature. The concentrated residue was partitioned between sat. aq. NaHCO3 (15 mL) and EtOAc (15 mL). The aqueous layer was further extracted with EtOAc (2 × 15 mL). The combined organic layer was dried (Na2SO4), filtered, and evaporated. The residue was subjected to chromatography on silica gel eluting with 50% EtOAc in hexanes to afford the intermediate tert-butyl {(1S)-2-methyl-1-[(4-thiophen-3-ylpiperidin-1-yl)carbonyl]propyl}carbamate as a light yellow oil (1.34 g, 88%): 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20–7.38 (m, 1H), 6.89–7.07 (m, 2H), 5.40 (d, J = 9.04 Hz, 1H), 4.63–4.84 (m, 1H), 4.54 (br. s., 1H), 3.92–4.20 (m, 1H), 3.10–3.29 (m, 1H), 2.90 (t, J = 11.68 Hz, 1H), 2.65–2.80 (m, 1H), 1.85–2.18 (m, 3H), 1.52–1.77 (m, 1H), 1.45 (s, 9H), 0.82–1.07 (m, 7H). The Boc-intermediate was dissolved in CH3CN (10 mL) then treated with HCl (4 N in dioxane, 10 mL). After 1 h at ambient temperature, the solution was evaporated to afford a quantitative yield (1.09 g) of the (2S)-3-methyl-1-oxo-1-(4-thiophen-3-ylpiperidin-1-yl)butan-2-amine hydrochloride salt: 1H NMR (300 MHz, DMSO-d6) δ 8.21 (d, J = 11.30 Hz, 3H), 7.38–7.58 (m, 1H), 7.21 (d, J = 2.83 Hz, 1H), 6.96–7.14 (m, 1H), 4.50 (d, J = 12.62 Hz, 1H), 4.27 (br. s., 1H), 4.04 (d, J = 10.36 Hz, 1H), 3.17 (t, J = 12.43 Hz, 1H), 2.91 (t, J = 10.46 Hz, 1H), 2.75 (t, J = 12.53 Hz, 1H), 1.81–2.14 (m, 3H), 1.30–1.78 (m, 2H), 0.83–1.09 (m, 6H); MS (ESI) m/z 267.2 (M+H)+. The intermediate amide was dissolved in THF (25 mL) then treated with borane dimethyl sulfide complex (1.8 mL, 18 mmol) at 0 °C. The reaction mixture was warmed to ambient temperature and stirred overnight. The reaction mixture was refluxed for 2 h, cooled in ice, treated with methanol (20 mL) and stirred for 1 h at ambient temperature. The reaction mixture was cooled in ice and treated with 2M hydrogen chloride in ether (40 mL) then refluxed overnight. The solvent was evaporated and the resulting residue was subjected to chromatography on silica gel eluting with a gradient up to 30% CMA80 in CHCl3 to afford 680 mg (75%) of 13d as a white solid: 1H NMR (300 MHz, CHLOROFORM-d) d 7.13–7.25 (m, 1H), 6.82–6.98 (m, 2H), 4.91 (br. s., 2H), 2.78–3.03 (m, 3H), 2.48–2.62 (m, 1H), 2.36–2.45 (m, 2H), 2.29 (dt, J = 2.54, 11.54 Hz, 1H), 1.53–2.07 (m, 6H), 1.03 (d, J = 6.78 Hz, 3H), 0.93 (d, J = 6.78 Hz, 3H); 13C NMR (75 MHz, CHLOROFORM-d) d 146.9, 126.8, 125.3, 118.9, 59.7, 55.6, 54.4, 52.9, 37.4, 33.3, 32.9, 30.3, 19.3, 18.8; MS (ESI) m/z 253.2 (M+H)+.
5.18. (2S)-3-Methyl-1-(4-methyl-4-pyridin-3-ylpiperidin-1-yl)butan-2-amine (13e)
A solution of 9e (152 mg, 0.86 mmol) and Boc-L-valine (230 mg, 1.06 mmol) in acetonitrile (30 mL) was treated with EDC•HCl (340 mg, 1.8 mmol) and triethylamine (0.7 mL, 5 mmol) and stirred 12 h. The concentrated residue was partitioned between EtOAc and water. The organic layer was washed (aq. NaHCO3 then brine) then dried (Na2SO4). The concentrated residue was eluted from silica gel using ethyl acetate to afford an oil which was dissolved in methanol (10 mL) and treated with HCl (12 N, 2 mL), stirred 1 h and then concentrated. The residue was subjected to chromatography on silica gel eluting with a gradient up to 50% CMA80 in CH2Cl2 to afford 201 mg (84% over two steps) of the intermediate amide: 1H NMR (300 MHz, CHLOROFORM-d) δ 8.64 (br. s., 1H), 8.40–8.57 (m, 1H), 7.55–7.74 (m, 1H), 7.18–7.39 (m, 1H), 1.60–4.27 (m, 12H), 1.33 (d, J = 11.87 Hz, 3H), 0.83–1.11 (m, 6H); MS (ESI) m/z 276.5 (M+H)+. A solution of the intermediate amide (201 mg, 0.73 mmol) in THF (3 mL) was treated with BH3•THF (1 M, 10 mL) then heated to reflux overnight. The reaction mixture was quenched with methanol and concentrated. The residue was subjected to chromatography on silica gel eluting with 25% CMA80 in CH2Cl2 to afford 117 mg (62%) of the desired 13e: 1H NMR (300 MHz, CHLOROFORM-d) δ 8.55 (s, 1H), 8.39–8.50 (m, 1H), 7.95 (d, J = 7.91 Hz, 1H), 7.50 (dd, J = 5.93, 7.82 Hz, 1H), 4.56 (br. s., 2H), 3.58–3.72 (m, 1H), 2.73–2.96 (m, 1H), 2.49–2.73 (m, 2H), 2.21–2.50 (m, 3H), 1.95–2.21 (m, 2H), 1.54–1.94 (m, 3H), 1.15–1.42 (m, 3H), 0.72–1.11 (m, 6H).
5.19. (2S)-3-Methyl-1-[4-methyl-4-(pyridin-4-yl)piperidin-1-yl]butan-2-amine (13f)
Compound 13f was prepared according to procedure analogous to that of 13e from amine 9f (100 mg, 0.57 mmol) to afford 78 mg (52% over three steps) of 13f: 1H NMR (300 MHz, CDCl3) δ 8.53 (dd, J = 2.3, 4.0 Hz, 2H), 7.25 (d, J = 3.2 Hz, 2H), 2.33–2.76 (m, 4H), 1.96–2.33 (m, 7H), 1.67–1.88 (m, 2H), 1.41–1.61 (m, 1H), 1.15–1.27 (m, 3H), 0.81–0.99 (m, 6H).
5.20. [35S]GTPγS assay
The [35S]GTPγS assays were conducted using the methods previously reported.[13]
5.21. Calculated pharmacokinetic properties
The topological polar surface area (TPSA) and calculated lipophilicity (clogP) values were calculated using the ChemAxon Instant JChem package. Predictions of the logarithm of the in vivo blood-brain ratio (logBB) were based on the Clark and Pickett model (Eq.3).[11, 14]
Supplementary Material
Acknowledgments
This research was supported by the National Institute on Drug Abuse Grant No. DA09045. We thank Tiffany Langston and Keith Warner for conducting the in vitro testing.
Abbreviations
- [35S]GTPγS
sulfur-35 guanosine-5′-O-(3-thio)triphosphate
- DAMGO
[D-Ala2,MePhe4,Gly-ol5]enkephalin
- DPDPE
[d-Pen2,d-Pen5]enkephalin
- U69
593, (5α,7α,8β)-(-)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide
Footnotes
Supplementary data: Supplementary data ssociated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carroll FI, Carlezon J, William A. J Med Chem. 2013;56:2178. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2003;46:3127. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 3.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2001;44:2687. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 4.Carroll FI, Gichinga MG, Kormos CM, Maitra R, Runyon SP, Thomas JB, Mascarella SW, Decker AM, Navarro HA. Bioorg Med Chem. 2015;23:6379. doi: 10.1016/j.bmc.2015.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kormos CM, Gichinga MG, Maitra R, Runyon SP, Thomas JB, Brieaddy LE, Mascarella SW, Navarro HA, Carroll FI. J Med Chem. 2014;57:7367. doi: 10.1021/jm5008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel MV, Kolasa T, Mortell K, Matulenko MA, Hakeem AA, Rohde JJ, Nelson SL, Cowart MD, Nakane M, Miller LN, Uchic ME, Terranova MA, El-Kouhen OF, Donnelly-Roberts DL, Namovic MT, Hollingsworth PR, Chang R, Martino BR, Wetter JM, Marsh KC, Martin R, Darbyshire JF, Gintant G, Hsieh GC, Moreland RB, Sullivan JP, Brioni JD, Stewart AO. J Med Chem. 2006;49:7450. doi: 10.1021/jm060662k. [DOI] [PubMed] [Google Scholar]
- 7.Cid JM, Tresadern G, Vega JA, de Lucas AI, Matesanz E, Iturrino L, Linares ML, Garcia A, Andres JI, Macdonald GJ, Oehlrich D, Lavreysen H, Megens A, Ahnaou A, Drinkenburg W, Mackie C, Pype S, Gallacher D, Trabanco AA. J Med Chem. 2012;55:8770. doi: 10.1021/jm3010724. [DOI] [PubMed] [Google Scholar]
- 8.Atkins RJ, Banks A, Bellingham RK, Breen GF, Carey JS, Etridge SK, Hayes JF, Hussain N, Morgan DO, Oxley P, Passey SC, Walsgrove TC, Wells AS. Org Process Res Dev. 2003;7:663. [Google Scholar]
- 9.Summerfeld SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. J Pharmacol Exp Ther. 2007;322:205. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- 10.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. ACS Chem Neurosci. 2012;3:50. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark DE. J Pharm Sci. 1999;88:815. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 12.Wager TT, Hou X, Verhoest PR, Villalobos A. ACS Chem Neurosci. 2010;1:435. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll FI, Chaudhari S, Thomas JB, Mascarella SW, Gigstad KM, Deschamps J, Navarro HA. J Med Chem. 2005;48:8182. doi: 10.1021/jm058261c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark DE, Pickett SD. Drug Discov Today. 2000;5:49. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.