

Abstract
Two [18F]fluoroalkyl substituted amino acids differing only by the presence or absence of a methyl group on the α-carbon, (S)-2-amino-7-[18F]fluoro-2-methylheptanoic acid ((S)-[18F]FAMHep, (S)-[18F]14) and (S)-2-amino-7-[18F]-fluoroheptanoic acid ((S)-[18F]FAHep, (S)-[18F]15), were developed for brain tumor imaging and compared to the well-established system L amino acid tracer, O-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET), in the delayed brain tumor (DBT) mouse model of high-grade glioma. Cell uptake, biodistribution, and PET/CT imaging studies showed differences in amino acid transport of these tracer by DBT cells. Recognition of (S)-[18F]15 but not (S)-[18F]14 by system L amino acid transporters led to approximately 8–10-fold higher uptake of the α-hydrogen substituted analogue (S)-[18F]15 in normal brain. (S)-[18F]15 had imaging properties similar to those of (S)-[18F]FET in the DBT tumor model while (S)-[18F]14 afforded higher tumor to brain ratios due to much lower uptake by normal brain. These results have important implications for the future development of α-alkyl and α,α-dialkyl substituted amino acids for brain tumor imaging.
Graphical abstract
INTRODUCTION
Radiolabeled amino acid tracers are known to have superior brain tumor imaging properties compared to the most widely used PET tracer in oncology, the glucose analogue 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG).1–3 This advantage is due to a combination of lower uptake in the normal brain of amino acid tracers compared to [18F]FDG and the upregulation of amino acid transporters in many gliomas and other neoplasms. Natural and many non-natural amino acids enter cells through transport mediated by specific cell membrane proteins. Over 20 amino acid transporters, which differ in terms of their preferred substrates, tissue expression, and mechanisms of transport, have been identified in mammalian cells. Certain amino acid transporters are upregulated in human cancers, making them useful targets for molecular imaging and therapy.4–7
The system L family of amino acid transporters has received the most attention for brain tumor imaging. System L (so named from leucine preferring) is a major nutrient transport system responsible for the cellular uptake of large neutral branched and aromatic amino acids including L-leucine, L-phenylalanine, L-tyrosine, L-tryptophan, and L-methionine. There are four known members of the system L amino acid transporter family: LAT1, LAT2, LAT3, and LAT4. Important properties of system L transport for brain tumor imaging include activity at the luminal side of the normal blood–brain barrier (BBB) and upregulation in gliomas. Thus, unlike conventional contrast agents used for computed tomography and magnetic resonance imaging (MRI), system L substrates are able to reach the entire brain tumor volume even when the BBB is not disrupted. This property of amino acids targeting system L is important because many gliomas, including the most aggressive form glioblastoma, have tumor regions with intact BBB that do not enhance with conventional contrast and are not accessible to PET tracers that do not cross the BBB.
The L-type amino acid transporters LAT1 and LAT2 are sodium-independent transporters that function through an exchange mechanism which couples influx of extracellular amino acids with efflux of intracellular amino acids in a 1:1 ratio. In contrast to LAT1 and LAT2, LAT3 and LAT4 function through a facilitated diffusion mechanism. Because system L transporters cannot directly concentrate their substrates, tumor to brain ratios of system L selective tracers are limited, in comparison with tracers that are substrates of transport systems that can concentrate substrates intracellularly. Despite this inherent limitation, LAT1 is an important imaging and therapeutic target because of increased expression in many human neoplasms including gliomas, and increased LAT1 levels are associated with decreased survival.4,8–10 LAT1 also mediates transport of large neutral amino acids across the BBB into the brain.11–13
Currently, a number of PET and single photon emission computed tomography (SPECT) tracers targeting system L transporters have been used for human brain tumor imaging. These include O-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET), 6-[18F]fluoro-3,4-dihydroxy-L-phenylalanine ([18F]FDOPA), L-[11C]methionine ([11C]MET), and 3-[123I]iodo-α-methyl-L-tyrosine ([123I]IMT) as shown in Figure 1.1,3,14–18 The vast majority of radiolabeled amino acids targeting system L contain aromatic rings. [11C]MET does not have an aromatic substituent, but the 20 min half-life of carbon-11 presents logistical challenges for batch production and remote distribution. In contrast, the 110 min half-life of fluorine-18 is suitable for this purpose as exemplified by the success of [18F]FDG for clinical oncologic imaging.
Figure 1.
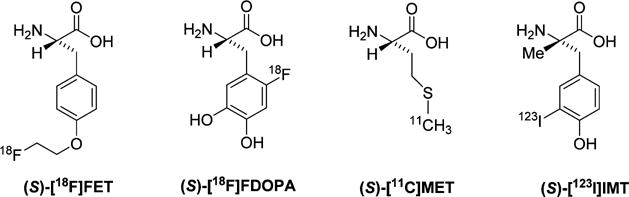
Selected radiolabeled amino acids that undergo system L amino acid transport.
As shown in Figure 1, three of the radiolabeled system L substrates have a hydrogen on the α-carbon as it is the case for the standard amino acid substrates for system L. However, the substitution of the α-carbon hydrogen with an α-methyl group for amino acids with aromatic side chains is reported to be well-tolerated by system L. For example (S)-[123I]IMT has been shown to be a good system L substrate in vitro.19 Recently, Wiriyasermkul et al. showed that compounds with an α-methyl group such as L-3-[18F]fluoro-α-methyl tyrosine ([18F]FAMT, also abbreviated as [18F]FMT) were transported by LAT1 but not LAT2,20 while tracers like [11C]MET and [18F]FET were transported by both LAT1 and LAT2. By comparing the selectivity of a few compounds for LAT1 versus LAT2, the authors concluded that the α-methyl group was responsible for the selectivity for LAT1 versus LAT2.8,13,21–23
Recently, we developed a new α,α-dialkylated amino acid tracer, (S)-2-amino-5-[18F]fluoro-2-methylpentanoic acid ((S)-[18F]FAMPe), which contains an α-methyl group substitution as shown in Figure 2.24 Our primary goal in the prior work was to develop an amino acid based tracer that crosses the BBB via system L transport and also undergoes uptake by other amino acid transporters to provide higher tumor to brain ratios than can be achieved with system L selective substrates. (S)-[18F]FAMPe uses a mixture of the system L and other neutral amino acid transporters that recognize glutamine to enter delayed brain tumor (DBT) cells, a mouse cell line that is a model for high grade glioma.25 (S)-[18F]FAMPe showed very high uptake by DBT tumors in biodistribution and PET imaging studies. Additionally, comparisons of the biological properties of the (S)- and the (R)-enantiomers indicate that the (S)-enantiomer is a better tracer for brain tumor imaging, which is consistent with reports for other amino acid (AA) tracers including (S)-[18F]FET.26–28 Although tumor uptake was high, the normal brain uptake of (S)-[18F]FAMPe was relatively low, and system L transport was a minor route of uptake of this compound in vitro by DBT glioma cells.
Figure 2.
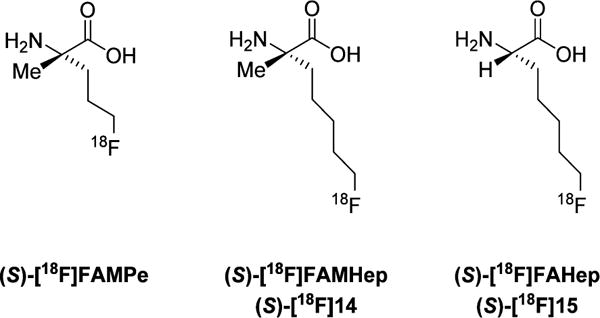
18F-labeled fluoroalkyl amino acids.
In order to develop analogues with increased brain availability and to further elucidate the structure–activity relationship for this class of fluorinated α,α-dialkyl amino acids, we subsequently developed two analogues of (S)-[18F]FAMPe with a longer fluoroalkyl side chain. These analogues, (S)-2-amino-7-[18F]fluoro-2-methylheptanoic acid ((S)-[18F]FAMHep ((S)-[18F]14) and (S)-2-amino-7-[18F]-fluoroheptanoic acid ((S)-[18F]FAHep, (S)-[18F]15), have two more carbons in their side chain than (S)-[18F]FAMPe, and their structures are shown in Figure 2. (S)-[18F]14 was designed as a substrate for system L amino acid transporter with more brain availability than (S)-[18F]FAMPe due to its longer alkyl chain. However, the lack of system L transport in vitro and the unexpectedly low brain uptake observed in vivo with (S)-[18F]14 prompted us to subsequently synthesize (S)-[18F]15 to determine whether the fluoroalkyl group or the α-methyl group was responsible for the low brain uptake.
In this study, the novel 18F-labeled amino acids (S)-[18F]14 and (S)-[18F]15 were synthesized and evaluated through cell uptake assays as well as through in vivo biodistribution and PET/CT imaging studies in the mouse DBT model of high grade glioma. The biodistribution and imaging properties of these new tracers were compared to the established system L amino acid, (S)-[18F]FET, which has been used extensively for brain tumor imaging in patients. Although (S)-[18F]14 and (S)-[18F]15 differ only by the substitution at the α-carbon (α-methyl vs α-hydrogen substitution, respectively), our results demonstrate that these compounds differ markedly in terms of system L transport by mouse DBT cells in vitro and normal brain uptake in vivo. The α-hydrogen substituted analogue (S)-[18F]15, but not the α-methyl substituted analogue (S)-[18F]14, was a selective system L substrate in vitro with corresponding high uptake in normal brain, consistent with system L transport at the BBB in vivo. These findings have important implications for the design of α-fluoroalkyl substituted amino acids and may also be relevant to α-alkyl substituted aromatic amino acids.
RESULTS AND DISCUSSION
1. Chemistry
The labeling precursors for (S)-[18F]14 and (S)-[18F]15 were obtained through a multistep synthesis as shown in Schemes 1 and 2. Commercially available starting material Fmoc-(S)-2-(4-pentenyl)alanine was used for the synthesis of the α-methyl Boc-protected amino ester precursor (S)-4 for the radiosynthesis of (S)-[18F]14. For the α-hydrogen substituted amino acid tracer (S)-[18F]15, synthesis of the corresponding precursor (S)-8 started with the commercially available (S)-2-amino-6-heptenoic acid.
Scheme 1.
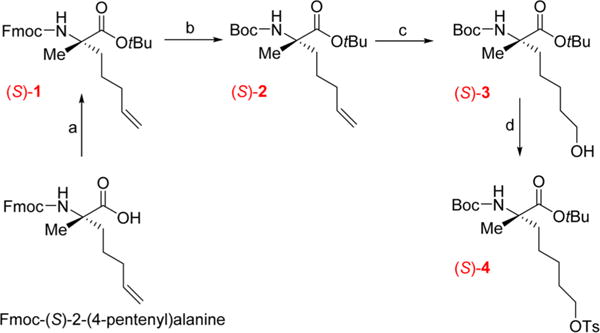
Multistep Synthesis of the Radiolabeling Precursor (S)-4a
aReagents and conditions: (a) tert-butyl 2,2,2-trichloroacetimidate, CH2Cl2, rt, 18 h; (b) Boc2O, KF, NEt3, DMF, rt, 18 h; (c) BH3·THF, 0 °C, 2 h then 1 M NaOH, 30% H2O2, 0 °C, rt, 18 h; (d) TsCl, NaOtBu, CH2Cl2, 0 °C then rt 18 h.
Scheme 2.
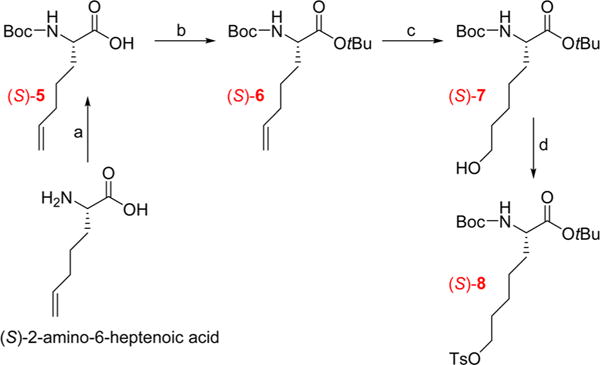
Multistep Synthesis of the Radiolabeling Precursor (S)-8a
aReagents and conditions: (a) Boc2O, MeOH/NEt3/NaOH 1 M for 18 h; (b) tert-butyl 2,2,2-trichloroacetimidate, CH2Cl2, rt, 18 h; (c) BH3·THF, 0 °C, 2 h then 1 M NaOH, 30% H2O2, 0 °C, rt, 18 h; (d) TsCl, NaOtBu, CH2Cl2, 0 °C then rt 18 h.
The first step to synthesize (S)-4 was the protection of the carboxylic acid of Fmoc-(S)-2-(4-pentenyl)alanine by a tert-butyl ester using tert-butyl 2,2,2-trichloroacetate in anhydrous dichloromethane at rt for 18 h to provide the full protected amino acid (S)-1 in a good yield (79%). In the second step, the Fmoc group of the protected amino acid (S)-1 was converted into a Boc group to allow simultaneous removal of all the protecting groups at the final step of the multistep synthesis.29 Thus, the Boc-amino ester, compound (S)-2, was obtained in good yield (78%) by successively adding potassium fluoride, trimethylamine, and di-tert-butyl dicarbonate to a stirred solution of the Fmoc protected compound (S)-1. The subsequent steps were based on the previously reported synthesis of (S)-[18F]FAMPe.24 Thus, the alkene on the alkyl side chain of the amino acid was converted into a terminal alcohol to obtain compound (S)-3 (54%). The alcohol group of (S)-3 was in turn converted into a good leaving group to provide the tosylate labeling precursor (S)-4 in moderate yield (36%) with some unreacted starting material remaining (32%).
To obtain precursor (S)-8, a similar procedure was used as for (S)-4, except that the two first protection steps were changed due to the use of the unprotected amino acid (S)-2-(4′-pentenyl)glycine as the starting material. The four-step synthesis shown in Scheme 2 afforded (S)-8 with similar yields to (S)-4.
As the R- and S-enantiomers of amino acids typically have distinct biological and imaging properties, establishing the enantiomeric purity of an amino acid tracer is important.18,30,31 For this purpose, the racemic nonradioactive forms of both amino acids (R,S)-14 and (R,S)-15 were prepared from glycine tert-butyl ester hydrochloride as shown in Scheme 3. These racemic compounds provided a reference standard containing equal amounts of both enantiomers for chiral high performance liquid chromatography (HPLC) separation and confirmation of the enantiomeric purity of the final products (S)-[18F]14 and (S)-[18F]15. From the α-imine ester (R,S)-9 obtained in quantitative yield during the first step, alkylation was conducted without further purification using lithium diisopropyl amide (LDA) and 1-bromo-5-fluoropentane to provide the tert-butyl (R,S)-2-((diphenylmethylene)amino)-7-fluoroheptanoate (R,S)-10, which served as the intermediate for (R,S)-14 as well as (R,S)-15. To prepare (R,S)-14, a second alkylation was performed with (R,S)-10 in the presence of methyl iodide which provided the α,α-dialkylated compound (R,S)-11. From the crude intermediates (R,S)-10 and (R,S)-11, a simple hydrolysis of the benzophenone imine in the presence of hydroxylamine hydrochloride provided the corresponding amino esters (R,S)-12 and (R,S)-13, in good yield and purity. Finally, the ester group of (R,S)-12 or (R,S)-13 was removed in the presence of hydrochloric acid to provide the corresponding final racemic nonradioactive product (R,S)-15 or (R,S)-14 in good yields (48% and 55% respectively).
Scheme 3.
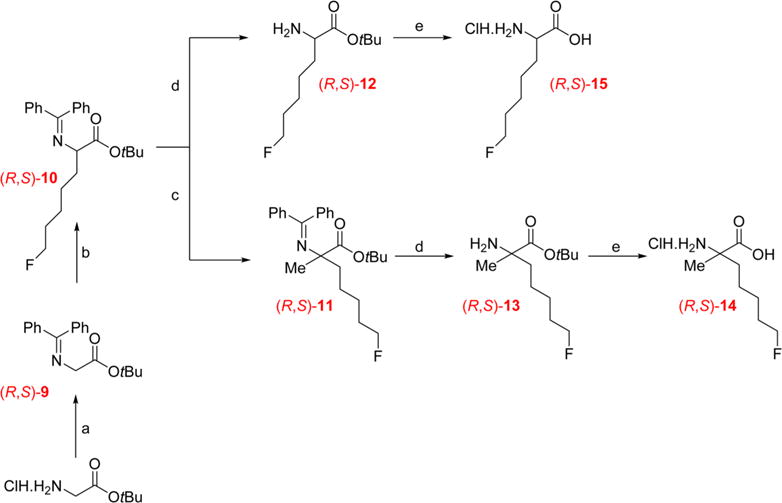
Multistep Synthesis of the Racemic Nonradioactive Amino Acids (R,S)-14 and (R,S)-15a
aReagents and conditions: (a) diphenylmethanimine, CH2Cl2, rt, 2 h; (b) LDA, 1-bromo-5-fluoropentane, THF, −78 °C, 1 h, then rt, 18 h; (c) LDA, methyl iodide, THF, −78 °C, 1 h, then rt, 18 h; (d) NH2OH, MeOH, rt, 2 h; (e) 4 M HCl, 60 °C, 3 h.
In addition to the racemic forms of 14 and 15, the nonradioactive (S)-enantiomers of both compounds were prepared starting from the respective precursors (S)-4 and (S)-8 in two steps as shown in Scheme 4. The selection of the (S)-enantiomers for this study was based on the observation in a prior study that (S)-FAMPe had superior tumor uptake and imaging properties in the DBT tumor model.24 First, the nucleophilic substitution of the tosylate precursors by fluoride was performed using cesium fluoride in the protic solvent 2-methylbutan-2-ol to provide the desired products (S)-16 and (S)-17 in good yields (73% and 75%).24,32 Then, deprotection in acidic conditions provided the nonradioactive amino acids (S)-14 and (S)-15 as hydrochloride salts in good yields (54 and 75% respectively).
Scheme 4.
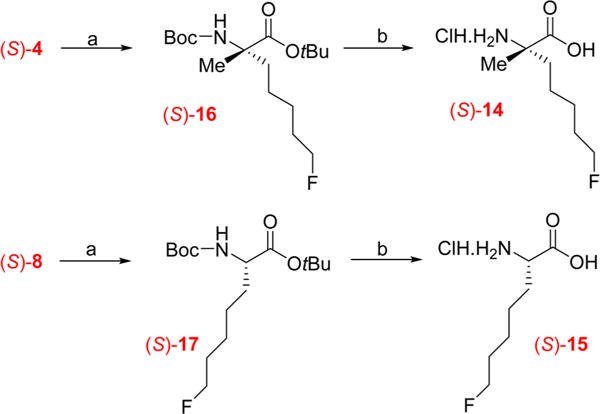
Syntheses of the (S)-Enantiomers of Nonradioactive Amino Acids (S)-14 and (S)-15a
aReagents and conditions: (a) CsF, 2-methylbutan-2-ol, 100 °C, 1 h; (b) 4 M HCl, 60 °C, 3 h.
2. Radiochemistry
The radiosyntheses of the two new tracers (S)-[18F]14 and (S)-[18F]15 were performed as shown in Scheme 5 based on the previously reported procedure for (S)-[18F]FAMPe with minor modifications.24
Scheme 5.
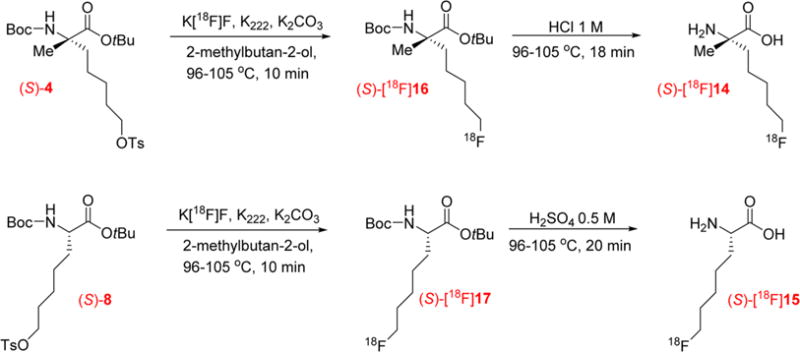
Two-Step Synthesis of the Amino Acid Tracers (S)-[18F]14 and (S)-[18F]15
The initially aqueous [18F]fluoride ion was azeotropically dried with acetonitrile in standard fashion in the presence of Kryptofix 222 (K222) and potassium carbonate (K2CO3). After the drying, nucleophilic [18F]fluorination was conducted with 3–4 mg of the appropriate precursor in 2-methylbutan-2-ol at 102–104 °C for 10 min. The [18F]fluoride incorporation for each precursor, (S)-4 or (S)-8, provided the corresponding intermediates (S)-[18F]16 or (S)-[18F]17 in good radio-chemical yield (>90%, n = 5) as estimated by radiometric thin-layer chromatography (radio-TLC). Then, acetonitrile and water were added directly to the reaction mixture to prepare the sample for purification by reverse-phase HPLC. The use of the C18 cartridge before the HPLC as described previously for FAMPe was eliminated.24 Omitting that step reduced the loss of the intermediate, (S)-[18F]16 or (S)-[18F]17, in the reaction vessel and increased the overall yield of final radiolabeled product. The collected fractions containing the desired 18F-labeled product eluted from the HPLC were then combined and passed through a light hydrophilic–lipophilic-balanced (HLB) cartridge that retained the intermediate (S)-[18F]16 or (S)-[18F]17 and allowed removal of the acetonitrile from the mobile phase. After elution of (S)-[18F]16 or (S)-[18F]17 with small volumes of ethanol, the deprotection was conducted in 1 M HCl for the α-methylated intermediate (S)-[18F]16 and in 0.5 M H2SO4 for the α-hydrogenated intermediate (S)-[18F]17 to provide (S)-[18F]14 or (S)-[18F]15, respectively. This change in the acid was made because the deprotection of (S)-[18F]17 in 1 M HCl produced a small radioactive impurity (~3% of the total activity present in the final solution) at early retention time which increased with longer deprotection time and increasing concentration of HCl (e.g., 18% impurity with 4 M HCl). By comparing the results of a deprotection conducted in 1 M HCl and 1 M H2SO4, we observed that, in the first case, deprotection was not complete whereas, with H2SO4, all the intermediate (S)-[18F]17 was deprotected but the radioactive impurity was still present in small amounts. However, when the concentration of H2SO4 was reduced 2-fold to 0.5 M, the radioactive impurity decreased to approximately 1%, and the deprotection of (S)-[18F]17 was still complete.
After deprotection, sterile water was added to the acidic reaction mixture, and the resulting solution was passed through a strong anion exchange cartridge, neutralizing the solution and providing the final products in a form and a pH suitable for the cell uptake studies. For the animal studies, the final product was diluted with 0.9% sodium chloride. At the end of the synthesis, the final products (S)-[18F]14 or (S)-[18F]15 were obtained in 47–83% radiochemical and in 22–37% uncorrected yields (n = 5), with a radiochemical purity over 97%. For (S)-[18F]14 as well as for (S)-[18F]15, less than 2% of the corresponding (R)-enantiomer was observed. During the [18F]fluoride incorporation step of the radiosynthesis of (S)-[18F]14, the α-carbon of the corresponding precursor (S)-4 and intermediate (S)-[18F] 16 cannot undergo racemization due to the presence of the α-methyl group; consequently the 2% of the (R)-enantiomer found in the end product arose from the starting material, Fmoc-(S)-2-(4-pentenyl)alanine (enantiomeric excess >97%) used for the synthesis of the precursor. In contrast, during radiosynthesis of (S)-[18F]15, both the precursor (S)-8 and the intermediate (S)-[18F]17 can potentially racemize during the [18F]fluorination step due to the presence of the α-hydrogen. The basic conditions and high temperatures used for the nucleophilic substitution with [18F]fluoride could reversibly deprotonate the α-carbon of the precursor, leading to racemization of the α-carbon of the protected amino acid. However, the small amount (approximately 2%) of (R)-[18F]15 observed at the end of synthesis also arose from the starting material, (S)-2-amino-6-heptenoic acid (enantiomeric excess >98%), indicating that minimal if any racemization occurred under these radiolabeling conditions. The radiochemical purity of the final 18F-labeled product was determined by analytical HPLC as shown in Figure 3 and by radio-TLC. As a representative example, from 46.8 mCi of [18F]fluoride after the drying process, 19.2 mCi of (S)-[18F]14 in ~2 mL solution was obtained at the EOS in approximately 120 min using ~4.4 mg of the tosylate precursor (S)-4.
Figure 3.
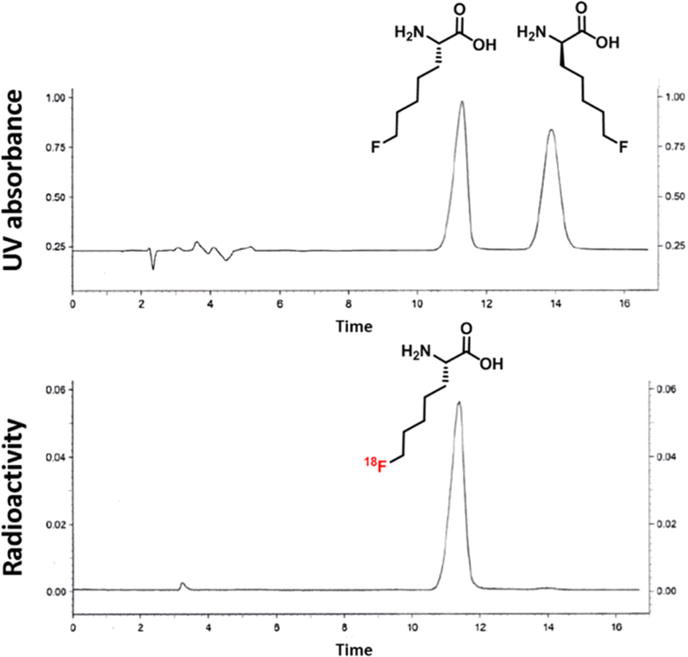
Analytical chiral HPLC coinjections of a mixture of the nonradioactive racemic amino acid (R,S)-15 (seen with UV detection) and (S)-[18F]15 (seen with radiometric detection).
In order to compare the biological properties of (S)-[18F]14 and (S)-[18F]15 to a well-established system L substrate used for brain tumor imaging, the radiosynthesis of the tyrosine analogue [18F]FET was conducted following the published method of Hamacher and colleagues with minor modifications as described below.33
The radiosynthesis was carried out in two steps starting from the commercially available precursor, O-(2-tosyloxyethyl)-N-trityl-L-tyrosine tert-butyl ester as shown in Scheme 6. Instead of using tetra-n-butyl ammonium hydrogen carbonate during the [18F]fluoride drying step, a mixture of K222 and K2CO3 as described for the synthesis of (S)-[18F]14 and (S)-[18F]15 was used. Then, the fluorination was conducted in acetonitrile at 104 °C for 5 min, which provided the [18F]fluorinated intermediate in good radiochemical yield as estimated by radio-TLC (>93%, n = 2). After evaporation of the reaction mixture solution, deprotection was conducted in 1,2-dichloroethane and trifluoroacetic acid for 2 min at rt and then at 80 °C for 5 min to provide the desired product (S)-[18F]FET. After dichloromethane was added to the crude reaction mixture, the solution was passed through a silica cartridge which was washed with a solution of diethyl ether/pentane as described in the published procedure. The tracer was then eluted with two aliquots of 1 mL of water rather than a warm sodium glycinate solution and then purified by HPLC using a different HPLC column: a Chirobiotic TAG column with a solution of 95:5 water:ethanol as the mobile phase which provided the tracer, (S)-[18F]FET, in a form suitable for animal studies. The HPLC eluate was passed through a 0.22 μm nylon filter and then diluted with 0.9% sodium chloride. The uncorrected end of synthesis as well as the radiochemical yields were good (35–52% and 60–82% respectively, n = 2). The radiochemical purity determined by the analytical HPLC and TLC was greater than 99%. The identity of the final product was confirmed by coinjection of the nonradioactive hydrochloride salt of (S)-FET and the final radiolabeled product (S)-[18F]FET on an analytical HPLC system.
Scheme 6.
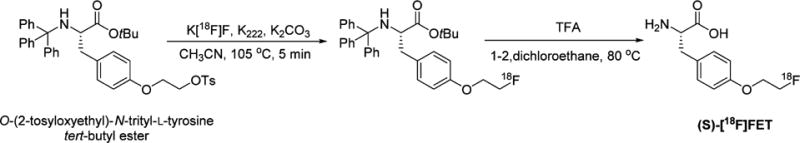
Radiosynthesis of (S)-[18F]FET
3. Cell Uptake Assays
The mechanisms of transport of the two novel amino acid tracers, (S)-[18F]14 and (S)-[18F]15, were assessed through in vitro uptake assays with mouse DBT glioma cells in the absence and presence of amino acid transport inhibitors, and the results are depicted in Figure 4. Three different competitive inhibition conditions were assessed: system A inhibition (alanine preferring, small neutral amino acids) using N-methyl α-aminoisobutyric acid (MeAIB), system L inhibition (leucine preferring, large neutral amino acids) using 2-aminobicyclo[2.2.1]heptane-2-carboxylic acid (BCH), and a broad range neutral amino acid inhibitory conditions consisting of the amino acids alanine, serine, and cysteine (ASC). The well-established inhibitors used for system A and system L were MeAIB and BCH, respectively. In each condition, the same concentration of inhibitor was used. In the case where no inhibitor was used (control conditions), sucrose was added in order maintain consistent osmolality between the different conditions. Some amino acid transporters including system A and system ASC are sodium-dependent, while other transporters, including system L, are sodium-independent. Consequently, assays were carried out both in the presence of sodium ions and in the absence of sodium ions with choline substituted for sodium. Additionally, to evaluate the initial influx of the amino acid tracers and minimize the potential for efflux which occurs with some amino acid transporters such as system L and system ASC, the uptake assays were performed using a short uptake time (60 s).
Figure 4.
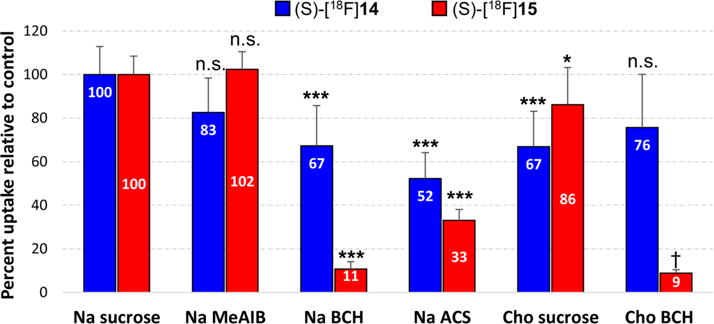
In vitro uptake of (S)-[18F]14 and (S)-[18F]15 in DBT glioma cells in the presence and absence of competitive inhibitors of amino acid transport. The uptake data are normalized based on the amount of activity added to each well and the total amount of protein in each well. The data are expressed as percentage uptake relative to the sodium control condition, and the values for each condition are noted in the appropriate bars. Each condition was assessed with 8 replicates and is depicted as the mean value with standard deviation. Na control and Cho control contain 10 mM sucrose to provide an osmolarity consistent with the inhibitory conditions. Na = assay buffer containing sodium ions; Cho = assay buffer containing choline ions; MeAIB = 10 mM N-methyl α-aminoisobutyric acid (system A inhibitor); BCH = 10 mM 2-amino-bicyclo[2.2.1]heptane-2-carboxylic acid (system L inhibitor); ASC = 3.3 mM each of L-Ala, L-Ser, L-Cys. p values associated with asterisks represent comparisons of uptake in the presence of inhibitor to the sodium control uptake for each radiotracer (1-way ANOVA) with Dunnett’s multiple comparison post-tests. *, p < 0.05; ***, p < 0.001. The choline sucrose control condition was compared to the choline BCH condition using two-tailed t tests (†, p < 0.0001).
In the presence of the system A inhibitor MeAIB, only a small amount of FAMHep uptake inhibition was observed (17 ± 16% reduction from control), which was not statistically significant. For FAHep, no inhibition by MeAIB was observed. These data indicate that these two amino acid tracers did not use system A to enter DBT cells, as expected due to the long fluoroalkyl side chains of these compounds. In the presence of BCH, a competitive antagonist of system L, only 33 ± 18% of the uptake of (S)-[18F]14 was inhibited relative to the sodium control (p < 0.001), suggesting that only a portion of (S)-[18F] 14 uptake is mediated by system L. However, as BCH can also inhibit other amino acid transporters including the sodium-dependent B0,+ and B0 transport systems, and as the system L amino acid transporter is sodium-independent, an assessment of BCH inhibition of uptake performed in the absence of sodium ions is more specific for system L transport. Comparison of the uptake of (S)-[18F]14 in the presence and absence of BCH in the sodium-free choline condition did not demonstrate inhibition of (S)-[18F]14 uptake by BCH. These results indicate that, despite its long fluoroalkyl side chain, (S)-[18F]14 was not an effective substrate for in vitro system L transport in DBT cells. In contrast, the cell uptake assays demonstrated that (S)-[18F]15 was an excellent substrate for in vitro system L transport. In the presence of sodium, BCH inhibited 89 ± 3% of the uptake of (S)-[18F]15 (p < 0.001); under sodium-free choline conditions, BCH inhibited 90% of the uptake of (S)-[18F]15 (90 ± 2% reduction relative to choline control, p < 0.0001).
In the presence of a mixture of alanine, serine, and cysteine, used as inhibitors of a broad range of neutral amino acid transporters including system ASC, 48 ± 12% and 77 ± 5% of the uptake of (S)-[18F]14 and (S)-[18F]15 were respectively inhibited (p < 0.001 for each tracer). The lack of inhibition of (S)-[18F]14 uptake by MeAIB but the substantial inhibition by ASC conditions suggests that neutral amino acid transport systems other than system L and system A mediate a portion of the uptake of this tracer by DBT cells. Substituting choline for sodium inhibited only 14 ± 17% of (S)-[18F]15 uptake (p < 0.05), indicating that the uptake of (S)-[18F]15 is predominantly sodium-independent. 33 ± 16% of the uptake of (S)-[18F]14 was inhibited by substituting sodium ions by choline (p < 0.001), indicating a minor sodium-dependent component of transport.
These cell uptake data showed that (S)-[18F]14 and (S)-[18F]15, which differ structurally only by the presence of an α-methyl versus a, α-hydrogen group, have very different mechanisms of in vitro transport by DBT cells, which is consistent with the results of studies in DBT tumor-bearing mice described in sections 4 and 5. The α-hydrogen substituted amino acid, (S)-[18F]15, was a very good substrate for system L, with an in vitro transport profile very similar to that of [18F]FET as previously reported using the same assay conditions and cell line.24 (S)-[18F]14 is not a good substrate for system L, which is somewhat surprising given that aromatic substituted amino acids such as [18F]FAMT and [123I]IMT have been reported by other groups to be effective system L substrates.20,34,35 In contrast, our in vitro cell uptake studies demonstrate that the presence or absence of an α-methyl group on the amino acid can dramatically change the mechanism of transport of this class of α-alkyl amino acids.
Although it is not clear if these results can be extended to aromatic amino acids, our previous finding that α-[18F]-fluoromethyl phenylalanine (FMePhe) was a poor system L substrate in the same DBT model suggests that these results may be applicable to amino acids with aromatic side chains.36 It is also possible that a species difference (mouse vs human system L transporters) in substrate recognition may in part explain this discrepancy with reports by other groups, but the limited data available do not indicate that there are known species differences for substrate recognition by system L transporters.37,38
4. Biodistribution Studies in Mice with Subcutaneous DBT Glioma
Table 1 shows the biodistribution of (S)-[18F]14 and (S)-[18F]15 in BALB/c mice implanted with subcutaneous DBT tumors at 5, 30, and 60 min postinjection (p.i.). There was good tumor uptake at all time points, with the highest uptake observed with the α-hydrogen substituted amino acid, [18F]15, at 60 min p.i. At each time point, the tumor uptake was statistically significantly higher than the normal brain uptake (p = 0.02 or less) with the exception of the 5 min time point for (S)-[18F]15 due to variability in the tumor uptake between mice.
Table 1.
Biodistribution of (S)-[18F]14 and (S)-[18F]15 in BALB/c Mice with Subcutaneous DBT Tumorsa
| organ | 5 min
|
30 min
|
60 min
|
|||
|---|---|---|---|---|---|---|
| (S)-[18F]15 | (S)-[18F]14 | (S)-[18F]15 | (S)-[18F]14 | (S)-[18F]15 | (S)-[18F]14 | |
| blood | 4.8 ± 0.4 | 6.0 ± 0.9 | 3.8 ± 0.2 | 4.8 ± 2.6 | 3.7 ± 0.2 | 2.4 ± 0.8 |
| bone | 2.9 ± 0.2 | 1.9 ± 0.5 | 2.4 ± 0.5 | 2.0 ± 0.3 | 2.9 ± 0.3 | 1.8 ± 0.6 |
| brain | 3.6 ± 0.6 | 0.4 ± 0.1 | 4.3 ± 0.4 | 0.4 ± 0.03 | 4.2 ± 0.3 | 0.5 ± 0.2 |
| fat | 0.8 ± 0.1 | 1.1 ± 0.3 | 0.8 ± 0.1 | 0.8 ± 0.3 | 0.8 ± 0.1 | 0.5 ± 0.2 |
| heart | 6.9 ± 0.3 | 3.0 ± 0.5 | 5.3 ± 0.4 | 2.4 ± 0.5 | 4.9 ± 0.3 | 2.4 ± 0.5 |
| kidney | 7.4 ± 1.3 | 51.8 ± 5.2 | 7.4 ± 0.7 | 31.2 ± 5.8 | 7.0 ± 0.5 | 13.1 ± 2.3 |
| liver (all) | 5.4 ± 0.6 | 5.2 ± 1.1 | 4.0 ± 0.3 | 4.3 ± 0.7 | 4.2 ± 0.1 | 2.7 ± 0.9 |
| lung | 4.8 ± 0.4 | 6.4 ± 1.0 | 3.7 ± 0.3 | 3.6 ± 0.6 | 3.6 ± 0.3 | 2.3 ± 0.6 |
| muscle | 3.5 ± 0.3 | 1.6 ± 0.2 | 3.8 ± 0.2 | 1.6 ± 0.4 | 3.6 ± 0.2 | 1.5 ± 0.3 |
| pancreas | 32.7 ± 2.8 | 31.1 ± 3.0 | 28.9 ± 4.1 | 39.9 ± 10.5 | 29.3 ± 2.6 | 27.6 ± 6.9 |
| spleen | 7.0 ± 0.4 | 5.3 ± 1.0 | 4.6 ± 0.4 | 4.3 ± 0.2 | 4.6 ± 0.6 | 2.9 ± 0.7 |
| thyroid | 4.4 ± 0.7 | 3.4 ± 0.7 | 4.9 ± 0.6 | 2.1 ± 0.4 | 5.4 ± 1.0 | 2.2 ± 0.6 |
| tumor | 5.2 ± 2.1 | 4.2 ± 1.0 | 6.3 ± 1.4 | 5.4 ± 0.5 | 9.1 ± 1.0 | 4.8 ± 1.6 |
32 μCi (1.18 MBq) of (S)-[18F]14 and 28 μCi (1.04 MBq) of (S)-[18F]15 were administrated via tail vein injection without anesthesia. The animals were euthanized after 5, 30, and 60 min. Each time point is the average of 5 mice except for the 30 min time point for (S)-[18F]14 (n = 3) and the 60 min time point for (S)-[18F]15 (n = 4). The data are expressed as mean percent of total injected dose per gram of tissue (%ID/g) with standard deviation.
The tumor uptake of (S)-[18F]15 was rapid with 5.2 ± 2.2 % ID/g at 5 min and increased over the course of the study to 6.3 ± 1.4 %ID/g at 30 min and 9.1 ± 1.0 %ID/g at 60 min. For (S)-[18F]14, similar tumor uptake was observed at 5 min with a value of 4.2 ± 0.98 %ID/g. However, the uptake of (S)-[18F]14 did not increase to the same extent over time as with (S)-[18F] 15, and was relatively constant over time with 5.4 ± 0.54 %ID/g at 30 min p.i. and 4.8 ± 1.6 %ID/g at 60 min p.i. At 1 h p.i. the tumor uptake of (S)-[18F]15 was almost 2-fold higher than that of (S)-[18F]14 (p < 0.001).
While their tumor uptakes were similar, (S)-[18F]15 showed much higher uptake in normal brain (3.6 to 4.3 %ID/g) than (S)-[18F]14 (0.4 to 0.5 %ID/g) with an approximately 8- to 10-fold difference throughout the study (p < 0.001 at all time points). These biodistribution results are in agreement with the cell uptake study results, which demonstrated that (S)-[18F]15 was a good substrate for system L amino acid transport and thus was expected to cross the BBB. In contrast, (S)-[18F]14 was not an in vitro substrate for system L transport and would not be predicted to cross the BBB as was borne out in the biodistribution and small animal PET studies.
A large difference in uptake between (S)-[18F]15 and (S)-[18F]14 was also observed in the kidneys. Renal uptake of (S)-[18F]15 was essentially constant over time at 7.0 to 7.4 %ID/g. However, for (S)-[18F]14, the renal uptake was 52 ± 5.2 %ID/g at 5 min p.i. and decreased to 31 ± 5.8 %ID/g at 30 min p.i. and to 13 ± 2.3 %ID/g at 60 min p.i. The blood uptake of (S)-[18F]14 was 6.0 ± 0.9 %ID/g at 5 min p.i. and decreased over time to 2.4 ± 0.8 %ID/g at 60 min p.i. In contrast, the blood activity associated with (S)-[18F]15 was fairly constant with 3.7 and 3.8 %ID/g at 30 and 60 min p.i., respectively. Although not tested directly, these results suggest that (S)-[18F]15 is reabsorbed into the blood after renal filtration with slower elimination than with (S)-[18F]14.
The bone uptake of (S)-[18F]15 was slightly higher than with (S)-[18F]14 but was constant up to 60 min p.i., indicating that minimal in vivo defluorination occurred over the time course of the study. These results observed with the α-hydrogen amino acid (S)-[18F]15 differ from a previously reported analogue with a shorter aliphatic chain, 3-(1-[18F]fluoromethyl)-L-alanine (L-[18F]FMA), which showed rapid defluorination with progressively increasing bone uptake.39 The uptake of (S)-[18F]15 was approximately 2-fold higher in the heart, muscle, and thyroid compared to (S)-[18F]14, while the uptake of both tracers was similar in the liver, lung, and spleen. The relatively high uptake observed in the pancreas for both tracers is typical of radiolabeled amino acids.
To further evaluate the suitability of (S)-[18F]15 as a system L tracer, a comparison was made with (S)-[18F]FET, a well-established system L substrate. (S)-[18F]FET is a metabolically stable 18F-labeled tyrosine derivative that has been used extensively for human neuro-oncologic imaging.40–42 The results of the biodistribution study with (S)-[18F]FET in BALB/c mice with subcutaneous DBT tumors at 5, 30, and 60 min p.i. are presented in Table 2.
Table 2.
Biodistribution of (S)-[18F]FET in BALB/c Mice with Subcutaneous DBT Tumorsa
| organ | [18F]FET
|
||
|---|---|---|---|
| 5 min | 30 min | 60 min | |
| blood | 5.4 ± 0.3 | 3.7 ± 0.2 | 3.4 ± 3.3 |
| bone | 2.2 ± 0.3 | 1.4 ± 0.1 | 1.5 ± 1.3 |
| brain | 1.5 ± 0.3 | 2.8 ± 0.2 | 2.5 ± 2.4 |
| fat | 1.0 ± 0.2 | 0.8 ± 0.2 | 0.8 ± 0.6 |
| heart | 5.3 ± 0.3 | 3.3 ± 0.1 | 3.0 ± 3.0 |
| kidney | 5.6 ± 0.1 | 4.1 ± 0.3 | 4.4 ± 4.2 |
| liver(all) | 4.6 ± 0.3 | 3.1 ± 0.1 | 2.9 ± 2.9 |
| lung | 5.0 ± 0.4 | 3.3 ± 0.2 | 2.9 ± 2.9 |
| muscle | 3.9 ± 0.2 | 3.2 ± 0.1 | 3.1 ± 2.9 |
| pancreas | 26.9 ± 5.5 | 38.2 ± 7.9 | 20.1 ± 18.0 |
| spleen | 6.5 ± 0.3 | 4.1 ± 0.2 | 3.5 ± 3.2 |
| thyroid | 4.4 ± 0.3 | 3.4 ± 0.2 | 2.8 ± 2.7 |
| tumor | 5.9 ± 0.7 | 6.7 ± 2.0 | 7.4 ± 7.4 |
34 μCi (1.26 MBq) of (S)-[18F]FET was administrated via tail vein injection without anesthesia. The animals were euthanized at 5 min (n = 5), 30 min (n = 5), and 60 min (n = 4) after injection. The data are expressed as mean %ID/g with standard deviation.
(S)-[18F]FET and (S)-[18F]15 showed a similar biodistribution pattern at all time points with the absolute uptake of (S)-[18F]15 modestly higher than (S)-[18F]FET in most tissues. The normal brain uptake of (S)-[18F]15 was significantly higher than that of (S)-[18F]FET at all time points (p < 0.001). The reason for this difference in normal brain uptake is not clear but could reflect differences in recognition of 15 and FET by system L transporter family members at the BBB. The higher normal brain uptake of (S)-[18F]15 led to slightly better tumor to brain ratios for (S)-[18F]FET, particularly at early time points, although this difference did not reach statistical significance at any time point in the study. In contrast, the tumor to blood and tumor to muscle ratios observed with (S)-[18F]15 and (S)-[18F]FET were very similar at all time points. The tumor to brain ratios observed with (S)-[18F]14 were much higher than with (S)-[18F]15 and (S)-[18F]FET (p < 0.01 at all time points for both compounds) due to the much lower uptake of (S)-[18F]14 in normal brain. The tumor to blood, tumor to brain, and tumor to muscle ratios are presented in Table 3.
Table 3.
Tumor to Blood, Tumor to Brain, and Tumor to Muscle Ratios Observed after Injection of (S)-[18F]FET, (S)-[18F]15, and (S)-[18F]14 Calculated from the Biodistribution Studies in Mice with Subcutaneous DBT Tumorsa
| time, min | [18F]FET | (S)-[18F]15 | (S)-[18F]14 | |
|---|---|---|---|---|
| tumor to blood ratio | 5 | 1.1 ± 0.2 | 1.1 ± 0.4 | 0.7 ± 0.2 |
| 30 | 1.8 ± 0.5 | 1.7 ± 0.4 | 1.4 ± 0.7 | |
| 60 | 2.2 ± 0.2 | 2.4 ± 0.2 | 2.1 ± 0.9 | |
| tumor to brain ratio | 5 | 3.9 ± 0.8 | 1.4 ± 0.6 | 11.1 ± 3.2 |
| 30 | 2.4 ± 0.7 | 1.4 ± 0.3 | 14.2 ± 2.4 | |
| 60 | 3.0 ± 0.2 | 2.2 ± 0.2 | 9.6 ± 4.4 | |
| tumor to muscle ratio | 5 | 1.5 ± 0.2 | 1.4 ± 0.5 | 2.7 ± 0.7 |
| 30 | 2.1 ± 0.6 | 1.7 ± 0.4 | 3.7 ± 1.4 | |
| 60 | 2.5 ± 0.2 | 2.5 ± 0.3 | 3.2 ± 1.0 |
5. Small Animal PET/CT Studies in Mice with Intracranial DBT Tumors
Small animal PET/CT studies were performed with (S)-[18F]14, (S)-[18F]15, and (S)-[18F]FET to confirm the biodistribution results and to compare the imaging properties of these compounds in orthotopic brain tumors. The (S)-[18F]FET-PET data used for comparison were collected and reported previously.18 Overall, the results from the small animal PET studies were very similar to the results from the biodistribution studies. The time–activity curves (TACs) for [18F]14 and [18F]15 are depicted in Figure 5. (S)-[18F]15 showed a peak tumor uptake of radioactivity within 20 min and then slowly decreased over time. In contrast, (S)-[18F] 14 had peak tumor uptake of radioactivity within the first 5 min followed by slow washout. The absolute uptake of 15 was higher in the intracranial DBT gliomas than for 14 at all time points and in keeping with the biodistribution studies within mice with subcutaneous DBT tumors.
Figure 5.
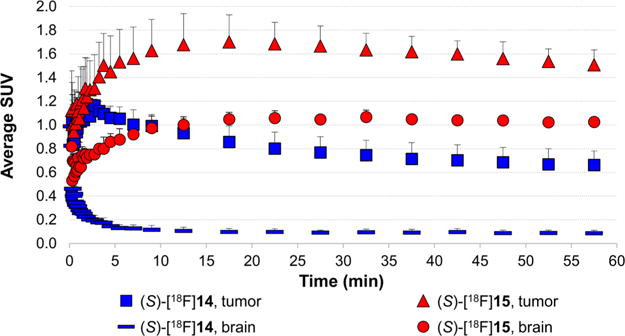
Time–activity curves of (S)-[18F]14 and (S)-[18F]15 uptake in intracranial DBT tumors and contralateral normal brain. Mice were anesthetized with 1% isoflurane/oxygen for 0–60 min dynamic small animal PET/CT scans. Intravenous injection of 150–200 μCi (5.55–7.4 MBq) of (S)-[18F]14 (n = 4) or (S)-[18F]15 (n = 3) was done in different sets of mice. The data are displayed as average SUVs.
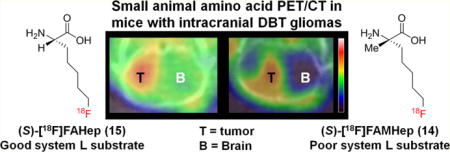
The mean standardized uptake value (SUV) in the tumor with (S)-[18F]15 at 5–15 min p.i. was 1.6 ± 0.3 and was significantly higher than with (S)-[18F]FET (mean SUV = 1.1 ± 0.02, p < 0.05) and with (S)-[18F]14 (mean SUV = 1.0 ± 0.1, p < 0.01). At 45–60 min after injection, the mean SUV in the tumor with (S)-[18F]15 was 1.5 ± 0.1 and was also significantly higher than with (S)-[18F]FET (mean SUV = 1.1 ± 0.1, p < 0.01) and with (S)-[18F]14 (mean SUV = 0.7 ± 0.1, p < 0.01). In the normal brain, the highest uptake at 5–15 min and 45–60 min p.i. was observed with (S)-[18F]15 with values of 1.0 ± 0.1 and 1.0 ± 0.02. The normal brain values were significantly higher for (S)-[18F]15 compared to (S)-[18F]FET at both of these time points (p < 0.001). Both of these system L substrates had much higher normal brain uptake than (S)-[18F]14 (p < 0.01 at both time points). At both 5–15 min and 45–60 min p.i., (S)-[18F]14 demonstrated much higher tumor to brain ratios than (S)-[18F]15 and (S)-[18F]FET due to the very low normal brain uptake of (S)-[18F]14. As in the biodistribution studies, (S)-[18F]FET had slightly higher tumor to brain ratios at 5–15 min and 45–60 min p.i. (mean ratios of 2.9 and 2.0, respectively) compared to (S)-[18F]15 (mean ratios of 1.7 and 1.5, respectively), but these differences did not reach statistical significance, likely due to the small sample size. Representative images at 45–60 min after tracer injection are shown in Figures 6 and 7. These data are depicted graphically as mean SUVs in Figure 8 and as tumor to brain ratios in Figure 9.
Figure 6.
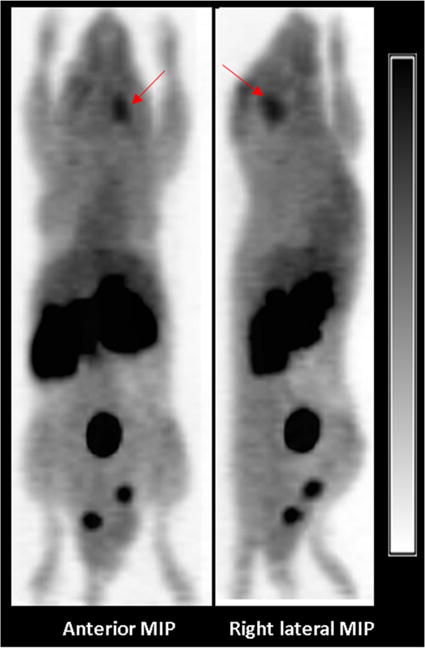
Representative anterior and right lateral maximum intensity projection (MIP) PET images of obtained 50–60 min after injection of (S)-[18F]15. The intracranial DBT tumor is designated by the red arrow. High levels of activity are seen in the abdomen and pelvis corresponding to the kidneys, pancreas, and the urinary bladder.
Figure 7.
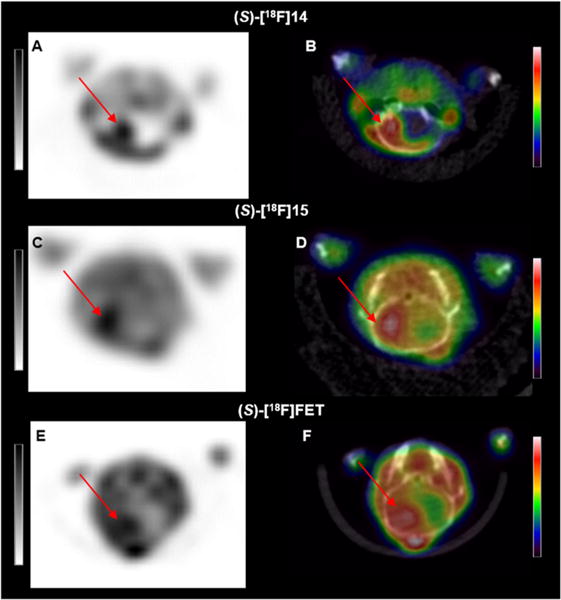
Representative small animal PET (A, C, E) and fused PET/CT (B, D, F) axial images of mice with intracranial DBT gliomas acquired 45–60 min after injection of (S)-[18F]14 (A, B), (S)-[18F]15 (C, D), or (S)-[18F]FET (E, F). All three tracers had relatively high uptake in the glioma (red arrows), but (S)-[18F]14 had substantially less uptake in the contralateral normal brain. Note that intensity scales are different and optimized for each tracer, reflecting the different concentrations of each tracer in the tumor and normal brain.
Figure 8.
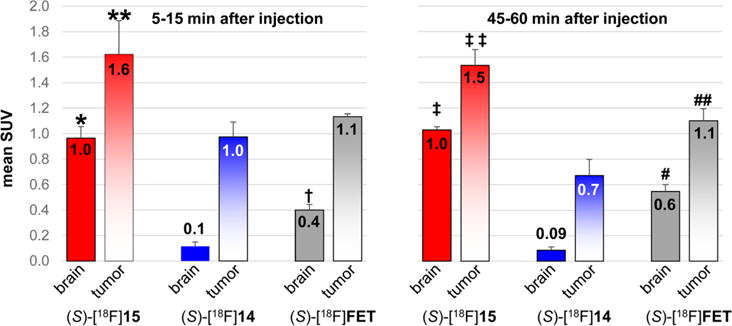
Mean SUVs of the tumor and normal brain uptake from small animal PET studies performed with (S)-[18F]14 in four mice, (S)-[18F]15 in three mice, and (S)-[18F]FET in three mice at early (5–15 min) and late (45–60 min) time points after injection. Errors bars show standard deviation. p values represent comparisons of tumor and brain uptakes at early and late time points for (S)-[18F]14, (S)-[18F]15, and (S)-[18F]FET through 1-way ANOVA with Tukey post-tests.*, p < 0.001 for 15 vs FET, p < 0.001 for 15 vs 14, **, p < 0.05 for 15 vs FET, p < 0.01 for 15 vs 14, †, p < 0.01 for FET vs 14, ‡, p < 0.001 for 15 vs FET and 15 vs 14, ‡‡, p < 0.01 for 15 vs FET, p < 0.001 for 15 vs 14, #, p < 0.001 for FET vs 14, ##, p < 0.01 for FET vs 14.
Figure 9.
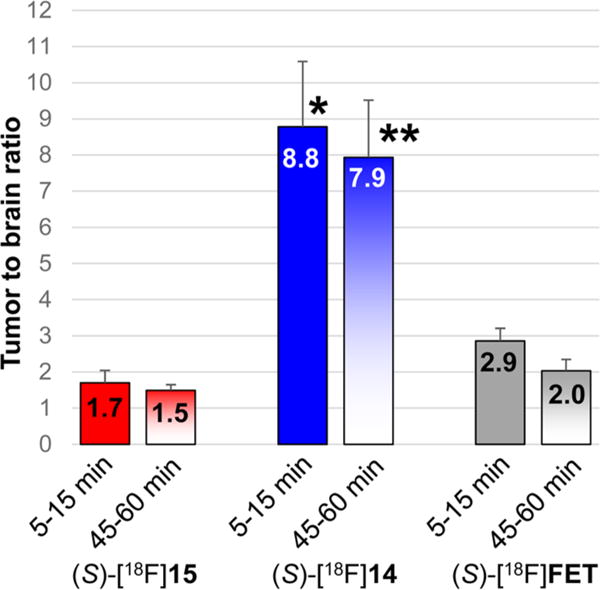
Tumor to brain ratios from small animal PET studies performed with (S)-[18F]14 in four animals, (S)-[18F]15 in three animals, and (S)-[18F]FET in three mice, at early (5–15 min) and late (45–60 min) time points after injection. Errors bars show standard deviations. p values represent comparisons of the tumor:brain ratios for (S)-[18F]14, (S)-[18F]15, and (S)-[18F]FET through 1-way ANOVA with Tukey post-tests. *, p < 0.001 for 14 vs 15 and vs FET, **, p < 0.001 for 14 vs 15 and FET.
Although (S)-[18F]14 demonstrated good tumor uptake and much higher tumor to normal brain ratios than (S)-[18F]15 and (S)-[18F]FET in both the biodistribution and the PET studies, this very low brain uptake likely will prevent visualization of regions of gliomas with intact BBBs and may limit its clinical utility for imaging gliomas. The higher normal brain uptake of (S)-[18F]15 and (S)-[18F]FET is consistent with transport by system L, and this property allows visualization of the entire tumor volume including nonenhancing regions of tumor. Of all the tracers evaluated, (S)-[18F]15 had the highest uptake in both normal brain and DBT gliomas at the early and late time points in the PET studies. Interestingly, (S)-[18F]FET had slightly higher tumor to brain ratios than (S)-[18F]15 due to higher uptake of (S)-[18F]15 in normal brain, although this difference did not reach statistical significance.
CONCLUSION
Biological comparison of the novel 18F-labeled amino acids (S)-[18F]14 and (S)-[18F]15 demonstrate that the α-methyl group plays a key role in the recognition of this class of amino acids by system L amino acid transporters. The in vitro results demonstrated that (S)-[18F]15 was a very good substrate for system L amino acid while (S)-[18F]14 was not, and these results were supported by the biodistribution and small animal imaging studies in mice with DBT gliomas. This result is somewhat surprising given the reported brain availability of radiolabeled aromatic amino acids with α-methyl substitutions. (S)-[18F]15 had much higher uptake in normal brain than (S)-[18F]14 as well as higher absolute tumor uptake, although [18F] 14 had higher tumor to brain ratios due to the very low normal brain uptake of [18F]14. The biodistribution and imaging properties of (S)-[18F]15 were very similar to those of (S)-[18F]FET, a well-established system L substrate. (S)-[18F]15 had higher normal brain and tumor uptake than (S)-[18F]FET in the PET studies, but the tumor to brain ratios were slightly higher with (S)-[18F]FET due to the higher normal brain uptake of (S)-[18F]15. These data indicate that the presence of an α-methyl group on α,α-dialkyl substituted amino acids decreases recognition by system L transporters compared to their α-hydrogen analogues.
EXPERIMENTAL SECTION
Materials and Instrumentation
All reagents and materials were purchased from commercially available sources. Chemicals and solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA), Sigma Chemical Co. (St. Louis, MO, USA), Ryan Scientific Inc. (Mount Pleasant, SC, USA), Ark Pharm (Libertyville, IL, USA), and Advanced Chemtech Co. (Louisville, KY, USA). Chromatography was carried out using silica gel 60 (0.040–0.063 mm) from EMD Millipore (EMD Millipore USA). TLC analyses were performed with 200 μm UV254 silica gel backing on aluminum sheets (EMD Millipore USA). The TLC plates were revealed with ninhydrin and/or iodide stains. Sep-Pak C18 Plus Short Cartridge, Sep-Pak Plus Silica Cartridge, and Oasis HLB Plus Light cartridge were purchased from Waters, Inc. (Milford, MA, USA). Dionex OnGuard II A cartridge was purchased from Fisher Scientific (Pittsburgh, PA USA). Melting points were measured with an IA9100X1 series digital melting point apparatus (ThermoFisher Scientific, USA) in capillary tubes and are uncorrected.
1H and 13C NMR spectra were recorded on either a 300 or 400 MHz NMR spectrometer (Varian/Agilent, Santa Clara, CA, USA) maintained by the Washington University High Resolution NMR Facility. Chemical shifts (δ values) are reported as parts per million (ppm), and coupling values are reported in hertz. Elemental analyses, performed by Atlantic Microlabs, Inc. (Norcross, GA, USA), were conducted on certain key nonradioactive intermediates in the multistep synthesis as well as on the hydrochloride salts of the nonradioactive final amino acid products (R,S)-6 and (R,S)-12, and the results were within 0.4% of theoretical values unless otherwise stated. High resolution mass spectra (HRMS) were obtained with a Bruker Maxis Q-ToF mass spectrometer using high-resolution electron ionization at the Chemistry Department of Washington University in St. Louis. The purity of the final 18F-labeled amino acids used for biological studies was measured using radio-TLC and analytical HPLC. Chiral HPLC was performed using a Chirex 3126 D-penicillamine column (15 cm × 4.6 mm) with a mobile phase consisting of 85:15 3 mM copper sulfate (CuSO4):acetonitrile, a flow rate of 1 mL/min, detection at λ = 254 nm, and a column temperature of 25 °C. This analysis demonstrated that the 18F-labeled tracers used in biological studies were at least 95% pure. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA) with p values of 0.05 or less considered statistically significant.
Chemistry
tert-Butyl (S)-2-((((9H-Fluoren-9-yl)methoxy)-carbonyl)amino)-2-methylhept-6-enoate, (S)-1
tert-Butyl 2,2,2-trichloroacetate (0.37 mL, 2.04 mmol) was added to a solution of Fmoc-(S)-2-(4-pentenyl)alanine (257 mg, 0.68 mmol) in 4 mL of anhydrous DCM. The mixture was stirred at rt for 18 h, and a precipitate was formed. The mixture was concentrated to dryness under reduced pressure, and the crude compound was purified by silica gel column chromatography eluted with 10% ethyl acetate in hexane to give (S)-1 as a colorless oil (79%).
1H NMR (CDCl3)
δ 1.03–1.34 (m, 2H), 1.48 (s, 9H), 1.57 (s, 3H), 1.57–1.82 (m, 1H), 1.93–2.30 (m, 3H), 4.18–4.44 (m, 3H), 4.92–5.04 (m, 2H), 5.70–5.84 (m, 1H), 7.31 (t, J = 7.55 Hz, 2H), 7.40 (t, J = 7.55 Hz, 2H), 7.61 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H).
13C NMR (CDCl3)
δ 23.7 (CH2), 23.8 (CH3), 28.0 (3CH3), 33.6 (CH2), 35.9 (CH2), 47.4 (CH), 60.3 (C), 66.3 (CH2), 82.2 (C), 115.0 (CH2═), 120.1 (2CH), 125.2 (CH═), 127.2 (2CH), 127.8 (2CH), 138.4 (2CH), 141.4 (2C), 144.1 (2C), 154.4 (C═O), 173.7 (C═O).
HRMS (ESI)
m/z calculated for C27H33NO4 + Na [M + Na]+: 458.23025. Found: 458.2301.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-2-methylhept-6-enoate, (S)-2
Compound 1 (229 mg, 0.53 mmol) was solubilized in 4 mL of DMF. Under a nitrogen atmosphere, first potassium fluoride (215.5 mg, 3.71 mmol) and then trimethylamine (0.15 mL, 1.11 mmol) were added to the solution. Then, di-tert-butyl dicarbonate (151 mg, 0.69 mmol) was added. The reaction was followed by TLC, and after several hours the reaction mixture was diluted with ethyl acetate (40 mL) and washed with water (20 mL) three times. Then, the organic layer was washed with 5% HCl, 5% sodium bicarbonate (NaHCO3), and saturated sodium chloride (NaCl) solutions, dried over sodium sulfate (Na2SO4), filtered, and concentrated to dryness under reduced pressure. The crude compound was purified by silica gel column chromatography eluted with 10% ethyl acetate in hexane to give (S)-2 as a colorless oil (78%).
1H NMR (CDCl3)
δ 1.12–1.37 (m, 2H), 1.41 (s, 9H), 1.44 (s, 9H), 1.48 (s, 3H), 1.62–1.77 (m, 1H), 1.92–2.18 (m, 3H), 4.85–5.04 (m, 2H), 5.34 (bs, 1H), 5.65–5.82 (m, 1H).
13C NMR (CDCl3)
δ 23.5 (CH3, CH2), 28.0 (3CH3), 28.5 (3CH3), 33.6 (CH2), 36.3 (CH2), 59.8 (C), 81.6 (2C), 114.9 (CH2═), 138.4 (CH═), 154.3 (C═O), 173.8 (C═O).
Elemental analysis calculated (%) for C17H31NO4: C 65.14, H 9.97, N 4.47. Found: C 65.41, H 9.98, N 4.40.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-7-hydroxy-2-methylheptanoate, (S)-3
Borane–THF solution (1.48 mL of 1.0 M solution, 1.48 mmol) was slowly added to a solution of 2 (124 mg, 0.40 mmol) in 5 mL of anhydrous THF under nitrogen at 0 °C. The resulting solution was stirred for 2 h at 0 °C. Then, still at 0 °C, a solution of 1 M sodium hydroxide (NaOH) (1.48 mL, 1.48 mmol) was added slowly, followed by aqueous hydrogen peroxide 30% (0.63 mL, 5.48 mmol). The mixture was stirred overnight and allowed to return to rt. Ethyl acetate (20 mL) and brine (20 mL) were added, and the two layers were separated. The aqueous phase was extracted with ethyl acetate (2 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated to dryness under reduced pressure. The crude compound was purified by silica gel column chromatography eluted with 40% ethyl acetate in hexane to give (S)-3 as a colorless oil (54%).
1H NMR (CDCl3)
δ 1.25–1.39 (m, 4H), 1.43 (s, 9H), 1.45 (s, 9H), 1.49 (s, 3H), 1.52–1.74 (m, 3H), 2.08 (bs, 1H), 3.62 (t, J = 6.5 Hz, 2H), 5.35 (bs, 1H).
13C NMR (CDCl3)
δ 23.6 (CH2), 23.9 (CH3), 25.8 (CH2), 28.0 (3CH3), 28.5 (3CH3), 32.6 (CH2), 36.7 (CH2), 59.8 (C), 62.8 (CH2), 81.6 (2C), 154.3 (C═O), 173.8 (C═O).
Elemental analysis calculated (%) for C17H33NO5
C 61.60, H 10.04, N 4.23. Found: C 61.67, H 10.10, N 4.04.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-2-methyl-7-(tosyloxy)heptanoate, (S)-4
Sodium tert-butoxide (142 mg, 1.48 mmol) was added to a solution of the alcohol 3 (222 mg, 0.67 mmol) and p-toulenesulfonyl chloride (192 mg, 1.0 mmol) in 8 mL of anhydrous DCM at 0 °C under a nitrogen atmosphere. After 5 min of stirring at 0 °C, the ice bath was removed, and the reaction mixture was stirred overnight at rt. The mixture was then concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography eluted with 20% ethyl acetate in hexane to provide (S)-4 as a colorless oil (36%). Some starting material was recovered after the overnight reaction (32%).
1H NMR (CDCl3)
δ 1.15–1.33 (m, 4H), 1.41 (s, 9H), 1.44 (s, 9H), 1.45 (s, 3H), 1.57–1.69 (m, 3H), 2.03 (bs, 1H), 2.45 (s, 3H), 3.99 (t, J = 6.5 Hz, 2H), 5.31 (bs, 1H), 7.34 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.3 Hz, 2H).
13C NMR (CDCl3)
δ 21.8 (CH3), 23.6 (CH3, CH2), 25.4 (CH2), 28.0 (3CH3), 28.5 (3CH3), 28.8 (CH2), 59.7 (C), 70.5 (CH2), 81.8 (2C), 128.0 (2CH), 130.0 (2CH), 133.2 (C), 144.8 (C), 154.3 (C═O), 173.7 (C═O).
HRMS (ESI)
m/z calculated for C24H39NO7S + Na [M + Na]: 508.2339. Found: 508.2359.
(S)-2-((tert-Butoxycarbonyl)amino)hept-6-enoic Acid, (S)-5
The starting material (S)-2-amino-6-heptenoic acid (500 mg, 3.49 mmol) was suspended in 11 mL of methanol:trimethylamine:1 N NaOH (9/1/1: v/v/v), and di-tert-butyl dicarbonate (1.52 g, 6.96 mmol) was added in one portion. The reaction mixture was stirred at rt overnight. The organic solvent was removed under reduced pressure, and 20 mL of ethyl acetate and 15 mL of water were added. The pH of the aqueous phase was adjusted to 2 with 3 N HCl with stirring. The organic layer was retained while the aqueous layer was saturated with NaCl and extracted with ethyl acetate (3 × 10 mL). The combined organic phases were dried over sodium sulfate, filtered, and concentrated to dryness under reduced pressure. The product (S)-5 was obtained as a colorless oil (66%) and was used without further purification.
1H NMR (CDCl3)
δ 1.45 (s, 3H), 1.46–1.57 (m, 2H), 1.59–1.75 (m, 1H), 1.77–1.95 (m, 1H), 2.02–2.14 (m, 2H), 4.25–4.37 (m, 1H), 4.94–5.06 (m, 2H), 5.68–5.85 (m, 1H).
13C NMR (CDCl3)
δ 24.7 (CH2), 28.4 (3CH3), 31.9 (CH2), 33.3 (CH2), 53.4 (CH), 80.4 (C), 115.3 (CH2═), 138.0 (CH═), 155.8 (C═O), 177.4 (C═O).
HRMS (ESI)
m/z calculated for C12H21NO4 + Na [M + Na]+: 266.1363. Found: 266.1382.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)hept-6-enoate, (S)-6
To a solution of the N-Boc acid (S)-1 (528 mg, 2.17 mmol) in 10 mL of anhydrous DCM was added tert-butyl-2,2,2-trichloroacetimidate (1.17 mL, 6.51 mmol). After overnight stirring at rt, the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography eluted with 20% ethyl acetate in hexane to give (S)-6 as a colorless oil (97%).
1H NMR (CDCl3)
δ 1.43 (s, 9H), 1.45 (s, 9H), 1.47–1.81 (m, 4H), 2.00–2.13 (m, 2H), 4.09–4.21 (m, 1H), 4.91–5.04 (m, 3H), 5.68–5.83 (m, 1H).
13C NMR (CDCl3)
δ 24.5 (CH2), 28.1 (3CH3), 28.4 (3CH3), 32.5 (CH2), 33.3 (CH2), 53.9 (CH), 79.7 (C), 81.8 (C), 115.0 (CH2═), 138.2 (CH═), 155.5 (C═O), 172.1 (C═O).
HRMS (ESI)
m/z calculated for C16H29NO4 + Na [M + Na]+: 322.1989. Found: 322.2019.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-7-hydroxyheptanoate, (S)-7
(S)-7 was obtained with the same procedure that provided (S)-3 except that the crude compound (S)-7 was purified by silica gel column chromatography eluted initially with 20% ethyl acetate in hexane followed by 40% ethyl acetate in hexane to give (S)-7 as a colorless oil (84%).
1H NMR (CDCl3)
δ 1.26–1.41 (m, 4H), 1.44 (s, 9H), 1.46 (s, 9H), 1.49–1.66 (m, 3H), 1.70–1.84 (m, 1H), 3.63 (t, J = 6.5 Hz, 2H), 4.12–4.22 (m, 1H), 5.03 (d, J = 7.7 Hz, 1H).
13C NMR (CDCl3)
δ 24.9 (CH2), 25.3 (CH2), 28.2 (3CH3), 28.5 (3CH3), 32.6 (CH2), 33.2 (CH2), 53.8 (CH), 62.7 (CH2), 79.8 (C), 81.9 (C), 155.6 (C═O), 172.2 (C═O).
Elemental analysis calculated (%) for C16H31NO5
C 60.54, H 9.84, N 4.41. Found: C 60.50, H 9.98, N 4.24.
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-7-(tosyloxy)-heptanoate, (S)-8
(S)-8 was obtained with the same procedure that provided (S)-4. The crude product (S)-8 was also purified by silica gel column chromatography eluted with 20% ethyl acetate in hexane to provide (S)-8 as a colorless oil (37%). Some starting material was recovered after the overnight reaction (45%).
1H NMR (CDCl3)
δ 1.22–1.37 (m, 4H), 1.43 (s, 9H), 1.45 (s, 9H), 1.51–1.77 (m, 4H), 2.45 (s, 3H), 4.01 (t, J = 6.4 Hz, 2H), 4.08–4.15 (m, 1H), 4.98 (d, J = 7.7 Hz, 1H), 7.34 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H).
13C NMR (CDCl3)
δ 21.8 (CH3), 24.7 (CH2), 25.2 (CH2), 28.2 (3CH3), 28.5 (3CH3), 28.8 (CH2), 32.9 (CH2), 53.9 (CH), 70.5 (CH2), 79.8 (C), 82.0 (C), 110.2 (C), 128.0 (2CH), 130.0 (2CH), 133.3 (C), 144.8 (C═O), 172.0 (C═O).
Elemental analysis calculated (%) for C23H37NO7S
C 58.58, H 7.91, N 2.97. Found: C 58.46, H 7.96, N 2.96.
tert-Butyl 2-((Diphenylmethylene)amino)acetate, (R,S)-9
Benzophenone imine (1.0 mL, 5.96 mmol) was added to a solution of glycine tert-butyl ester hydrochloride (1.0 g, 5.96 mmol) in 15 mL of DCM. The mixture was stirred at rt for 2 h. A white precipitate was formed. The mixture was diluted with water, and two phases were separated. The aqueous phase was extracted with DCM (2 × 20 mL). The combined organic layers were washed with a saturated solution of NaCl (1 × 25 mL), dried over Na2SO4, filtered, and finally concentrated to dryness under reduced pressure. (R,S)-9 was obtained as a white solid (95%), which was used without further purification. Mp: 104–106 °C.
1H NMR (CDCl3)
δ 1.46 (s, 9H), 4.12 (s, 2H), 7.17–7.20 (m, 2H), 7.29–7.49 (m, 6H), 7.65–7.67 (m, 2H).
13C NMR (CDCl3)
28.2 (3CH3), 56.5 (CH2), 81.2 (C), 127.8 (2CH), 128.1 (2CH), 128.7 (2CH), 128.9 (2CH), 130.5 (2CH), 136.3 (C), 139.5 (C), 170.0 (C═N), 171.6 (C═O).
HRMS (ESI)
m/z calculated for C19H21NO2 + H [M + H]: 296.1645. Found: 296.1729.
tert-Butyl (R,S)-2-((Diphenylmethylene)amino)-7-fluoroheptanoate, (R,S)-10
Imine (R,S)-9 (500 mg, 1.69 mmol) solubilized in 1 mL of THF was added to a solution of LDA (1.86 mL of 2.0 M solution in THF, heptane, ethylbenzene, 3.72 mmol) in 6 mL of anhydrous THF at −78 °C under nitrogen. An orange enolate solution resulted. The mixture was stirred at −78 °C for 1 h, and then the alkylating agent 1-bromo-5-fluoropentane (0.46 mL, 3.72 mmol) was added. The solution was stirred overnight and allowed to return to rt. The reaction was quenched with the addition of a saturated solution of ammonium chloride (NH4Cl) (10 mL) and diluted with Et2O (15 mL). Two phases were separated. The aqueous phase was extracted with Et2O (2 × 15 mL). The combined organic layers were washed with a saturated solution of NaCl (1 × 20 mL), dried over Na2SO4, filtered, and concentrated to dryness under reduced pressure. The crude compound was purified by silica gel column chromatography eluted with 2% ethyl acetate in hexane to give (R,S)-10 as a colorless oil (43%).
1H NMR (CDCl3)
δ 1.27–1.37 (m, 4H), 1.45 (s, 9H), 1.55–1.73 (m, 2H), 1.87–1.93 (m, 2H), 3.92 (t, J = 6.5 Hz, 1H), 4.40 (dt, 2J (H,F) = 47.5 Hz, 3J (H,H) = 6.1 Hz, 2H), 7.16–7.19 (m, 2H), 7.30–7.47 (m, 6H), 7.64–7.67 (m, 2H).
13C NMR (CDCl3)
δ 25.0 (d, 3J (C,F) = 5.5 Hz, CH2), 25.7 (CH2), 28.2 (3CH3), 30.3 (d, 2J (C,F) = 19.7 Hz, CH2), 33.6 (CH2), 66.0 (CH), 81.0 (C), 84.1 (d, 1J (C,F) = 164.4 Hz, CH2), 127.9 (2CH), 128.1 (2CH), 128.5 (2CH), 128.8 (2CH), 130.3 (2CH), 136.8 (C), 139.8 (C), 170.0 (C═N), 171.6 (C═O).
tert-Butyl (R,S)-2-Amino-7-fluoroheptanoate, (R,S)-12
Hydroxylamine hydrochloride (557 mg, 8.01 mmol) was added to a solution of (R,S)-10 (846 mg, 2.29 mmol) in 20 mL of anhydrous methanol under nitrogen at rt. After 2 h, the reaction was complete. The mixture was then concentrated under reduced pressure. Water (20 mL) and DCM (40 mL) were added, and the two phases were separated. The aqueous phase was basified to pH = 7.5 by using a saturated solution of K2CO3 and then extracted with DCM (2 × 40 mL). The combined organic layers were washed with a solution of NaCl saturated (20 mL), dried over Na2SO4, filtered, and concentrated to dryness under reduced pressure. The crude compound was purified by silica gel column chromatography; it was eluted first with 20% ethyl acetate in hexane and then with 10% methanol in DCM to provide the free amine (R,S)-12 as a yellow oil (64%).
1H NMR (CDCl3)
δ 1.39–1.44 (m, 4H), 1.46 (s, 9H), 1.51–1.75 (m, 6H), 3.29–3.32 (m, 1H), 4.43 (dt, 2J (H,F) = 47.4 Hz, 3J (H,H) = 6.1 Hz, 2H).
tert-Butyl (R,S)-2-Amino-7-fluoro-2-methylheptanoate, (R,S)-13
(R,S)-13 was obtained in two steps. First, (R,S)-10 (384 mg, 1.0 mmol) solubilized in 1 mL of THF was added to a solution of LDA (1.0 mL of 2.0 M solution in THF, heptane, ethylbenzene, 2.2 mmol) in 6 mL of anhydrous THF at −78 °C under nitrogen. An orange enolate solution resulted. The mixture was stirred at −78 °C for 1 h, and then the alkylating agent methyl iodide (0.17 mL, 2.2 mmol) was added. The solution was stirred overnight and allowed to return to rt. The reaction was quenched with the addition of a saturated solution of NH4Cl (10 mL) and diluted with Et2O (15 mL). Two phases were separated. The aqueous phase was extracted with Et2O (2 × 15 mL). The combined organic layers were washed with a saturated solution of NaCl (1 × 20 mL), dried over Na2SO4, filtered, and concentrated to dryness under reduced pressure. The crude compound was partially purified on silica gel column chromatography eluted with 10% ethyl acetate in hexane to give (R,S)-11 (115 mg) as a crude oil, which was used without any further purification for the second step. In the second step, hydroxylamine hydrochloride (70 mg, 1.01 mmol) was added to a solution of (R,S)-11 (115 mg) solubilized in 5 mL of anhydrous methanol under nitrogen. The solution was stirred at rt and for 2 h. The mixture was then concentrated under reduced pressure. Water (10 mL) and DCM (20 mL) were added, and the two phases were separated. The aqueous phase was basified to pH = 7.5 by using a saturated solution of K2CO3 and then extracted with DCM (2 × 20 mL). The combined organic layers were washed with a saturated solution of NaCl (20 mL), dried over Na2SO4, filtered, and concentrated to dryness under reduced pressure. The crude compound was purified by silica gel column chromatography first eluted with 10% ethyl acetate in hexane followed by 10% methanol in DCM to provide the free amine (R,S)-13 as a colorless oil (total yield was 17%).
1H NMR (CDCl3)
δ 1.27 (s, 3H), 1.31–1.43 (m, 4H), 1.45 (s, 9H), 1.61–1.76 (m, 6H), 4.43 (dt, 2J (H,F) = 47.4 Hz, 3J (H,H) = 6.1 Hz, 2H).
13C NMR (CDCl3)
δ 23.9 (CH2), 25.6 (d, 3J (C,F) = 6.3 Hz, CH2), 26.6 (CH3), 28.1 (3CH3), 30.4 (d, 2J (C,F) = 20.5 Hz, CH2), 40.9 (CH2), 58.0 (C), 80.9 (C), 84.1 (d, 1J (C,F) = 162.6 Hz, CH2), 177.0 (C═O).
HRMS (ESI)
m/z calculated for C12H24NO2F + H [M + H]+: 234.1864. Found: 234.1924.
(R,S)-2-Amino-7-fluoro-2-methylheptanoic Acid Hydrochloride, (R,S)-14
The amino ester (R,S)-13 (69 mg, 0.29 mmol) was stirred in 1 mL of 4 M HCl at 60 °C for 3 h. The mixture was then concentrated under reduced pressure. The residue obtained was triturated in Et2O to precipitate the desired compound. After filtration, product (R,S)-14 was obtained as a white solid (34 mg, 55%). Mp: 165–167 °C.
1H NMR (D2O)
δ 1.27–1.53 (m, 4H), 1.58 (s, 3H), 1.66–2.02 (m, 4H), 4.53 (dt, 2J (H,F) = 47.3 Hz, 3J (H,H) = 6.1 Hz, 2H).
13C NMR (D2O)
δ 21.7 (CH3), 22.5 (CH2), 24.2 (d, 3J (C,F) = 5.5 Hz, CH2), 29.0 (d, 2J (C,F) = 18.8 Hz, CH2), 36.5 (CH2), 60.2 (C), 85.2 (d, 1J (C,F) = 157.3 Hz, CH2F), 174.5 (C).
HRMS (ESI)
m/z calculated for C8H17NO2F - Cl [M - Cl ]: 178.1238. Found: 178.1286.
Elemental analysis calculated (%) for C8H17ClFNO2
C 44.97, H 8.02, N 6.56. Found: C 44.97, H 8.05, N 6.42.
(S)-14 was obtained from (S)-16 following the same procedure that provided (R,S)-14. The 1H NMR spectrum of (S)-14 agrees with the 1H NMR of (R,S)-14 as shown below.
1H NMR (D2O)
δ 1.21–1.47 (m, 4H), 1.52 (s, 3H), 1.62–1.97 (m, 4H), 4.49 (dt, 2J (H,F) = 47.3 Hz, 3J (H,H) = 6.0 Hz, 2H).
(R,S)-2-Amino-7-fluoroheptanoic Acid Hydrochloride, (R,S)-15
(R,S)-15 was obtained from (R,S)-12 following the same procedure that provided (R,S)-14. After filtration, product (R,S)-15 was obtained (28 mg, 48%) as a yellow solid. Mp: 227 °C.
1H NMR (D2O)
δ 1.37–1.56 (m, 4H), 1.66–1.83 (m, 2H), 1.84–2.02 (m, 2H), 3.91 (t, J = 6.2 Hz, 1H), 4.54 (dt, 2J (H,F) = 47.0 Hz, 3J (H,H) = 6.0 Hz, 2H).
HRMS (ESI)
m/z calculated for C7H15NO2F – Cl [M – Cl ]: 164.1081. Found: 164.1108.
Elemental analysis calculated (%) for C7H15ClFNO2
C 42.11, H 7.57, N 7.02. Found: C 41.89, H 7.20, N 6.52.
(S)-15 was obtained from (S)-17 following the same procedure that provided (R,S)-15. The 1H NMR spectrum of (S)-15 agrees with the 1H NMR of (R,S)-15 as shown below.
1H NMR (D2O)
δ 1.35–1.56 (m, 4H), 1.65–1.83 (m, 2H), 1.85–2.06 (m, 2H), 3.97 (t, J = 6.3 Hz, 1H), 4.54 (dt, 2J (H,F) = 47.2 Hz, 3J (H,H) = 6.0 Hz, 2H).
13C NMR (D2O)
δ 23.7 (CH2), 24.0 (d, 3J (C,F) = 5.9 Hz, CH2), 29.1 (d, 2J (C,F) = 18.2 Hz, CH2), 29.7 (CH2), 53.4 (CH), 85.3 (d, 1J (C,F) = 154.4 Hz, CH2), 173.1 (C═O).
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-5-fluoro-2-methyl-heptanoate, (S)-16
Cesium fluoride (139 mg, 1.20 mmol) was added to a solution of the tosylate (S)-4 (118 mg, 0.24 mmol) in 2–4 mL of anhydrous 2-methylbutan-2-ol. The mixture was stirred at 100 °C for 1h. Then, the reaction mixture was triturated with diethyl ether (Et2O) to remove most of the ionic salts. The filtrate was concentrated under reduced pressure, and the crude product was purified on silica gel column chromatography and eluted with 10% ethyl acetate in hexane to provide (S)-16 as a colorless oil (73%).
1H NMR (CDCl3)
δ 1.24–1.41 (m, 4H), 1.42 (s, 9H), 1.45 (s, 9H), 1.49 (s, 3H), 1.61–1.75 (m, 3H), 2.11 (bs, 1H), 4.38 (dt, 2J (H,F) = 47.4 Hz, 3J (H,H) = 6.8 Hz, 2H), 5.36 (bs, 1H).
13C NMR (CDCl3)
δ 23.7 (CH2), 23.8 (CH3), 25.2 (d, 3J (C,F) = 5.1 Hz, CH2), 28.0 (3CH3), 28.5 (3CH3), 30.3 (d, 2J (C,F) = 18.9 Hz, CH2), 36.5 (CH2), 59.8 (C), 81.7 (2C), 84.1 (d, 1J (C,F) = 162.1 Hz, CH2), 154.3 (C═O), 173.8 (C═O).
tert-Butyl (S)-2-((tert-Butoxycarbonyl)amino)-7-fluorohepta-noate, (S)-17
(S)-17 was obtained with the same procedure that provided (S)-16. The crude product was also purified on silica gel column chromatography and eluted with 10% ethyl acetate in hexane to provide (S)-17 as a colorless oil (75%).
1H NMR (CDCl3)
δ 1.28–1.42 (m, 4H), 1.44 (s, 9H), 1.46 (s, 9H), 1.56–1.84 (m, 4H), 4.10–4.21 (m, 1H), 4.43 (dt, 2J (H,F) = 47.1 Hz, 3J (H,H) = 6.2 Hz, 2H), 5.01 (d, J = 7.7 Hz, 1H).
13C NMR (CDCl3)
δ 24.9 (CH2), 25.0 (d, 3J (C,F) = 5.4 Hz, CH2), 28.1 (3CH3), 28.5 (3CH3), 30.3 (d, 2J (C,F) = 19.8 Hz, CH2), 33.0 (CH2), 54.0 (CH), 79.7 (C), 81.9 (C), 84.1 (d, 1J (C,F) = 164.3 Hz, CH2), 155.5 (C═O), 172.1 (C═O).
HRMS (ESI)
m/z calculated for C16H30NO4F + Na [M + Na]: 342.2051. Found: 342.2094.
Radiochemistry
[18F]fluoride was produced using an RDS-11 cyclotron or a TR-19 cyclotron in the Washington University Cyclotron Facility. [18F]fluoride was produced from [18O]water and the 18O(p,n)18F reaction. Typical radiosyntheses began with approximately 50 mCi of [18F]fluoride. First, the [18F]fluoride was dried by azeotropic distillation using acetonitrile (2 × 1 mL, then 2 × 0.5 mL) in the presence of K2CO3 (0.5 mg in 25 μL of water) and K2.2.2 (3 mg in 50 μL of anhydrous acetonitrile) between 96 and 105 °C under a flow of nitrogen. Then, a solution of the appropriate tosylate precursor (S)-4 or (S)-8 (4–5 mg in 500 μL of anhydrous 2-methylbutan-2-ol) was added, and the mixture was heated for 10 min between 96 and 105 °C in a sealed vessel. Incorporation of [18F]fluoride was estimated at greater than 90% for both tracers (n = 5) using radio-TLC developed in 100% methanol. Then, 1 mL of acetonitrile and 1 mL of water were added to the mixture, and the solution was purified using an Agilent Zorbax SB-C18 semipreparative high-performance liquid chromatography (HPLC) column (25 cm × 9.4 mm × 5 μm) eluted with a gradient mobile phase: solution A (30% acetonitrile, 70% water, 0.1% trifluoroacetic acid) and solution B (80% acetonitrile, 20% water, 0.1% trifluoroacetic acid) starting from 50% solution A, 50% solution B and transitioning to 100% solution B in 30 min, with a flow rate of 4 mL/min and λ = 230 nm. In these conditions, intermediates (S)-[18F]16 and (S)-[18F]17 had retention times of 25 and 18 min, respectively. The eluted HPLC fractions containing the labeled intermediate were combined and diluted with ~40 mL of water and passed through an Oasis HLB Plus Light cartridge; after 2 more washes, (S)-[18F]16 or (S)-[18F]17 was eluted with very small volumes of ethanol (between 200 and 300 μL). Then, the deprotection was performed by heating in the presence of aqueous acid at 96 to 105 °C for 20 min. In the case of (S)-[18F]16, a 500 μL solution of 1 M HCl was used for deprotection, but for (S)-[18F]17 a 500 μL solution of 0.5 M sulfuric acid was used. These conditions provided the desired products (S)-[18F]14 and (S)-[18F]15, respectively. After cooling, the acidic solution was diluted in 0.5 mL of sterile water and passed through a Dionex OnGuard II A cartridge. The cartridge was then eluted one or two times with 0.5 mL aliquots of sterile water until all the tracer was recovered from the cartridge. This method provided the final products (S)-[18F]14 or (S)-[18F]15 at pH = 5–7 and in a form suitable for cell uptake studies. For the animal studies, the final product was formulated in a 0.9% saline solution prior to injection.
The final product identity as well as the radiochemical and enantiomeric purity was assessed by coinjection of the racemic nonradioactive standard, either (R,S)-14 or (R,S)-15 with the (S)-[18F]14 or (S)-[18F]15 respectively onto a chiral analytical column: the Chirex 3126 D-penicillamine column (15 cm × 4.6 mm) using a solution of 85:15 3 mM CuSO4:acetonitrile as mobile phase with a flow rate of 1 mL/min and λ = 254 nm. (R,S)-14 and (R,S)-15 had retention times of 16 and 12 min, respectively. The specific activity was also determined using these analytical HPLC conditions. The mass associated with 18F-labeled product was determined by comparison of the integrated UV absorbance with a calibrated mass/UV absorbance standard curve of the nonradioactive standard (S)-14 for (S)-[18F]14 and (S)-15 for (S)-[18F]15. The specific activity of the final product was greater than 1 Ci/μmol at end of synthesis.
The radiosynthesis of (S)-[18F]FET was performed based on the procedure of K. Hamacher et al.33 with minor modifications of the drying step of [18F]fluoride, the elution of (S)-[18F]FET from the silica cartridge, and the HPLC purification. [18F]fluoride was produced and dried as it was done for (S)-[18F]14 and (S)-[18F]15. To the reaction vessel containing dried [18F]fluoride, a solution of 2.5 mg of O-(2-tosyloxyethyl)-N-trityl-L-tyrosine tert-butyl ester solubilized in 0.5 mL of acetonitrile was added, and the reaction mixture was heated between 96 and 105 °C for 5 min. The [18F]fluoride incorporation was superior to 96% determined by TLC. After evaporating the acetonitrile of the reaction mixture under a nitrogen flow maintaining the temperature between 96 and 105 °C, the deprotection was performed in a solution of 0.65 mL of 1,2-dichloroethane and 0.3 mL of trifluoroacetic acid for 2 min at rt and then 4 min at 80 °C. Then, 5 mL of DCM was added to the reaction mixture, and the solution was passed through a silica cartridge which trapped the crude (S)-[18F]FET. By using 8 mL of a solution of pentane/diethyl ether, 1/1, the cartridge was washed from any residual of trifluoroacetic acid, triphenylcarbinol, and halogenated solvents. 10 mL of air was then passed through the cartridge, and crude (S)-[18F]FET was eluted with two 1 mL aliquots of water. The collected solution, at pH = 5, was passed through a 0.45 μm filter and injected into the HPLC system for purification using a Chirobiotic TAG column (25 cm × 10 mm × 5 μm) with a mobile phase consisting of a solution of 95:5 water:ethanol with a flow of 3 mL/min and λ = 220 nm. The fractions containing (S)-[18F]FET collected from the HPLC purification had a retention time of 15–17 min and was adjusted to a solution of 0.9% NaCl, passed through a 0.2 μm filter and used directly for animal studies. The pH of the final product was at 7.
The identity as well as the radiochemical and enantiomeric purity of the final (S)-[18F]FET product was assessed by coinjection of the nonradioactive FET onto a chiral analytical column: the Chirex 3126 D-penicillamine column (15 cm × 4.6 mm) using a solution of 85:15 3 mM CuSO4:acetonitrile as mobile phase with a flow rate of 1.5 mL/min and λ = 254 nm. (S)-[18F]FET had a retention time of 25 min under these conditions. The specific activity was also determined using these analytical HPLC conditions. The mass associated with 18F-labeled product was determined by comparison of the integrated UV absorbance with a calibrated mass/UV absorbance standard curve of the nonradioactive standard. The specific activity of (S)-[18F]FET was greater than 1 Ci/μmol at end of synthesis.
Cell Uptake Assays
Cell uptake assays were performed using our previously reported procedure.24,43 Mouse DBT glioma cells were used in these assays. Two buffer conditions (one with and one without sodium) were used for the assays. The following inhibitors were used for the cell uptake assays: N-methyl-α-aminoisobutyric acid (MeAIB, 10 mM), a mixture of L-alanine/L-serine/L-cysteine (ASC, 3.3 mM of each amino acid to make the 10 mM solution, or 0.33 mM of each amino acid to make the 1 mM solution), and finally (R,S)-(endo,exo)-2-aminobicyclo(2,2,1)-heptane-2-carboxylic acid (BCH, 10 mM). The control conditions contained 10 mM of sucrose to maintain consistent osmolality. The assays were performed as at pH 7.40 with each condition performed in 8 replicates. Briefly, cells were washed twice with 37 °C assay buffer (2 mL) prior to initiating the assay. For both (S)-[18F]14 and (S)-[18F]15, solutions containing approximately 2.0 mCi/mL (64 MBq/mL) were prepared in the appropriate assay buffer, and then 20 μL of the tracer was added to appropriate buffer with or without inhibitors. Cells were incubated with radiotracers in assay buffer (0.4 mL total volume) under control or inhibitor conditions for 60 s at 37 °C. The assay buffer was then discarded from each well followed by washes (3 × 2 mL) with ice-cold buffer to remove extracellular radiotracer. The cells were lysed with 0.2% sodium dodecyl sulfate (SDS)/0.2 M NaOH (0.3 mL) at rt for 30 min. A 100 μL portion of the lysate from each well was counted to determine the amount of radioactivity taken up by the cells, and 3 × 20 μL portions were used for determination of protein content using the bicinchoninic acid (BCA) method (Pierce, BCA Protein Assay Kit). Standard dilutions of each assay condition were counted to determine the amount of activity added to each well. The amount of radioactivity per well was normalized based on the amount of radioactivity added and the protein content of each cell. The uptake data were expressed as percent of uptake relative to control, and the uptake of each tracer in the various conditions was compared to the sodium sucrose control with a one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison post-tests using GraphPad Prism software. The choline sucrose control condition was compared to the choline BCH condition using a 2-sided t test to evaluate specificity for system L transport.
DBT Tumor Model
All animal experiments were conducted following the Institutional Animal Care and Use Committee approved protocols in compliance with the Guide for the Care and Use of Research Animals. For the unilateral intracranial tumors, mouse DBT tumor cells (1 × 104 cells suspended in a volume of 8 μL) were implanted in the right midcerebrum of male BALB/C mice (24–26 g) as described previously.44,45 For subcutaneous tumors, DBT cells (5 × 105 cells suspended in a volume of 50 μL) were injected subcutaneously into the flanks of male BALB/c mice (23–30 g).46 Tumor-bearing animals were used for imaging and biodistribution studies 14 days after implantation.
Biodistribution Studies with (S)-[18F]14 and (S)-[18F]15 in BALB/c Mice with Subcutaneous DBT Glioma
The same procedure was used to evaluate the biodistribution of (S)-[18F]14, (S)-[18F]15, and (S)-[18F]FET in tumor-bearing mice. Approximately 32 μCi (1.2 MBq) of (S)-[18F]14, 28 μCi (1.0 MBq) of (S)-[18F]15, or 34 μCi (1.3 MBq) of (S)-[18F]FET was administrated via tail vein injection in conscious animals. Groups of five animals were euthanized at 5, 30, and 60 min p.i. In a few animals, the subcutaneous tumors did not grow, and these animals were excluded from analysis. The tumor and tissues of interest were collected and weighed, and the radioactivity was measured using an automated Beckman Gamma 8000 well counter with a standard dilution of the injection. The raw counts were corrected for background and radioactive decay, and the results were reported as the percent of total injected dose per gram of tissue (%ID/g). For each tracer at each time point, the tumor uptake versus normal brain uptake was compared using two-tailed paired t tests with p values less than 0.05 considered significant. The %ID/g in tumor and normal brain as well as tumor to brain ratios for all three tracers were compared separately at each time point using one-way ANOVAs with Tukey post-tests using GraphPad Prism Software with p values less than 0.05 considered statistically significant.
Small Animal PET/CT Studies in Mice with Intracranial DBT Tumors
Male BALB/c mice with unilateral intracranial DBT tumors were placed in an induction chamber containing ~2% isoflurane/oxygen and then secured to a custom 4 mouse bed for placement of tail vein catheters; anesthesia was maintained via nose cone at ~1% isoflurane/oxygen for the imaging procedures. The mice underwent dynamic small animal PET imaging from 0 to 60 min after intravenous tail injection of 150–200 μCi (5.55–7.4 MBq) of (S)-[18F]14 (n = 4 mice) or (S)-[18F]15 (n = 3 mice) as well as computed tomography (CT) images using an Inveon PET/CT system (Siemens Medical Solutions Inc.). Body temperature was maintained with a heating lamp. At the conclusion of the imaging studies, the animals were euthanized, and their brains were fixed in 4% paraformaldehyde for histologic analysis with hematoxylin and eosin staining to verify the presence and location of tumor.
The small animal PET data obtained with (S)-[18F]14 and (S)-[18F] 15 were analyzed by manually drawing three-dimensional regions of interest (ROIs) over the areas of tumor identified on the PET studies and the contralateral normal brain using the Inveon Research Workplace software package (Siemens, Inc.). Previously reported PET data for (S)-[18F]FET was also included in this analysis.18 The uptake data were expressed as average standardized uptake values (SUVs) for each ROI. The average SUVs and the tumor to brain ratios at 5–15 and 45–60 min p.i. after injection of (S)-[18F]14, (S)-[18F]15, and (S)-[18F]FET were used for comparative analysis. The uptake in the brain and tumor and the tumor to brain ratios obtained with the three tracers were compared individually at both time points compared through one-way ANOVA with Tukey post-tests using the GraphPad prism software with p values less than 0.05 considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful for the assistance of the personnel of the Washington University Cyclotron Facility for [18F]fluoride production. This research was supported through grants from the National Cancer Institute (K08CA154790) and Department of Energy (SC0004832). We thank also the Siteman Cancer Center Small Animal Imaging Core for the use of the Pre-Clinical PET/CT Imaging Facility, which provided biodistribution and small animal PET/CT services. The Siteman Cancer Center is funded in part through the National Cancer Institute (P30CA091842). The Siemens Inveon scanner was acquired through an NIH High-End Instrumentation grant (S10 RR 025097). We would like to thank the Department of Chemistry staff and the Washington University High Resolution NMR Facility for assistance with NMR spectra; purchase of the 400 MHz NMR instrument was partially supported by S10 RR027207 from the NIH Shared Instrument Grant program. The mass spectral data from Washington University Mass Spectrometry Resource were supported by grants from the National Institute of General Medical Sciences (8 P41 GM103422-35) from the National Institutes of Health.
ABBREVIATIONS
- ANOVA
analysis of variance
- ASC
alanine, cysteine, and serine
- BCA
bicinchoninic acid
- BBB
blood–brain barrier
- BCH
2-aminobicyclo[2.2.1]heptane-2-carboxylic acid
- Boc
tert-butoxycarbonyl
- bs
broad signal
- CDCl3
deuterated chloroform
- CT
computed tomography
- CuSO4
copper sulfate
- D2O
deuterated water
- DMEM
Dulbecco’s modified Eagle’s medium
- DBT
delayed brain tumor
- DCM
dichloromethane
- EOS
end of synthesis
- Et2O
diethyl ether
- FAHep
2-amino-7-fluoroheptanoic acid
- FAMHep
2-amino-7-fluoro-2-methylheptanoic acid
- FAMPe
2-amino-5-fluoro-2-methylpentanoic acid
- FBS
fetal bovine serum
- FDG
2-deoxy-2-fluoro-D-glucose
- FDOPA
6-fluoro-3,4-dihydroxy-L-phenylalanine
- FET
O-(2-fluoroethyl)-L-tyrosine
- Fmoc
9-fluorenylmethoxycarbonyl
- HCl
hydrochloric acid
- HLB
hydrophilic–lipophilic-balanced
- HPLC
high performance liquid chromatography
- ID
injected dose
- IMT
3-iodo-α-methyl-L-tyrosine
- K2.2.2
Kryptofix 2.2.2
- K2CO3
potassium carbonate
- LDA
lithium diisopropyl amide
- MeAIB
N-methyl α-aminoisobutyric acid
- MET
L-methionine
- MIP
maximum intensity projection
- NaCl
sodium chloride
- NaHCO3
sodium bicarbonate
- NaOH
sodium hydroxide
- Na2SO4
sodium sulfate
- NH4Cl
ammonium chloride
- PET
positron emission tomography
- p.i
postinjection
- ROIs
regions of interest
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- SUV
standardized uptake values
- TACs
time–activity curves
- TLC
thin-layer chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b00189.
SMILES data (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.McConathy J, Yu W, Jarkas N, Seo W, Schuster DM, Goodman MM. Radiohalogenated nonnatural amino acids as PET and SPECT tumor imaging agents. Med Res Rev. 2012;32:868–905. doi: 10.1002/med.20250. [DOI] [PubMed] [Google Scholar]
- 2.McConathy J, Goodman MM. Non-natural amino acids for tumor imaging using positron emission tomography and single photon emission computed tomography. Cancer Metastasis Rev. 2008;27:555–573. doi: 10.1007/s10555-008-9154-7. [DOI] [PubMed] [Google Scholar]
- 3.Huang C, McConathy J. Fluorine-18 labeled amino acids for oncologic imaging with positron emission tomography. Curr Top Med Chem. 2013;13:871–891. doi: 10.2174/1568026611313080002. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin. Cancer Biol. 2005;15:254–266. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Shennan DB, Thomson J. Inhibition of system L (LAT1/CD98hc) reduces the growth of cultured human breast cancer cells. Oncol Rep. 2008;20:885–889. [PubMed] [Google Scholar]
- 6.Nawashiro H, Otani N, Shinomiya N, Fukui S, Ooigawa H, Shima K, Matsuo H, Kanai Y, Endou H. L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int J Cancer. 2006;119:484–492. doi: 10.1002/ijc.21866. [DOI] [PubMed] [Google Scholar]
- 7.Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, Cha SH, Matsuo H, Fukushima J, Fukasawa Y, Tani Y, Taketani Y, Uchino H, Kim JY, Inatomi J, Okayasu I, Miyamoto K, Takeda E, Goya T, Endou H. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochim Biophys Acta, Biomembr. 2001;1514:291–302. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 8.Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98) J Biol Chem. 1998;273:23629–23632. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- 9.Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. 1999;274:19745–19751. doi: 10.1074/jbc.274.28.19745. [DOI] [PubMed] [Google Scholar]
- 10.Kanai Y, Endou H. Heterodimeric amino acid transporters: molecular biology and pathological and pharmacological relevance. Curr Drug Metab. 2001;2:339–354. doi: 10.2174/1389200013338324. [DOI] [PubMed] [Google Scholar]
- 11.Killian DM, Chikhale PJ. Predominant functional activity of the large, neutral amino acid transporter (LAT1) isoform at the cerebrovasculature. Neurosci Lett. 2001;306:1–4. doi: 10.1016/s0304-3940(01)01810-9. [DOI] [PubMed] [Google Scholar]
- 12.Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci USA. 1999;96:12079–12084. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Yan AT, Cwirla SE, Grindstaff KK. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008;155:423–438. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Qu W, Lieberman BP, Plössl K, Kung HF. Synthesis, uptake mechanism characterization and biological evaluation of 18F labeled fluoroalkyl phenylalanine analogs as potential PET imaging agents. Nucl Med Biol. 2011;38:53–62. doi: 10.1016/j.nucmedbio.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tripathi M, Sharma R, D’Souza M, Jaimini A, Panwar P, Varshney R, Datta A, Kumar N, Garg G, Singh D, Grover RK, Mishra AK, Mondal A. Comparative evaluation of F-18 FDOPA, F-18 FDG, and F-18 FLT-PET/CT for metabolic imaging of low grade gliomas. Clin Nucl Med. 2009;34:878–883. doi: 10.1097/RLU.0b013e3181becfe0. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Silverman DHS, Delaloye S, Czernin J, Kamdar N, Pope W, Satyamurthy N, Schiepers C, Cloughesy T. 18F-FDOPA PET imaging of brain tumors: Comparison study with 18F-FDG PET and evaluation of diagnostic accuracy. J Nucl Med. 2006;47:904–911. [PubMed] [Google Scholar]
- 17.Pauleit D, Stoffels G, Bachofner A, Floeth FW, Sabel M, Herzog H, Tellmann L, Jansen P, Reifenberger G, Hamacher K, Coenen HH, Langen KJ. Comparison of 18F-FET and 18F-FDG PET in brain tumors. Nucl Med Biol. 2009;36:779–787. doi: 10.1016/j.nucmedbio.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Sai KS, Huang C, Yuan L, Zhou D, Piwnica-Worms D, Garbow JR, Engelbach JA, Mach RH, Rich KM, McConathy J. 18F-AFETP, 18F-FET, and 18F-FDG imaging of mouse DBT gliomas. J Nucl Med. 2013;54:1120–1126. doi: 10.2967/jnumed.112.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchino H, Kanai Y, Kim DK, Wempe MF, Chairoungdua A, Morimoto E, Anders MW, Endou H. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): insights into the mechanisms of substrate recognition. Mol Pharmacol. 2002;61:729–737. doi: 10.1124/mol.61.4.729. [DOI] [PubMed] [Google Scholar]
- 20.Wiriyasermkul P, Nagamori S, Tominaga H, Oriuchi N, Kaira K, Nakao H, Kitashoji T, Ohgaki R, Tanaka H, Endou H, Endo K, Sakurai H, Kanai Y. Transport of 3-fluoro-L-alpha-methyltyrosine by tumor-upregulated L-type amino acid transporter 1: a cause of the tumor uptake in PET. J Nucl Med. 2012;53:1253–1261. doi: 10.2967/jnumed.112.103069. [DOI] [PubMed] [Google Scholar]
- 21.Morimoto E, Kanai Y, Kim DK, Chairoungdua A, Choi HW, Wempe MF, Anzai N, Endou H. Establishment and characterization of mammalian cell lines stably expressing human L-type amino acid transporters. J Pharmacol Sci. 2008;108:505–516. doi: 10.1254/jphs.08232fp. [DOI] [PubMed] [Google Scholar]
- 22.Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A, Giacomini KM. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc Natl Acad Sci USA. 2013;110:5480–5485. doi: 10.1073/pnas.1218165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.del Amo EM, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur J Pharm Sci. 2008;35:161–174. doi: 10.1016/j.ejps.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Bouhlel A, Zhou D, Li A, Yuan L, Rich KM, McConathy J. Synthesis, radiolabeling, and biological evaluation of (R)- and (S)-2-amino-5-[18F]fluoro-2-methylpentanoic acid ((R)-, (S)-[18F]FAMPe) as potential positron emission tomography tracers for brain tumors. J Med Chem. 2015;58:3817–3829. doi: 10.1021/jm502023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mourad PD, Farrell L, Stamps LD, Chicoine MR, Silbergeld DL. Why are systemic glioblastoma metastases rare? Systemic and cerebral growth of mouse glioblastoma. Surg Neurol. 2005;63:511–519. doi: 10.1016/j.surneu.2004.08.062. and discussion 519. [DOI] [PubMed] [Google Scholar]
- 26.Langen KJ, Hamacher K, Weckesser M, Floeth F, Stoffels G, Bauer D, Coenen HH, Pauleit D. O-(2-[18F]fluoroethyl)-L-tyrosine: uptake mechanisms and clinical applications. Nucl Med Biol. 2006;33:287–294. doi: 10.1016/j.nucmedbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Wester HJ, Herz M, Weber W, Heiss P, Senekowitsch-Schmidtke R, Schwaiger M, Stocklin G. Synthesis and radio-pharmacology of O-(2-[18F]fluoroethyl)-L-tyrosine for tumor imaging. J Nucl Med. 1999;40:205–212. [PubMed] [Google Scholar]
- 28.Wang L, Lieberman BP, Plossl K, Qu W, Kung HF. Synthesis and comparative biological evaluation of L- and D-isomers of 18F-labeled fluoroalkyl phenylalanine derivatives as tumor imaging agents. Nucl Med Biol. 2011;38:301–312. doi: 10.1016/j.nucmedbio.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Wen-Ren L, Jianjun J, Joullié MM. One-pot conversion of fluorenylmethyl carbamates into tert-butyl carbamates. Tetrahedron Lett. 1993;34:1413–1414. [Google Scholar]
- 30.Yu W, McConathy J, Williams L, Camp VM, Malveaux EJ, Zhang Z, Olson JJ, Goodman, M M. Synthesis, radiolabeling, and biological evaluation of (R)- and (S)-2-amino-3-[18F]fluoro-2-methylpropanoic acid (FAMP) and (R)- and (S)-3-[18F]fluoro-2-methyl-2-N-(methylamino)propanoic acid (NMeFAMP) as potential PET radioligands for imaging brain tumors. J Med Chem. 2010;53:876–886. doi: 10.1021/jm900556s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang T, Tang G, Wang H, Nie D, Tang X, Liang X, Hu K, Yi C, Yao B, Tang C. Synthesis and preliminary biological evaluation of S-11C-methyl-D-cysteine as a new amino acid PET tracer for cancer imaging. Amino Acids. 2015;47:719–727. doi: 10.1007/s00726-014-1899-4. [DOI] [PubMed] [Google Scholar]
- 32.Kim DW, Jeong HJ, Lim ST, Sohn MH, Katzenellenbogen JA, Chi DY. Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: Significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J Org Chem. 2008;73:957–962. doi: 10.1021/jo7021229. [DOI] [PubMed] [Google Scholar]
- 33.Hamacher K, Coenen HH. Efficient routine production of the 18F-labelled amino acid O-2-18F fluoroethyl-L-tyrosine. Appl Radiat Isot. 2002;57:853–856. doi: 10.1016/s0969-8043(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 34.Inoue T, Tomiyoshi K, Higuichi T, Ahmed K, Sarwar M, Aoyagi K, Amano S, Alyafei S, Zhang H, Endo K. Biodistribution studies on L-3-[fluorine-18]fluoro-alpha-methyl tyrosine: a potential tumor-detecting agent. J Nucl Med. 1998;39:663–667. [PubMed] [Google Scholar]
- 35.Prante O, Deichen JT, Hocke C, Kuwert T. Characterization of uptake of 3-[131I]iodo-alpha-methyl-L-tyrosine in human monocyte-macrophages. Nucl Med Biol. 2004;31:365–372. doi: 10.1016/j.nucmedbio.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Huang C, Yuan L, Rich KM, McConathy J. Radiosynthesis and biological evaluation of alpha-[F-18]fluoromethyl phenylalanine for brain tumor imaging. Nucl Med Biol. 2013;40:498–506. doi: 10.1016/j.nucmedbio.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain-Vakkalagadda B, Dey S, Pal D, Mitra AK. Identification and functional characterization of a Na+-independent large neutral amino acid transporter, LAT1, in human and rabbit cornea. Invest Ophthalmol Visual Sci. 2003;44:2919–2927. doi: 10.1167/iovs.02-0907. [DOI] [PubMed] [Google Scholar]
- 38.Wipf D, Ludewig U, Tegeder M, Rentsch D, Koch W, Frommer WB. Conservation of amino acid transporters in fungi, plants and animals. Trends Biochem Sci. 2002;27:139–147. doi: 10.1016/s0968-0004(01)02054-0. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Zha Z, Qu W, Qiao H, Lieberman BP, Plossl K, Kung HF. Synthesis and evaluation of 18F labeled alanine derivatives as potential tumor imaging agents. Nucl Med Biol. 2012;39:933–943. doi: 10.1016/j.nucmedbio.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gotz I, Grosu AL. [18F]FET-PET Imaging for Treatment and Response Monitoring of Radiation Therapy in Malignant Glioma Patients - A Review. Front Oncol. 2013;3:104. doi: 10.3389/fonc.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plotkin M, Blechschmidt C, Auf G, Nyuyki F, Geworski L, Denecke T, Brenner W, Stockhammer F. Comparison of F-18 FET-PET with F-18 FDG-PET for biopsy planning of non-contrast-enhancing gliomas. Eur Radiol. 2010;20:2496–2502. doi: 10.1007/s00330-010-1819-2. [DOI] [PubMed] [Google Scholar]
- 42.Weckesser M, Langen KJ, Rickert CH, Kloska S, Straeter R, Hamacher K, Kurlemann G, Wassmann H, Coenen HH, Schober O. O-(2-[18F]fluorethyl)-L-tyrosine PET in the clinical evaluation of primary brain tumours. Eur J Nucl Med Mol Imaging. 2005;32:422–429. doi: 10.1007/s00259-004-1705-8. [DOI] [PubMed] [Google Scholar]
- 43.McConathy J, Zhou D, Shockley SE, Jones LA, Griffin EA, Lee H, Adams SJ, Mach RH. Click synthesis and biologic evaluation of (R)- and (S)-2-amino-3-[1-(2-[18F]fluoroethyl)-1H-[1,2,3]triazol-4-yl]propanoic acid for brain tumor imaging with positron emission tomography. Mol Imaging. 2010;9:329–342. [PubMed] [Google Scholar]
- 44.Jost SC, Wanebo JE, Song SK, Chicoine MR, Rich KM, Woolsey TA, Lewis JS, Mach RH, Xu J, Garbow JR. In vivo imaging in a murine model of glioblastoma. Neurosurgery. 2007;60:360–370. doi: 10.1227/01.NEU.0000249264.80579.37. and discussion 370 ff. [DOI] [PubMed] [Google Scholar]
- 45.Jost SC, Collins L, Travers S, Piwnica-Worms D, Garbow JR. Measuring brain tumor growth: combined bioluminescence imaging-magnetic resonance imaging strategy. Mol Imaging. 2009;8:245–253. [PMC free article] [PubMed] [Google Scholar]
- 46.Ploessl K, Wang L, Lieberman BP, Qu W, Kung HF. Comparative evaluation of 18F-labeled glutamic acid and glutamine as tumor metabolic imaging agents. J Nucl Med. 2012;53:1616–1624. doi: 10.2967/jnumed.111.101279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.